
The Multiple Faces of the MRN Complex: Roles in Medulloblastoma and Beyond

 , ,
, ,
Simple Summary
Abstract
1. Introduction
2. MRN Complex Functions in Cellular Homeostasis
2.1. MRN Complex in DSBs and Replication Stress Handling
2.1.1. MRN Complex Defects as Risk Factor for Cancer Development and Assumption for a Synthetic Lethal-Based Strategy
2.1.2. MRN Complex as a Critical Factor in Resistance to Oncogene- and Therapy-Induced DSBs/RS
2.2. MRN Complex in Innate and Adaptive Immune Response
2.3. MRN Complex in Telomere Homeostasis
2.4. MRN Complex in Centrosome Maintenance and Mitotic Spindle Dynamic
2.5. MRN Complex and Cancer-Related Pathways
3. The Prognostic Significance of the MRN Complex Expression in Cancer
4. Medulloblastoma as a Model for the Pleiotropic Role of MRN Complex in Cancer
5. Future Perspective/Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chamankhah, M.; Xiao, W. Formation of the yeast Mre11-Rad50-Xrs2 complex is correlated with DNA repair and telomere maintenance. Nucleic Acids Res. 1999, 27, 2072–2079. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xiao, Y. Conditional gene targeted deletion by Cre recombinase demonstrates the requirement for the double-strand break repair Mre11 protein in murine embryonic stem cells. Nucleic Acids Res. 1997, 25, 2985–2991. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Yao, M.S.; Bender, C.F.; Mills, M.; Bladl, A.R.; Bradley, A.; Petrini, J.H.J. Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc. Natl. Acad. Sci. USA 1999, 96, 7376–7381. [Google Scholar] [CrossRef]
- Zhu, J.; Petersen, S.; Tessarollo, L.; Nussenzweig, A. Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr. Biol. 2001, 11, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Delia, D. MRE11 mutations and impaired ATM-dependent responses in an Italian family with ataxia-telangiectasia-like disorder. Hum. Mol. Genet. 2004, 13, 2155–2163. [Google Scholar] [CrossRef]
- Fiévet, A.; Bellanger, D.; Valence, S.; Mobuchon, L.; Afenjar, A.; Giuliano, F.; Dubois d’Enghien, C.; Parfait, B.; Pedespan, J.; Auger, N.; et al. Three new cases of ataxia-telangiectasia-like disorder: No impairment of the ATM pathway, but S-phase checkpoint defect. Hum. Mutat. 2019, 40, 1690–1699. [Google Scholar] [CrossRef]
- Stewart, G.S.; Maser, R.S.; Stankovic, T.; Bressan, D.A.; Kaplan, M.I.; Jaspers, N.G.J.; Raams, A.; Byrd, P.J.; Petrini, J.H.J.; Taylor, A.M.R. The DNA Double-Strand Break Repair Gene hMRE11 Is Mutated in Individuals with an Ataxia-Telangiectasia-like Disorder. Cell 1999, 99, 577–587. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Miyamoto, T.; Sakamoto, H.; Izumi, H.; Nakazawa, Y.; Ogi, T.; Tahara, H.; Oku, S.; Hiramoto, A.; Shiiki, T.; et al. Two unrelated patients with MRE11A mutations and Nijmegen breakage syndrome-like severe microcephaly. DNA Repair 2011, 10, 314–321. [Google Scholar] [CrossRef]
- Maser, R.S.; Zinkel, R.; Petrini, J.H.J. An alternative mode of translation permits production of a variant NBS1 protein from the common Nijmegen breakage syndrome allele. Nat. Genet. 2001, 27, 417–421. [Google Scholar] [CrossRef]
- Kruger, L.; Demuth, I.; Neitzel, H.; Varon, R.; Sperling, K.; Chrzanowska, K.H.; Seemanova, E.; Digweed, M. Cancer incidence in Nijmegen breakage syndrome is modulated by the amount of a variant NBS protein. Carcinogenesis 2007, 28, 107–111. [Google Scholar] [CrossRef]
- Barbi, G.; Scheres, J.M.J.C.; Schindler, D.; Taalman, R.D.F.M.; Rodens, K.; Mehnert, K.; Müller, M.; Seyschab, H. Chromosome instability and X-ray hypersensitivity in a microcephalic and growth-retarded child. Am. J. Med. Genet. 1991, 40, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Waltes, R.; Kalb, R.; Gatei, M.; Kijas, A.W.; Stumm, M.; Sobeck, A.; Wieland, B.; Varon, R.; Lerenthal, Y.; Lavin, M.F.; et al. Human RAD50 Deficiency in a Nijmegen Breakage Syndrome-like Disorder. Am. J. Hum. Genet. 2009, 84, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Qiu, S.; Huang, J. MRN complex is an essential effector of DNA damage repair. J. Zhejiang Univ. Sci. B 2021, 22, 31–37. [Google Scholar] [CrossRef]
- Bian, L.; Meng, Y.; Zhang, M.; Li, D. MRE11-RAD50-NBS1 complex alterations and DNA damage response: Implications for cancer treatment. Mol. Cancer 2019, 18, 169. [Google Scholar] [CrossRef] [PubMed]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Agarwal, H.; Priya, S.; Batra, H.; Modi, P.; Pandey, M.; Saha, D.; Raghavan, S.C.; Sengupta, S. MRN complex-dependent recruitment of ubiquitylated BLM helicase to DSBs negatively regulates DNA repair pathways. Nat. Commun. 2018, 9, 1016. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.R.; Mirzoeva, O.K.; Morgan, W.F.; Lin, J.; Dunnick, W.; Petrini, J.H.J. A Murine Model of Nijmegen Breakage Syndrome. Curr. Biol. 2002, 12, 648–653. [Google Scholar] [CrossRef]
- Cerosaletti, K.; Concannon, P. Independent Roles for Nibrin and Mre11-Rad50 in the Activation and Function of Atm. J. Biol. Chem. 2004, 279, 38813–38819. [Google Scholar] [CrossRef]
- Difilippantonio, S.; Celeste, A.; Fernandez-Capetillo, O.; Chen, H.-T.; Martin, B.R.S.; Laethem, F.V.; Yang, Y.-P.; Petukhova, G.V.; Eckhaus, M.; Feigenbaum, L.; et al. Role of Nbs1 in the activation of the Atm kinase revealed in humanized mouse models. Nat. Cell Biol. 2005, 7, 675–685. [Google Scholar] [CrossRef]
- Schiller, C.B.; Lammens, K.; Guerini, I.; Coordes, B.; Feldmann, H.; Schlauderer, F.; Möckel, C.; Schele, A.; Strässer, K.; Jackson, S.P.; et al. Structure of Mre11–Nbs1 complex yields insights into ataxia-telangiectasia–like disease mutations and DNA damage signaling. Nat. Struct. Mol. Biol. 2012, 19, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Huang, J. DNA End Resection: Facts and Mechanisms. Genom. Proteom. Bioinform. 2016, 14, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, R.A.; Myler, L.R.; Soniat, M.M.; Makharashvili, N.; Lee, L.; Lees-Miller, S.P.; Finkelstein, I.J.; Paull, T.T. DNA-dependent protein kinase promotes DNA end processing by MRN and CtIP. Sci. Adv. 2020, 6, eaay0922. [Google Scholar] [CrossRef]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef]
- Gatei, M.; Kijas, A.W.; Biard, D.; Dörk, T.; Lavin, M.F. RAD50 phosphorylation promotes ATR downstream signaling and DNA restart following replication stress. Hum. Mol. Genet. 2014, 23, 4232–4248. [Google Scholar] [CrossRef]
