
Alterations in the Epigenetic Machinery Associated with Prostate Cancer Health Disparities
 , ,
, ,  , and
, and
Abstract
Simple Summary
Abstract
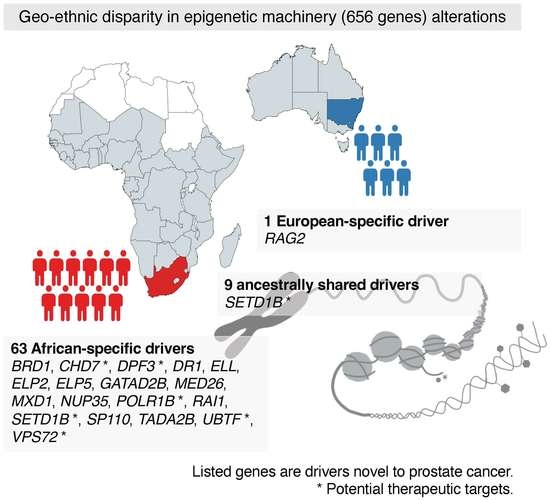
1. Introduction
2. Materials and Methods
2.1. Patient Clinical Characteristics and Genomic Data