- Oakley, G.G.; Tillison, K.; Opiyo, S.A.; Glanzer, J.G.; Horn, J.M.; Patrick, S.M. Physical Interaction between Replication Protein A (RPA) and MRN: Involvement of RPA2 Phosphorylation and the N-Terminus of RPA1. Biochemistry 2009, 48, 7473–7481. [Google Scholar] [CrossRef]
- Aze, A.; Zhou, J.C.; Costa, A.; Costanzo, V. DNA replication and homologous recombination factors: Acting together to maintain genome stability. Chromosoma 2013, 122, 401–413. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Ray Chaudhuri, A.; Lopes, M.; Costanzo, V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 2010, 17, 1305–1311. [Google Scholar] [CrossRef]
- Ying, S.; Hamdy, F.C.; Helleday, T. Mre11-Dependent Degradation of Stalled DNA Replication Forks Is Prevented by BRCA2 and PARP1. Cancer Res. 2012, 72, 2814–2821. [Google Scholar] [CrossRef]
- Trenz, K.; Smith, E.; Smith, S.; Costanzo, V. ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO J. 2006, 25, 1764–1774. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, C.P.; Petrini, J.H.J. ISG15: A link between innate immune signaling, DNA replication, and genome stability. BioEssays 2023, 45, 2300042. [Google Scholar] [CrossRef]
- Wardlaw, C.P.; Petrini, J.H.J. ISG15 conjugation to proteins on nascent DNA mitigates DNA replication stress. Nat. Commun. 2022, 13, 5971. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Soans, E.; Mishina, M.; Petricci, E.; Pommier, Y.; Nitiss, K.C.; Nitiss, J.L. Requirements for MRN endonuclease processing of topoisomerase II-mediated DNA damage in mammalian cells. Front. Mol. Biosci. 2022, 9, 1007064. [Google Scholar] [CrossRef] [PubMed]
- Hartsuiker, E.; Neale, M.J.; Carr, A.M. Distinct Requirements for the Rad32Mre11 Nuclease and Ctp1CtIP in the Removal of Covalently Bound Topoisomerase I and II from DNA. Mol. Cell 2009, 33, 117–123. [Google Scholar] [CrossRef]
- Aparicio, T.; Baer, R.; Gottesman, M.; Gautier, J. MRN, CtIP, and BRCA1 mediate repair of topoisomerase II–DNA adducts. J. Cell Biol. 2016, 212, 399–408. [Google Scholar] [CrossRef]
- Hoa, N.N.; Shimizu, T.; Zhou, Z.W.; Wang, Z.-Q.; Deshpande, R.A.; Paull, T.T.; Akter, S.; Tsuda, M.; Furuta, R.; Tsutsui, K.; et al. Mre11 Is Essential for the Removal of Lethal Topoisomerase 2 Covalent Cleavage Complexes. Mol. Cell 2016, 64, 580–592. [Google Scholar] [CrossRef]
- Bartek, J.; Lukas, J.; Bartkova, J. DNA Damage Response as an Anti-Cancer Barrier: Damage Threshold and the Concept of “Conditional Haploinsufficiency”. Cell Cycle 2007, 6, 2344–2347. [Google Scholar] [CrossRef]
- Stracker, T.H.; Couto, S.S.; Cordon-Cardo, C.; Matos, T.; Petrini, J.H.J. Chk2 Suppresses the Oncogenic Potential of DNA Replication-Associated DNA Damage. Mol. Cell 2008, 31, 21–32. [Google Scholar] [CrossRef]
- Taylor, A.M.R.; Rothblum-Oviatt, C.; Ellis, N.A.; Hickson, I.D.; Meyer, S.; Crawford, T.O.; Smogorzewska, A.; Pietrucha, B.; Weemaes, C.; Stewart, G.S. Chromosome instability syndromes. Nat. Rev. Dis. Primers 2019, 5, 64. [Google Scholar] [CrossRef]
- Seemanová, E. An increased risk for malignant neoplasms in heterozygotes for a syndrome of microcephaly, normal intelligence, growth retardation, remarkable facies, immunodeficiency and chromosomal instability. Mutat. Res./Rev. Genet. Toxicol. 1990, 238, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Steffen, J.; Varon, R.; Mosor, M.; Maneva, G.; Maurer, M.; Stumm, M.; Nowakowska, D.; Rubach, M.; Kosakowska, E.; Ruka, W.; et al. Increased cancer risk of heterozygotes with NBS1 germline mutations in poland. Int. J. Cancer 2004, 111, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Stumm, M.; Neubauer, S.; Keindorff, S.; Wegner, R.-D.; Wieacker, P.; Sauer, R. High frequency of spontaneous translocations revealed by FISH in cells from patients with the cancer-prone syndromes ataxia telangiectasia and Nijmegen breakage syndrome. Cytogenet. Genome Res. 2001, 92, 186–191. [Google Scholar] [CrossRef]
- Dumon-Jones, V.; Frappart, P.-O.; Tong, W.-M.; Sajithlal, G.; Hulla, W.; Schmid, G.; Herceg, Z.; Digweed, M.; Wang, Z.-Q. Nbn heterozygosity renders mice susceptible to tumor formation and ionizing radiation-induced tumorigenesis. Cancer Res. 2003, 63, 7263–7269. [Google Scholar] [PubMed]
- Demaria, O.; Cornen, S.; Daëron, M.; Morel, Y.; Medzhitov, R.; Vivier, E. Harnessing innate immunity in cancer therapy. Nature 2019, 574, 45–56. [Google Scholar] [CrossRef]
- Koppensteiner, R.; Samartzis, E.P.; Noske, A.; von Teichman, A.; Dedes, I.; Gwerder, M.; Imesch, P.; Ikenberg, K.; Moch, H.; Fink, D.; et al. Effect of MRE11 Loss on PARP-Inhibitor Sensitivity in Endometrial Cancer In Vitro. PLoS ONE 2014, 9, e100041. [Google Scholar] [CrossRef]
- McPherson, L.A.; Shen, Y.; Ford, J.M. Poly (ADP-ribose) polymerase inhibitor LT-626: Sensitivity correlates with MRE11 mutations and synergizes with platinums and irinotecan in colorectal cancer cells. Cancer Lett. 2014, 343, 217–223. [Google Scholar] [CrossRef]
- Vilar, E.; Bartnik, C.M.; Stenzel, S.L.; Raskin, L.; Ahn, J.; Moreno, V.; Mukherjee, B.; Iniesta, M.D.; Morgan, M.A.; Rennert, G.; et al. MRE11 Deficiency Increases Sensitivity to Poly(ADP-ribose) Polymerase Inhibition in Microsatellite Unstable Colorectal Cancers. Cancer Res. 2011, 71, 2632–2642. [Google Scholar] [CrossRef]
- Gaymes, T.J.; Mohamedali, A.M.; Patterson, M.; Matto, N.; Smith, A.; Kulasekararaj, A.; Chelliah, R.; Curtin, N.; Farzaneh, F.; Shall, S.; et al. Microsatellite instability induced mutations in DNA repair genes CtIP and MRE11 confer hypersensitivity to poly (ADP-ribose) polymerase inhibitors in myeloid malignancies. Haematologica 2013, 98, 1397–1406. [Google Scholar] [CrossRef]
- Takagi, M.; Yoshida, M.; Nemoto, Y.; Tamaichi, H.; Tsuchida, R.; Seki, M.; Uryu, K.; Hoshino, N.; Nishii, R.; Miyamoto, S.; et al. Loss of DNA Damage Response in Neuroblastoma and Utility of a PARP Inhibitor. JNCI J. Natl. Cancer Inst. 2017, 109, djx062. [Google Scholar] [CrossRef]
- Oplustilova, L.; Wolanin, K.; Mistrik, M.; Korinkova, G.; Simkova, D.; Bouchal, J.; Lenobel, R.; Bartkova, J.; Lau, A.; O’Connor, M.J.; et al. Evaluation of candidate biomarkers to predict cancer cell sensitivity or resistance to PARP-1 inhibitor treatment. Cell Cycle 2012, 11, 3837–3850. [Google Scholar] [CrossRef] [PubMed]
- Fagan-Solis, K.D.; Simpson, D.A.; Kumar, R.J.; Martelotto, L.G.; Mose, L.E.; Rashid, N.U.; Ho, A.Y.; Powell, S.N.; Wen, Y.H.; Parker, J.S.; et al. A P53-Independent DNA Damage Response Suppresses Oncogenic Proliferation and Genome Instability. Cell Rep. 2020, 30, 1385–1399.e7. [Google Scholar] [CrossRef] [PubMed]
- Daemen, A.; Wolf, D.M.; Korkola, J.E.; Griffith, O.L.; Frankum, J.R.; Brough, R.; Jakkula, L.R.; Wang, N.J.; Natrajan, R.; Reis-Filho, J.S.; et al. Cross-platform pathway-based analysis identifies markers of response to the PARP inhibitor olaparib. Breast Cancer Res. Treat. 2012, 135, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Lajud, S.A.; Nagda, D.A.; Yamashita, T.; Zheng, J.; Tanaka, N.; Abuzeid, W.M.; Civantos, A.; Bezpalko, O.; O’Malley, B.W.; Li, D. Dual Disruption of DNA Repair and Telomere Maintenance for the Treatment of Head and Neck Cancer. Clin. Cancer Res. 2014, 20, 6465–6478. [Google Scholar] [CrossRef]
- Brandt, S.; Samartzis, E.P.; Zimmermann, A.-K.; Fink, D.; Moch, H.; Noske, A.; Dedes, K.J. Lack of MRE11-RAD50-NBS1 (MRN) complex detection occurs frequently in low-grade epithelial ovarian cancer. BMC Cancer 2017, 17, 44. [Google Scholar] [CrossRef]
- Alblihy, A.; Ali, R.; Algethami, M.; Shoqafi, A.; Toss, M.S.; Brownlie, J.; Tatum, N.J.; Hickson, I.; Moran, P.O.; Grabowska, A.; et al. Targeting Mre11 overcomes platinum resistance and induces synthetic lethality in XRCC1 deficient epithelial ovarian cancers. NPJ Precis. Oncol. 2022, 6, 51. [Google Scholar] [CrossRef]
- Primo, L.M.F.; Teixeira, L.K. DNA replication stress: Oncogenes in the spotlight. Genet. Mol. Biol. 2020, 43, e20190138. [Google Scholar] [CrossRef]
- Bartkova, J.; Hořejší, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre’, M.; Giovanni Nuciforo, P.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.-V.F.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef]
- Polom, K.; Das, K.; Marrelli, D.; Roviello, G.; Pascale, V.; Voglino, C.; Rho, H.; Tan, P.; Roviello, F. KRAS Mutation in Gastric Cancer and Prognostication Associated with Microsatellite Instability Status. Pathol. Oncol. Res. 2019, 25, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.-C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [CrossRef] [PubMed]
- Nakayama, K.; Rahman, M.T.; Rahman, M.; Nakamura, K.; Ishikawa, M.; Katagiri, H.; Sato, E.; Ishibashi, T.; Iida, K.; Ishikawa, N.; et al. CCNE1 amplification is associated with aggressive potential in endometrioid endometrial carcinomas. Int. J. Oncol. 2016, 48, 506–516. [Google Scholar] [CrossRef]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef]
- Guerrero Llobet, S.; van der Vegt, B.; Jongeneel, E.; Bense, R.D.; Zwager, M.C.; Schröder, C.P.; Everts, M.; Fehrmann, R.S.N.; de Bock, G.H.; van Vugt, M.A.T.M. Cyclin E expression is associated with high levels of replication stress in triple-negative breast cancer. NPJ Breast Cancer 2020, 6, 40. [Google Scholar] [CrossRef]
- Luo, J. KRAS mutation in pancreatic cancer. Semin. Oncol. 2021, 48, 10–18. [Google Scholar] [CrossRef]
- Spehalski, E.; Capper, K.M.; Smith, C.J.; Morgan, M.J.; Dinkelmann, M.; Buis, J.; Sekiguchi, J.M.; Ferguson, D.O. MRE11 Promotes Tumorigenesis by Facilitating Resistance to Oncogene-Induced Replication Stress. Cancer Res. 2017, 77, 5327–5338. [Google Scholar] [CrossRef]
- Murakami, T.; Shoji, Y.; Nishi, T.; Chang, S.; Jachimowicz, R.D.; Hoshimoto, S.; Ono, S.; Shiloh, Y.; Takeuchi, H.; Kitagawa, Y.; et al. Regulation of MRE11A by UBQLN4 leads to cisplatin resistance in patients with esophageal squamous cell carcinoma. Mol. Oncol. 2021, 15, 1069–1087. [Google Scholar] [CrossRef]
- Yang, M.-H.; Chiang, W.-C.; Chou, T.-Y.; Chang, S.-Y.; Chen, P.-M.; Teng, S.-C.; Wu, K.-J. Increased NBS1 Expression Is a Marker of Aggressive Head and Neck Cancer and Overexpression of NBS1 Contributes to Transformation. Clin. Cancer Res. 2006, 12, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Hsu, D.S.-S.; Chang, S.-Y.; Liu, C.-J.; Tzeng, C.-H.; Wu, K.-J.; Kao, J.-Y.; Yang, M.-H. Identification of increased NBS1 expression as a prognostic marker of squamous cell carcinoma of the oral cavity. Cancer Sci. 2010, 101, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Petroni, M.; Sardina, F.; Infante, P.; Bartolazzi, A.; Locatelli, E.; Fabretti, F.; Di Giulio, S.; Capalbo, C.; Cardinali, B.; Coppa, A.; et al. MRE11 inhibition highlights a replication stress-dependent vulnerability of MYCN-driven tumors. Cell Death Dis. 2018, 9, 895. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.-C.; Teng, S.-C.; Su, Y.-N.; Hsieh, F.-J.; Wu, K.-J. c-Myc Directly Regulates the Transcription of the NBS1 Gene Involved in DNA Double-strand Break Repair. J. Biol. Chem. 2003, 278, 19286–19291. [Google Scholar] [CrossRef]
- Petroni, M.; Sardina, F.; Heil, C.; Sahún-Roncero, M.; Colicchia, V.; Veschi, V.; Albini, S.; Fruci, D.; Ricci, B.; Soriani, A.; et al. The MRN complex is transcriptionally regulated by MYCN during neural cell proliferation to control replication stress. Cell Death Differ. 2016, 23, 197–206. [Google Scholar] [CrossRef]
- Lee, J.; Dunphy, W.G. The Mre11-Rad50-Nbs1 (MRN) complex has a specific role in the activation of Chk1 in response to stalled replication forks. MBoC 2013, 24, 1343–1353. [Google Scholar] [CrossRef]
- Ltan, B.; Yokobori, T.; Ide, M.; Bai, T.; Yanoma, T.; Kimura, A.; Kogure, N.; Suzuki, M.; Bao, P.; Mochiki, E.; et al. High Expression of MRE11–RAD50–NBS1 Is Associated with Poor Prognosis and Chemoresistance in Gastric Cancer. Anticancer Res. 2016, 36, 5237–5248. [Google Scholar] [CrossRef]
- Alblihy, A.; Alabdullah, M.L.; Ali, R.; Algethami, M.; Toss, M.S.; Mongan, N.P.; Rakha, E.A.; Madhusudan, S. Clinicopathological and Functional Evaluation Reveal NBS1 as a Predictor of Platinum Resistance in Epithelial Ovarian Cancers. Biomedicines 2021, 9, 56. [Google Scholar] [CrossRef]
- Chen, C.; Wang, Y.; Mei, J.F.; Li, S.S.; Xu, H.X.; Xiong, H.P.; Wang, X.H.; He, X. Targeting RAD50 increases sensitivity to radiotherapy in colorectal cancer cells. Neoplasma 2018, 65, 75–80. [Google Scholar] [CrossRef]
- Alblihy, A.; Alabdullah, M.L.; Toss, M.S.; Algethami, M.; Mongan, N.P.; Rakha, E.A.; Madhusudan, S. RAD50 deficiency is a predictor of platinum sensitivity in sporadic epithelial ovarian cancers. Mol. Biomed. 2020, 1, 19. [Google Scholar] [CrossRef]
- Wang, Y.; Gudikote, J.; Giri, U.; Yan, J.; Deng, W.; Ye, R.; Jiang, W.; Li, N.; Hobbs, B.P.; Wang, J.; et al. RAD50 Expression Is Associated with Poor Clinical Outcomes after Radiotherapy for Resected Non–small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Huang, J.; Wang, K.; Li, J.; Yan, R.; Zhu, L.; Ye, J.; Wu, X.; Zhuang, S.; Li, D.; et al. Targeting Rad50 sensitizes human nasopharyngeal carcinoma cells to radiotherapy. BMC Cancer 2016, 16, 190. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.M.; Shi, G.; Li, G.; Carney, J.P.; O’Malley, B.; Li, D. Mutant Nbs1 Enhances Cisplatin-Induced DNA Damage and Cytotoxicity in Head and Neck Cancer. Otolaryngol.–Head Neck Surg. 2004, 131, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Dupré, A.; Boyer-Chatenet, L.; Sattler, R.M.; Modi, A.P.; Lee, J.-H.; Nicolette, M.L.; Kopelovich, L.; Jasin, M.; Baer, R.; Paull, T.T.; et al. A forward chemical genetic screen reveals an inhibitor of the Mre11–Rad50–Nbs1 complex. Nat. Chem. Biol. 2008, 4, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Guo, S.; Zhang, W.; Li, Z.; Xu, J.; Li, D.; Wang, Y.; Zhan, Q. A Comprehensive Analysis of Alterations in DNA Damage Repair Pathways Reveals a Potential Way to Enhance the Radio-Sensitivity of Esophageal Squamous Cell Cancer. Front. Oncol. 2020, 10, 575711. [Google Scholar] [CrossRef]
- Manic, G.; Sistigu, A.; Corradi, F.; Musella, M.; De Maria, R.; Vitale, I. Replication stress response in cancer stem cells as a target for chemotherapy. Semin. Cancer Biol. 2018, 53, 31–41. [Google Scholar] [CrossRef]
- Carruthers, R.D.; Ahmed, S.U.; Ramachandran, S.; Strathdee, K.; Kurian, K.M.; Hedley, A.; Gomez-Roman, N.; Kalna, G.; Neilson, M.; Gilmour, L.; et al. Replication Stress Drives Constitutive Activation of the DNA Damage Response and Radioresistance in Glioblastoma Stem-like Cells. Cancer Res. 2018, 78, 5060–5071. [Google Scholar] [CrossRef]
- Hill, S.J.; Decker, B.; Roberts, E.A.; Horowitz, N.S.; Muto, M.G.; Worley, M.J.; Feltmate, C.M.; Nucci, M.R.; Swisher, E.M.; Nguyen, H.; et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov. 2018, 8, 1404–1421. [Google Scholar] [CrossRef]
- Manic, G.; Musella, M.; Corradi, F.; Sistigu, A.; Vitale, S.; Soliman Abdel Rehim, S.; Mattiello, L.; Malacaria, E.; Galassi, C.; Signore, M.; et al. Control of replication stress and mitosis in colorectal cancer stem cells through the interplay of PARP1, MRE11 and RAD51. Cell Death Differ. 2021, 28, 2060–2082. [Google Scholar] [CrossRef]
- Paludan, S.R.; Bowie, A.G. Immune Sensing of DNA. Immunity 2013, 38, 870–880. [Google Scholar] [CrossRef]
- Shah, G.A.; O’Shea, C.C. Viral and Cellular Genomes Activate Distinct DNA Damage Responses. Cell 2015, 162, 987–1002. [Google Scholar] [CrossRef] [PubMed]
- Stracker, T.H.; Lee, D.V.; Carson, C.T.; Araujo, F.D.; Ornelles, D.A.; Weitzman, M.D. Serotype-Specific Reorganization of the Mre11 Complex by Adenoviral E4orf3 Proteins. J. Virol. 2005, 79, 6664–6673. [Google Scholar] [CrossRef] [PubMed]
- Araujo, F.D.; Stracker, T.H.; Carson, C.T.; Lee, D.V.; Weitzman, M.D. Adenovirus Type 5 E4orf3 Protein Targets the Mre11 Complex to Cytoplasmic Aggresomes. J. Virol. 2005, 79, 11382–11391. [Google Scholar] [CrossRef]
- Lilley, C.E.; Schwartz, R.A.; Weitzman, M.D. Using or abusing: Viruses and the cellular DNA damage response. Trends Microbiol. 2007, 15, 119–126. [Google Scholar] [CrossRef]
- Stracker, T.H.; Carson, C.T.; Weitzman, M.D. Adenovirus oncoproteins inactivate the Mre11–Rad50–NBS1 DNA repair complex. Nature 2002, 418, 348–352. [Google Scholar] [CrossRef]
- Wilkinson, D.E.; Weller, S.K. Recruitment of Cellular Recombination and Repair Proteins to Sites of Herpes Simplex Virus Type 1 DNA Replication Is Dependent on the Composition of Viral Proteins within Prereplicative Sites and Correlates with the Induction of the DNA Damage Response. J. Virol. 2004, 78, 4783–4796. [Google Scholar] [CrossRef]
- Lilley, C.E.; Carson, C.T.; Muotri, A.R.; Gage, F.H.; Weitzman, M.D. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc. Natl. Acad. Sci. USA 2005, 102, 5844–5849. [Google Scholar] [CrossRef]
- Roth, S.; Rottach, A.; Lotz-Havla, A.S.; Laux, V.; Muschaweckh, A.; Gersting, S.W.; Muntau, A.C.; Hopfner, K.-P.; Jin, L.; Vanness, K.; et al. Rad50-CARD9 interactions link cytosolic DNA sensing to IL-1β production. Nat. Immunol. 2014, 15, 538–545. [Google Scholar] [CrossRef]
- Kondo, T.; Kobayashi, J.; Saitoh, T.; Maruyama, K.; Ishii, K.J.; Barber, G.N.; Komatsu, K.; Akira, S.; Kawai, T. DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc. Natl. Acad. Sci. USA 2013, 110, 2969–2974. [Google Scholar] [CrossRef] [PubMed]
- Abdisalaam, S.; Mukherjee, S.; Bhattacharya, S.; Kumari, S.; Sinha, D.; Ortega, J.; Li, G.-M.; Sadek, H.A.; Krishnan, S.; Asaithamby, A. NBS1-CtIP–mediated DNA end resection suppresses cGAS binding to micronuclei. Nucleic Acids Res. 2022, 50, 2681–2699. [Google Scholar] [CrossRef] [PubMed]
- Woodbine, L.; Gennery, A.R.; Jeggo, P.A. The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair 2014, 16, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Helmink, B.A.; Bredemeyer, A.L.; Lee, B.-S.; Huang, C.-Y.; Sharma, G.G.; Walker, L.M.; Bednarski, J.J.; Lee, W.-L.; Pandita, T.K.; Bassing, C.H.; et al. MRN complex function in the repair of chromosomal Rag-mediated DNA double-strand breaks. J. Exp. Med. 2009, 206, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Kracker, S.; Imai, K.; Gardès, P.; Ochs, H.D.; Fischer, A.; Durandy, A.H. Impaired induction of DNA lesions during immunoglobulin class-switch recombination in humans influences end-joining repair. Proc. Natl. Acad. Sci. USA 2010, 107, 22225–22230. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.; Casellas, R.; Reina-San-Martin, B.; Chen, H.T.; Difilippantonio, M.J.; Wilson, P.C.; Hanitsch, L.; Celeste, A.; Muramatsu, M.; Pilch, D.R.; et al. AID is required to initiate Nbs1/γ-H2AX focus formation and mutations at sites of class switching. Nature 2001, 414, 660–665. [Google Scholar] [CrossRef]
- Kracker, S.; Bergmann, Y.; Demuth, I.; Frappart, P.-O.; Hildebrand, G.; Christine, R.; Wang, Z.-Q.; Sperling, K.; Digweed, M.; Radbruch, A. Nibrin functions in Ig class-switch recombination. Proc. Natl. Acad. Sci. USA 2005, 102, 1584–1589. [Google Scholar] [CrossRef]
- Pan, Q.; Petit-Frére, C.; Lähdesmäki, A.; Gregorek, H.; Chrzanowska, K.H.; Hammarström, L. Alternative end joining during switch recombination in patients with Ataxia-Telangiectasia. Eur. J. Immunol. 2002, 32, 1300. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F.; Teo, S.-H.; Jackson, S.P. Functional links between telomeres and proteins of the DNA-damage response. Genes Dev. 2004, 18, 1781–1799. [Google Scholar] [CrossRef]
- Chai, W.; Sfeir, A.J.; Hoshiyama, H.; Shay, J.W.; Wright, W.E. The involvement of the Mre11/Rad50/Nbs1 complex in the generation of G-overhangs at human telomeres. EMBO Rep. 2006, 7, 225–230. [Google Scholar] [CrossRef]
- Dimitrova, N.; de Lange, T. Cell Cycle-Dependent Role of MRN at Dysfunctional Telomeres: ATM Signaling-Dependent Induction of Nonhomologous End Joining (NHEJ) in G 1 and Resection-Mediated Inhibition of NHEJ in G 2. Mol. Cell. Biol. 2009, 29, 5552–5563. [Google Scholar] [CrossRef]
- Lamarche, B.J.; Orazio, N.I.; Weitzman, M.D. The MRN complex in double-strand break repair and telomere maintenance. FEBS Lett. 2010, 584, 3682–3695. [Google Scholar] [CrossRef]
- Fukasawa, K. Oncogenes and tumour suppressors take on centrosomes. Nat. Rev. Cancer 2007, 7, 911–924. [Google Scholar] [CrossRef]
- Petry, S. Mechanisms of Mitotic Spindle Assembly. Annu. Rev. Biochem. 2016, 85, 659–683. [Google Scholar] [CrossRef]
- Shimada, M.; Sagae, R.; Kobayashi, J.; Habu, T.; Komatsu, K. Inactivation of the Nijmegen Breakage Syndrome Gene Leads to Excess Centrosome Duplication via the ATR/BRCA1 Pathway. Cancer Res. 2009, 69, 1768–1775. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Xu, Y.; Huo, W.; Lv, Z.; Yuan, J.; Ning, S.; Wang, Q.; Hou, M.; Gao, G.; Ji, J.; et al. Mitosis-specific MRN complex promotes a mitotic signaling cascade to regulate spindle dynamics and chromosome segregation. Proc. Natl. Acad. Sci. USA 2018, 115, E10079–E10088. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi-Iwai, Y. Mre11 is essential for the maintenance of chromosomal DNA in vertebrate cells. EMBO J. 1999, 18, 6619–6629. [Google Scholar] [CrossRef] [PubMed]
- Rozier, L.; Guo, Y.; Peterson, S.; Sato, M.; Baer, R.; Gautier, J.; Mao, Y. The MRN-CtIP Pathway Is Required for Metaphase Chromosome Alignment. Mol. Cell 2013, 49, 1097–1107. [Google Scholar] [CrossRef][Green Version]
- Marshall, W.F.; Rosenbaum, J.L. Are there nucleic acids in the centrosome? In Current Topics in Developmental Biology; Elsevier: Amsterdam, The Netherlands, 1999; Volume 49, pp. 187–205. ISBN 978-0-12-153149-2. [Google Scholar]
- Zhou, Z.-W.; Kirtay, M.; Schneble, N.; Yakoub, G.; Ding, M.; Rüdiger, T.; Siniuk, K.; Lu, R.; Jiang, Y.-N.; Li, T.-L.; et al. NBS1 interacts with Notch signaling in neuronal homeostasis. Nucleic Acids Res. 2020, 48, 10924–10939. [Google Scholar] [CrossRef]
- Cheung, V.G.; Ewens, W.J. Heterozygous carriers of Nijmegen Breakage Syndrome have a distinct gene expression phenotype. Genome Res. 2006, 16, 973–979. [Google Scholar] [CrossRef][Green Version]
- Petroni, M.; Fabretti, F.; Di Giulio, S.; Nicolis di Robilant, V.; La Monica, V.; Moretti, M.; Belardinilli, F.; Bufalieri, F.; Coppa, A.; Paci, P.; et al. A gene dosage-dependent effect unveils NBS1 as both a haploinsufficient tumour suppressor and an essential gene for SHH-medulloblastoma. Neuropathol. Appl. Neurobiol. 2022, 48, e12837. [Google Scholar] [CrossRef]
- Hematulin, A.; Sagan, D.; Eckardt-Schupp, F.; Moertl, S. NBS1 is required for IGF-1 induced cellular proliferation through the Ras/Raf/MEK/ERK cascade. Cell. Signal. 2008, 20, 2276–2285. [Google Scholar] [CrossRef]
- Kuo, K.-T.; Chou, T.-Y.; Hsu, H.-S.; Chen, W.-L.; Wang, L.-S. Prognostic Significance of NBS1 and Snail Expression in Esophageal Squamous Cell Carcinoma. Ann. Surg. Oncol. 2012, 19, 549–557. [Google Scholar] [CrossRef]
- Espinosa-Diez, C.; Wilson, R.; Chatterjee, N.; Hudson, C.; Ruhl, R.; Hipfinger, C.; Helms, E.; Khan, O.F.; Anderson, D.G.; Anand, S. MicroRNA regulation of the MRN complex impacts DNA damage, cellular senescence, and angiogenic signaling. Cell Death Dis. 2018, 9, 632. [Google Scholar] [CrossRef]
- Yuan, J.; Song, Y.; Pan, W.; Li, Y.; Xu, Y.; Xie, M.; Shen, Y.; Zhang, N.; Liu, J.; Hua, H.; et al. LncRNA SLC26A4-AS1 suppresses the MRN complex-mediated DNA repair signaling and thyroid cancer metastasis by destabilizing DDX5. Oncogene 2020, 39, 6664–6676. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Y.; Chen, Y.-K.; Lo, S.; Chi, T.-C.; Chen, Y.-H.; Hu, S.C.-S.; Chen, Y.-W.; Jiang, S.S.; Tsai, F.-Y.; Liu, W.; et al. MRE11 promotes oral cancer progression through RUNX2/CXCR4/AKT/FOXA2 signaling in a nuclease-independent manner. Oncogene 2021, 40, 3510–3532. [Google Scholar] [CrossRef]
- Yuan, S.-S.F.; Hou, M.-F.; Hsieh, Y.-C.; Huang, C.-Y.; Lee, Y.-C.; Chen, Y.-J.; Lo, S. Role of MRE11 in Cell Proliferation, Tumor Invasion, and DNA Repair in Breast Cancer. JNCI J. Natl. Cancer Inst. 2012, 104, 1485–1502. [Google Scholar] [CrossRef]
- Yang, M.-H.; Chang, S.-Y.; Chiou, S.-H.; Liu, C.-J.; Chi, C.-W.; Chen, P.-M.; Teng, S.-C.; Wu, K.-J. Overexpression of NBS1 induces epithelial–mesenchymal transition and co-expression of NBS1 and Snail predicts metastasis of head and neck cancer. Oncogene 2007, 26, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-Y.; Lin, C.-T.; Wu, M.-Z.; Wu, K.-J. Induction of HSPA4 and HSPA14 by NBS1 overexpression contributes to NBS1-induced in vitro metastatic and transformation activity. J. Biomed. Sci. 2011, 18, 1. [Google Scholar] [CrossRef]
- Li, Y.; Wang, S.; Li, P.; Li, Y.; Liu, Y.; Fang, H.; Zhang, X.; Liu, Z.; Kong, B. Rad50 promotes ovarian cancer progression through NF-κB activation. J. Cell. Mol. Med. 2021, 25, 10961–10972. [Google Scholar] [CrossRef] [PubMed]
- Alblihy, A.; Shoqafi, A.; Toss, M.S.; Algethami, M.; Harris, A.E.; Jeyapalan, J.N.; Abdel-Fatah, T.; Servante, J.; Chan, S.Y.T.; Green, A.; et al. Untangling the clinicopathological significance of MRE11-RAD50-NBS1 complex in sporadic breast cancers. NPJ Breast Cancer 2021, 7, 143. [Google Scholar] [CrossRef] [PubMed]
- Mlody, B.; Wruck, W.; Martins, S.; Sperling, K.; Adjaye, J. Nijmegen Breakage Syndrome fibroblasts and iPSCs: Cellular models for uncovering disease-associated signaling pathways and establishing a screening platform for anti-oxidants. Sci. Rep. 2017, 7, 7516. [Google Scholar] [CrossRef]
- Berkel, C.; Cacan, E. Involvement of ATMIN-DYNLL1-MRN axis in the progression and aggressiveness of serous ovarian cancer. Biochem. Biophys. Res. Commun. 2021, 570, 74–81. [Google Scholar] [CrossRef]
- Lee, Y.-K.; Park, N.-H.; Lee, H. Clinicopathological values of NBS1 and DNA damage response genes in epithelial ovarian cancers. Exp. Mol. Med. 2015, 47, e195. [Google Scholar] [CrossRef]
- Ehlers, J.P.; Harbour, J.W. NBS1 Expression as a Prognostic Marker in Uveal Melanoma. Clin. Cancer Res. 2005, 11, 1849–1853. [Google Scholar] [CrossRef] [PubMed]
- Ihara, K.; Yamaguchi, S.; Ueno, N.; Tani, Y.; Shida, Y.; Ogata, H.; Domeki, Y.; Okamoto, K.; Nakajima, M.; Sasaki, K.; et al. Expression of DNA double-strand break repair proteins predicts the response and prognosis of colorectal cancer patients undergoing oxaliplatin-based chemotherapy. Oncol. Rep. 2016, 35, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Ho, V.; Chung, L.; Revoltar, M.; Lim, S.H.; Tut, T.-G.; Abubakar, A.; Henderson, C.J.; Chua, W.; Ng, W.; Lee, M.; et al. MRE11 and ATM Expression Levels Predict Rectal Cancer Survival and Their Association with Radiotherapy Response. PLoS ONE 2016, 11, e0167675. [Google Scholar] [CrossRef]
- Ho, V.; Chung, L.; Singh, A.; Lea, V.; Abubakar, A.; Lim, S.H.; Ng, W.; Lee, M.; de Souza, P.; Shin, J.-S.; et al. Overexpression of the MRE11-RAD50-NBS1 (MRN) complex in rectal cancer correlates with poor response to neoadjuvant radiotherapy and prognosis. BMC Cancer 2018, 18, 869. [Google Scholar] [CrossRef]
- Abad, E.; Civit, L.; Potesil, D.; Zdrahal, Z.; Lyakhovich, A. Enhanced DNA damage response through RAD50 in triple negative breast cancer resistant and cancer stem-like cells contributes to chemoresistance. FEBS J. 2021, 288, 2184–2202. [Google Scholar] [CrossRef]
- Laurberg, J.R.; Brems-Eskildsen, A.S.; Nordentoft, I.; Fristrup, N.; Schepeler, T.; Ulhøi, B.P.; Agerbaek, M.; Hartmann, A.; Bertz, S.; Wittlinger, M.; et al. Expression of TIP60 (tat-interactive protein) and MRE11 (meiotic recombination 11 homolog) predict treatment-specific outcome of localised invasive bladder cancer: PREDICTIVE VALUE OF TIP60 AND MRE11 EXPRESSION IN UCB. BJU Int. 2012, 110, E1228–E1236. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.M.; Kerr, M.; Teo, M.T.W.; Jevons, S.J.; Koritzinsky, M.; Wouters, B.G.; Bhattarai, S.; Kiltie, A.E. Post-transcriptional regulation of MRE11 expression in muscle-invasive bladder tumours. Oncotarget 2014, 5, 993–1003. [Google Scholar] [CrossRef]
- Choudhury, A.; Nelson, L.D.; Teo, M.T.W.; Chilka, S.; Bhattarai, S.; Johnston, C.F.; Elliott, F.; Lowery, J.; Taylor, C.F.; Churchman, M.; et al. MRE11 Expression Is Predictive of Cause-Specific Survival following Radical Radiotherapy for Muscle-Invasive Bladder Cancer. Cancer Res. 2010, 70, 7017–7026. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.-W.; Kopsida, M.; Liu, Y.-B.; Zhang, H.; Gao, J.-F.; Arbman, G.; Cao, S.-Y.-W.; Li, Y.; Zhou, Z.-G.; Sun, X.-F. Prognostic Heterogeneity of MRE11 Based on the Location of Primary Colorectal Cancer Is Caused by Activation of Different Immune Signals. Front. Oncol. 2020, 9, 1465. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zhang, H.; Arbman, G.; Sun, X.F. RAD50/MRE11/NBS1 proteins in relation to tumour development and prognosis in patients with microsatellite stable colorectal cancer. Histol. Histopathol. 2008, 23, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-J.; Ping, J.; Li, Y.; Holmqvist, A.; Adell, G.; Arbman, G.; Zhang, H.; Zhou, Z.-G.; Sun, X.-F. Prognostic Significance and Molecular Features of Colorectal Mucinous Adenocarcinomas: A Strobe-Compliant Study. Medicine 2015, 94, e2350. [Google Scholar] [CrossRef]
- Söderlund, K.; Stål, O.; Skoog, L.; Rutqvist, L.E.; Nordenskjöld, B.; Askmalm, M.S. Intact Mre11/Rad50/Nbs1 Complex Predicts Good Response to Radiotherapy in Early Breast Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2007, 68, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Fernández, J.A.; McArt, D.G.; Boyle, D.P.; Li, G.; Loughrey, M.B.; Irwin, G.W.; Harkin, D.P.; James, J.A.; McQuaid, S.; et al. Integrated tumor identification and automated scoring minimizes pathologist involvement and provides new insights to key biomarkers in breast cancer. Lab. Investig. 2018, 98, 15–26. [Google Scholar] [CrossRef]
- Attiyeh, E.F.; London, W.B.; Mossé, Y.P.; Wang, Q.; Winter, C.; Khazi, D.; McGrady, P.W.; Seeger, R.C.; Look, A.T.; Shimada, H.; et al. Chromosome 1p and 11q Deletions and Outcome in Neuroblastoma. N. Engl. J. Med. 2005, 353, 2243–2253. [Google Scholar] [CrossRef]
- Guo, C.; White, P.S.; Weiss, M.J.; Hogarty, M.D.; Thompson, P.M.; Stram, D.O.; Gerbing, R.; Matthay, K.K.; Seeger, R.C.; Brodeur, G.M.; et al. Allelic deletion at 11q23 is common in MYCN single copy neuroblastomas. Oncogene 1999, 18, 4948–4957. [Google Scholar] [CrossRef]
- Na, J.; Newman, J.A.; Then, C.K.; Syed, J.; Vendrell, I.; Torrecilla, I.; Ellermann, S.; Ramadan, K.; Fischer, R.; Kiltie, A.E. SPRTN protease-cleaved MRE11 decreases DNA repair and radiosensitises cancer cells. Cell Death Dis. 2021, 12, 165. [Google Scholar] [CrossRef]
- Hatton, B.A.; Villavicencio, E.H.; Tsuchiya, K.D.; Pritchard, J.I.; Ditzler, S.; Pullar, B.; Hansen, S.; Knoblaugh, S.E.; Lee, D.; Eberhart, C.G.; et al. The Smo/Smo Model: Hedgehog-Induced Medulloblastoma with 90% Incidence and Leptomeningeal Spread. Cancer Res. 2008, 68, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- Bakhshi, S.; Cerosaletti, K.M.; Concannon, P.; Bawle, E.V.; Fontanesi, J.; Gatti, R.A.; Bhambhani, K. Medulloblastoma With Adverse Reaction to Radiation Therapy in Nijmegen Breakage Syndrome. J. Pediatr. Hematol./Oncol. 2003, 25, 248–251. [Google Scholar] [CrossRef]
- Ciara, E.; Piekutowska-Abramczuk, D.; Popowska, E.; Grajkowska, W.; Barszcz, S.; Perek, D.; Dembowska-Bagińska, B.; Perek-Polnik, M.; Kowalewska, E.; Czajńska, A.; et al. Heterozygous germ-line mutations in the NBN gene predispose to medulloblastoma in pediatric patients. Acta Neuropathol. 2010, 119, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Viana-Pereira, M.; Almeida, I.; Sousa, S.; Mahler-Araújo, B.; Seruca, R.; Pimentel, J.; Reis, R.M. Analysis of microsatellite instability in medulloblastoma. Neuro-Oncology 2009, 11, 458–467. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Trubicka, J.; Żemojtel, T.; Hecht, J.; Falana, K.; Piekutowska- Abramczuk, D.; Płoski, R.; Perek-Polnik, M.; Drogosiewicz, M.; Grajkowska, W.; Ciara, E.; et al. The germline variants in DNA repair genes in pediatric medulloblastoma: A challenge for current therapeutic strategies. BMC Cancer 2017, 17, 239. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Grotzer, M.A.; Watanabe, T.; Hewer, E.; Pietsch, T.; Rutkowski, S.; Ohgaki, H. Mutations in the Nijmegen Breakage Syndrome Gene in Medulloblastomas. Clin. Cancer Res. 2008, 14, 4053–4058. [Google Scholar] [CrossRef]
- Petroni, M.; Sahùn Roncero, M.; Ramponi, V.; Fabretti, F.; Nicolis Di Robilant, V.; Moretti, M.; Alfano, V.; Corsi, A.; De Panfilis, S.; Giubettini, M.; et al. SMO-M2 mutation does not support cell-autonomous Hedgehog activity in cerebellar granule cell precursors. Sci. Rep. 2019, 9, 19623. [Google Scholar] [CrossRef]
- Yoon, J.W.; Lamm, M.; Iannaccone, S.; Higashiyama, N.; Leong, K.F.; Iannaccone, P.; Walterhouse, D. p53 modulates the activity of the GLI1 oncogene through interactions with the shared coactivator TAF9. DNA Repair 2015, 34, 9–17. [Google Scholar] [CrossRef]
- Stecca, B.; Ruiz I Altaba, A. A GLI1-p53 inhibitory loop controls neural stem cell and tumour cell numbers. EMBO J. 2009, 28, 663–676. [Google Scholar] [CrossRef]
- Mazzà, D.; Infante, P.; Colicchia, V.; Greco, A.; Alfonsi, R.; Siler, M.; Antonucci, L.; Po, A.; De Smaele, E.; Ferretti, E.; et al. PCAF ubiquitin ligase activity inhibits Hedgehog/Gli1 signaling in p53-dependent response to genotoxic stress. Cell Death Differ. 2013, 20, 1688–1697. [Google Scholar] [CrossRef]
- Martins, S.; Erichsen, L.; Datsi, A.; Wruck, W.; Goering, W.; Chrzanowska, K.; Adjaye, J. Impaired p53-mediated DNA damage response contributes to microcephaly in Nijmegen Breakage Syndrome patient-derived cerebral organoids. Cells 2022, 11, 802. [Google Scholar] [CrossRef]
- Halevy, T.; Akov, S.; Bohndorf, M.; Mlody, B.; Adjaye, J.; Benvenisty, N.; Goldberg, M. Chromosomal Instability and Molecular Defects in Induced Pluripotent Stem Cells from Nijmegen Breakage Syndrome Patients. Cell Rep. 2016, 16, 2499–2511. [Google Scholar] [CrossRef]
- Frappart, P.-O.; Lee, Y.; Russell, H.R.; Chalhoub, N.; Wang, Y.-D.; Orii, K.E.; Zhao, J.; Kondo, N.; Baker, S.J.; McKinnon, P.J. Recurrent genomic alterations characterize medulloblastoma arising from DNA double-strand break repair deficiency. Proc. Natl. Acad. Sci. USA 2009, 106, 1880–1885. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.T.; Kaushal, D.; Murphy, M.; Zhang, Y.; Datta, A.; Chen, C.; Monroe, B.; Mostoslavsky, G.; Coakley, K.; Gao, Y.; et al. XRCC4 suppresses medulloblastomas with recurrent translocations in p53-deficient mice. Proc. Natl. Acad. Sci. USA 2006, 103, 7378–7383. [Google Scholar] [CrossRef] [PubMed]
- Holcomb, V.B.; Vogel, H.; Marple, T.; Kornegay, R.W.; Hasty, P. Ku80 and p53 suppress medulloblastoma that arise independent of Rag-1-induced DSBs. Oncogene 2006, 25, 7159–7165. [Google Scholar] [CrossRef] [PubMed]
- Frappart, P.-O.; Lee, Y.; Lamont, J.; McKinnon, P.J. BRCA2 is required for neurogenesis and suppression of medulloblastoma. EMBO J. 2007, 26, 2732–2742. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.-M.; Ohgaki, H.; Huang, H.; Granier, C.; Kleihues, P.; Wang, Z.-Q. Null Mutation of DNA Strand Break-Binding Molecule Poly(ADP-ribose) Polymerase Causes Medulloblastomas in p53−/− Mice. Am. J. Pathol. 2003, 162, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hong, Y.; Li, M.; Long, J.; Zhao, Y.-P.; Zhang, J.-X.; Li, Q.; You, H.; Tong, W.-M.; Jia, J.-D.; et al. Mutation Inactivation of Nijmegen Breakage Syndrome Gene (NBS1) in Hepatocellular Carcinoma and Intrahepatic Cholangiocarcinoma. PLoS ONE 2013, 8, e82426. [Google Scholar] [CrossRef]
- Cho, S.; Kim, M.J.; Choi, Y.Y.; Yoo, S.S.; Lee, W.K.; Lee, E.J.; Jang, E.J.; Bae, E.Y.; Jin, G.; Jeon, H.-S.; et al. Associations between polymorphisms in DNA repair genes and TP53 mutations in non-small cell lung cancer. Lung Cancer 2011, 73, 25–31. [Google Scholar] [CrossRef]
- Watanabe, T.; Nobusawa, S.; Lu, S.; Huang, J.; Mittelbronn, M.; Ohgaki, H. Mutational Inactivation of the Nijmegen Breakage Syndrome Gene (NBS1) in Glioblastomas Is Associated With Multiple TP53 Mutations. J. Neuropathol. Exp. Neurol. 2009, 68, 210–215. [Google Scholar] [CrossRef]
- Reuss, D.E.; Downing, S.M.; Camacho, C.V.; Wang, Y.; Piro, R.M.; Herold-Mende, C.; Wang, Z.; Hofmann, T.G.; Sahm, F.; Von Deimling, A.; et al. Simultaneous Nbs1 and p53 inactivation in neural progenitors triggers High-Grade Gliomas (HGG). Neuropathol. Appl. Neurobiol. 2023, e12915. [Google Scholar] [CrossRef]
- Abe, Y.; Oda-Sato, E.; Tobiume, K.; Kawauchi, K.; Taya, Y.; Okamoto, K.; Oren, M.; Tanaka, N. Hedgehog signaling overrides p53-mediated tumor suppression by activating Mdm2. Proc. Natl. Acad. Sci. USA 2008, 105, 4838–4843. [Google Scholar] [CrossRef]
- Zhukova, N.; Ramaswamy, V.; Remke, M.; Pfaff, E.; Shih, D.J.H.; Martin, D.C.; Castelo-Branco, P.; Baskin, B.; Ray, P.N.; Bouffet, E.; et al. Subgroup-Specific Prognostic Implications of TP53 Mutation in Medulloblastoma. JCO 2013, 31, 2927–2935. [Google Scholar] [CrossRef] [PubMed]
- Petroni, M.; Giannini, G. A MYCN-MRN complex axis controls replication stress for the safe expansion of neuroprogenitor cells. Mol. Cell. Oncol. 2016, 3, e1079673. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Frappart, P.-O.; Tong, W.-M.; Demuth, I.; Radovanovic, I.; Herceg, Z.; Aguzzi, A.; Digweed, M.; Wang, Z.-Q. An essential function for NBS1 in the prevention of ataxia and cerebellar defects. Nat. Med. 2005, 11, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Williamson, C.T.; Kubota, E.; Hamill, J.D.; Klimowicz, A.; Ye, R.; Muzik, H.; Dean, M.; Tu, L.; Gilley, D.; Magliocco, A.M.; et al. Enhanced cytotoxicity of PARP inhibition in mantle cell lymphoma harbouring mutations in both ATM and p53. EMBO Mol. Med. 2012, 4, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, J.A. Targeting Replication Stress Response Pathways to Enhance Genotoxic Chemo- and Radiotherapy. Molecules 2022, 27, 4736. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, J.A.; Sharma, N.; Taylor, L.; Allen, S.J.; Hromas, R. The Safe Path at the Fork: Ensuring Replication-Associated DNA Double-Strand Breaks are Repaired by Homologous Recombination. Front. Genet. 2021, 12, 748033. [Google Scholar] [CrossRef]
- Shibata, A.; Moiani, D.; Arvai, A.S.; Perry, J.; Harding, S.M.; Genois, M.-M.; Maity, R.; van Rossum-Fikkert, S.; Kertokalio, A.; Romoli, F.; et al. DNA Double-Strand Break Repair Pathway Choice Is Directed by Distinct MRE11 Nuclease Activities. Mol. Cell 2014, 53, 7–18. [Google Scholar] [CrossRef]
- Bruhn, C.; Zhou, Z.-W.; Ai, H.; Wang, Z.-Q. The Essential Function of the MRN Complex in the Resolution of Endogenous Replication Intermediates. Cell Rep. 2014, 6, 182–195. [Google Scholar] [CrossRef][Green Version]
- Buis, J.; Wu, Y.; Deng, Y.; Leddon, J.; Westfield, G.; Eckersdorff, M.; Sekiguchi, J.M.; Chang, S.; Ferguson, D.O. Mre11 Nuclease Activity Has Essential Roles in DNA Repair and Genomic Stability Distinct from ATM Activation. Cell 2008, 135, 85–96. [Google Scholar] [CrossRef]
- da Costa, A.A.B.A.; Chowdhury, D.; Shapiro, G.I.; D’Andrea, A.D.; Konstantinopoulos, P.A. Targeting replication stress in cancer therapy. Nat. Rev. Drug Discov. 2023, 22, 38–58. [Google Scholar] [CrossRef]
- Gorecki, L.; Andrs, M.; Korabecny, J. Clinical Candidates Targeting the ATR–CHK1–WEE1 Axis in Cancer. Cancers 2021, 13, 795. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
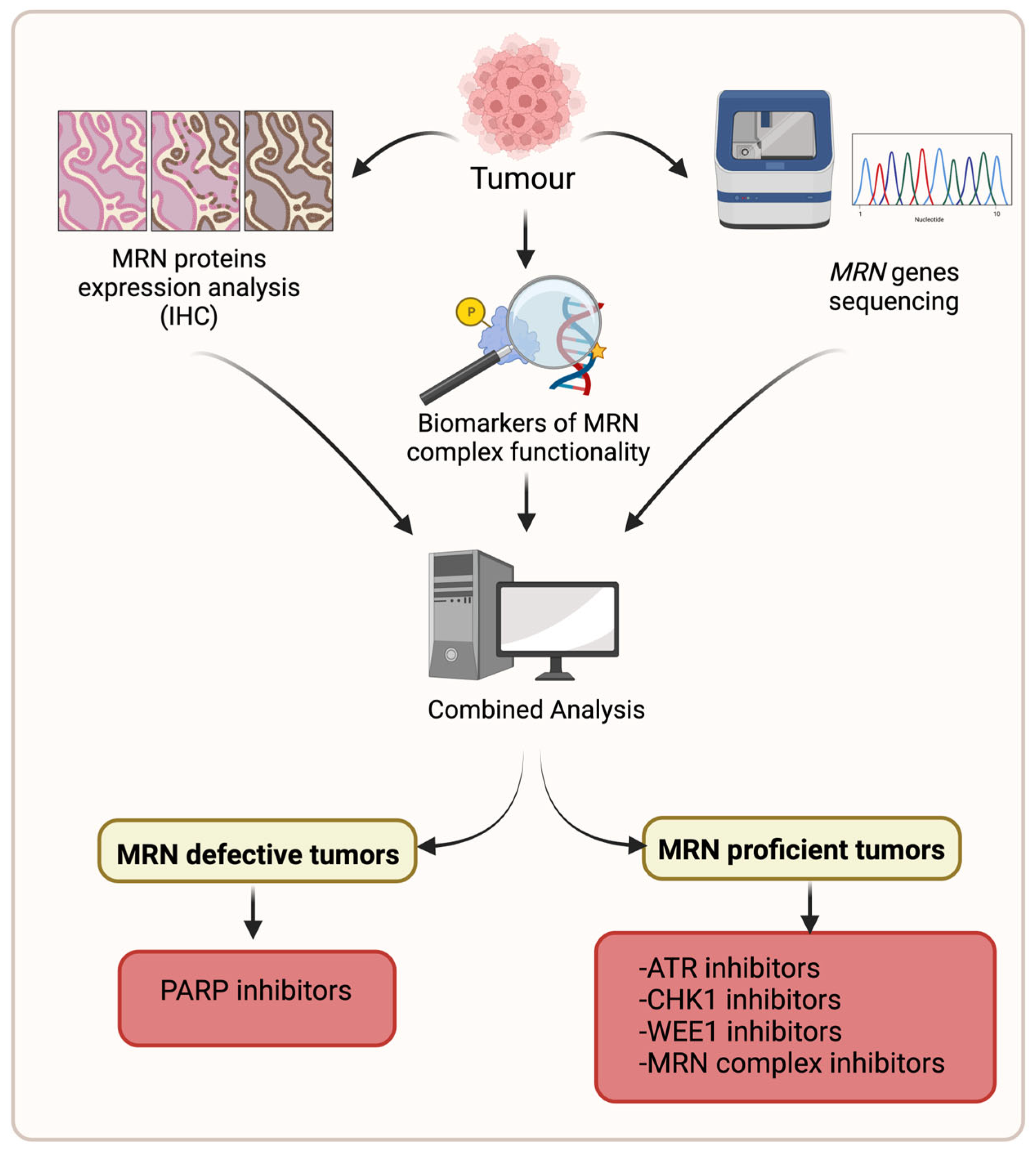{kind=link}
| Cancer | Mutation | Number of Patients with Cancer | % of Cancer Patients with NBN Mutation | Number of Healthy Subjects | % of Healthy Subjects with NBN Mutation | Statistical Analysis | PubMed IDentifier |
|---|---|---|---|---|---|---|---|
| Acute Lymphoblastic Leukaemia | p.I171V | 46 | 10.7 | 2400 | 0.5 | 0.0004 | 24093751 |
| Acute myeloid leukemia | p.I171V | 32 | 6.3 | 2400 | 0.5 | 0.0001 | 24093751 |
| Breast (familial) | c.657_661del5 | 80 | 1.3 | 530 | 0.6 | n.s | 12845677 |
| Breast | c.657_661del5 | 173 | 1.2 | 344 | 0 | 0.046 | 15578693 |
| c.657_661del5 | 224 | 1.8 | 1620 | 0.6 | n.s. | 15185344 | |
| c.657_661del5 | 150 | 3.7 | 530 | 0.6 | 0.037 | 12845677 | |
| c.657_661del5 | 562 | 2.0 | 1620 | 0.6 | 0.0107 | 16770759 | |
| c.657_661del5 | 700 | 0.7 | 344 | 0 | n.s. | 15578693 | |
| Colorectal | c.657_661del5 | 234 | 1.3 | 1620 | 0.6 | n.s. | 15185344 |
| I171V | 131 | 2.3 | 600 | 0.2 | 0.0196 | 18280732 | |
| IVS11+2insT | 472 | 0.6 | 2348 | 0.08 | 0.02 | 18056440 | |
| R215W | 234 | 1.3 | 1620 | 0.2 | 0.0472 | 15185344 | |
| Gastric cancer | IVS11+2insT | 472 | 0.4 | 2348 | 0.08 | 0.0001 | 18056440 |
| Gastrointestinal lymphoma | c.657_661del5 | 37 | 10.8 | 1620 | 0.6 | 0.0002 | 16998789 |
| Head and neck | I171V | 81 | 6.2 | 600 | 0.2 | 0.0001 | 18280732 |
| Larynx cancer | I171V | 176 | 2.3 | 500 | 0.2 | 0.0175 | 17894553 |
| Lung | c.551A>G | 453 | 3.8 | 2400 | 0.5 | <0.0001 | 26722329 |
| c.657_661del5 | 453 | 0.7 | 2090 | 0.2 | n.s. | 26722329 | |
| IVS11+2insT | 532 | 0.4 | 2348 | 0.08 | n.s. | 18056440 | |
| Medulloblastoma | c.511A>G | 104 | 3.8 | 4227 | 1.3 | 0.0241 | 19908051 |
| c.657_661del5 | 104 | 2.9 | 12,484 | 0.6 | 0.0028 | 19908051 | |
| Melanoma | c.657_661del5 | 376 | 0.3 | 866 | 0.1 | n.s. | 17496786 |
| c.657_661del5 | 80 | 2.5 | 530 | 0.6 | n.s. | 12883362 | |
| c.657_661del5 | 105 | 3.8 | 1620 | 0.6 | 0.0081 | 15185344 | |
| Non-Hodgkin Lymphoma | c.657_661del5 | 109 | 0 | 984 | 0.3 | n.s. | 10848790 |
| c.657_661del5 | 42 | 4.8 | 1620 | 0.6 | 0.0351 | 15185344 | |
| c.657_661del5 | 228 | 3.5 | 1620 | 0.6 | 0.0001 | 16998789 | |
| Prostate (familial) | c.657_661del5 | 56 | 9.0 | 1500 | 0.60 | <0.0001 | 14973119 |
| Prostate | c.657_661del5 | 305 | 2.2 | 1500 | 0.60 | 0.01 | 14973119 |
| Cancer | Mutation | Number of Patients with Cancer | % of Cancer Patients with RAD50 Mutation | Number of Healthy Subjects | % of Healthy Subjects with RAD50 Mutation | Statistical Analysis | PubMed IDentifier |
|---|---|---|---|---|---|---|---|
| Acute Lymphoblastic Leukaemia + Acute myeloid leukemia | - | 220 | - | 504 | 0.006 | 0.0019 | 24093751 |
| Breast | 687delT | 317 | 2.52 | 1000 | 0.6 | 0.008 | 16474176 |
| - | 7657 | 0.07 | 5000 | 0.02 | n.s. | 29726012 | |
| 687delT | 590 | 0.5 | 560 | 0.2 | n.s. | 16385572 | |
| Breast/Ovarian | 687delT | 151 | 1.3 | 1000 | 0.6 | n.d. | 14684699 |
| Esophageal squamous cell carcinoma | p.K722fs | 2088 | 0.14 | 2342 | 0 | 0.032 | 34572942 |
| p.Q672X/p.K722fs | 2088 | 0.19 | 2342 | 0 | 0.01 | 34572942 | |
| FH+ Esophageal squamous cell carcinoma | - | 372 | 1.1 | 19,954 | 0.15 | 0.0033 | 34572942 |
| Cancer | Mutation | Number of Patients with Cancer | % of Cancer Patients with MRE11 Mutation | Number of Healthy Subjects | % of Healthy Subjects with MRE11 Mutation | Statistical Analysis | PubMed IDentifier |
|---|---|---|---|---|---|---|---|
| Breast | - | 75,818 | 0.05 | 111,326 | 0.05 | n.s. | 31406321 |
| p.E506 * | 1925 | 0 | 2287 | 0.002 | n.s. | 33510186 | |
| p.R364X | 1 | n.d. | 100 | 0 | n.d. | 28559769 | |
| Colorectal | - | 1006 | 0.3 | 1609 | 0 | n.d. | 27329137 |
| - | 49 | 0.84 | n.d. | n.d. | n.d. | 15048091 | |
| Endometrium | - | 14 | 7 | n.d. | n.d. | n.d. | 15048091 |
| p.E506 * | 367 | 0 | 2287 | 0.002 | n.s. | 33510186 | |
| Gastric MSI | poly(T)11 | 27 | 0.81 | n.d. | n.d. | n.d. | 15319296 |
| Hereditary Breast and Ovarian Cancer | p.R305W | 151 | 0.3 | 1000 | 0 | n.d. | 14684699 |
| - | 708 | 0.4 | n.d. | n.d. | n.d. | 24549055 | |
| Medulloblastoma MSI | poly(T)11 | 4 | 0.25 | n.d. | n.d. | n.d. | 19179424 |
| Ovary | p.E506 * | 341 | 0 | 2287 | 0.002 | n.s. | 33510186 |
| - | 5 | 0.2 | n.d. | n.d. | n.d. | 15048091 | |
| Stomach | - | 1 | 1.0 | n.d. | n.d. | n.d. | 15048091 |
| Ureter | - | 1 | 0 | n.d. | n.d. | n.d. | 15048091 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petroni, M.; La Monica, V.; Fabretti, F.; Augusto, M.; Battaglini, D.; Polonara, F.; Di Giulio, S.; Giannini, G. The Multiple Faces of the MRN Complex: Roles in Medulloblastoma and Beyond. Cancers 2023, 15, 3599. https://doi.org/10.3390/cancers15143599
Petroni M, La Monica V, Fabretti F, Augusto M, Battaglini D, Polonara F, Di Giulio S, Giannini G. The Multiple Faces of the MRN Complex: Roles in Medulloblastoma and Beyond. Cancers. 2023; 15(14):3599. https://doi.org/10.3390/cancers15143599
Chicago/Turabian StylePetroni, Marialaura, Veronica La Monica, Francesca Fabretti, Mariaconcetta Augusto, Damiana Battaglini, Francesca Polonara, Stefano Di Giulio, and Giuseppe Giannini. 2023. "The Multiple Faces of the MRN Complex: Roles in Medulloblastoma and Beyond" Cancers 15, no. 14: 3599. https://doi.org/10.3390/cancers15143599
APA StylePetroni, M., La Monica, V., Fabretti, F., Augusto, M., Battaglini, D., Polonara, F., Di Giulio, S., & Giannini, G. (2023). The Multiple Faces of the MRN Complex: Roles in Medulloblastoma and Beyond. Cancers, 15(14), 3599. https://doi.org/10.3390/cancers15143599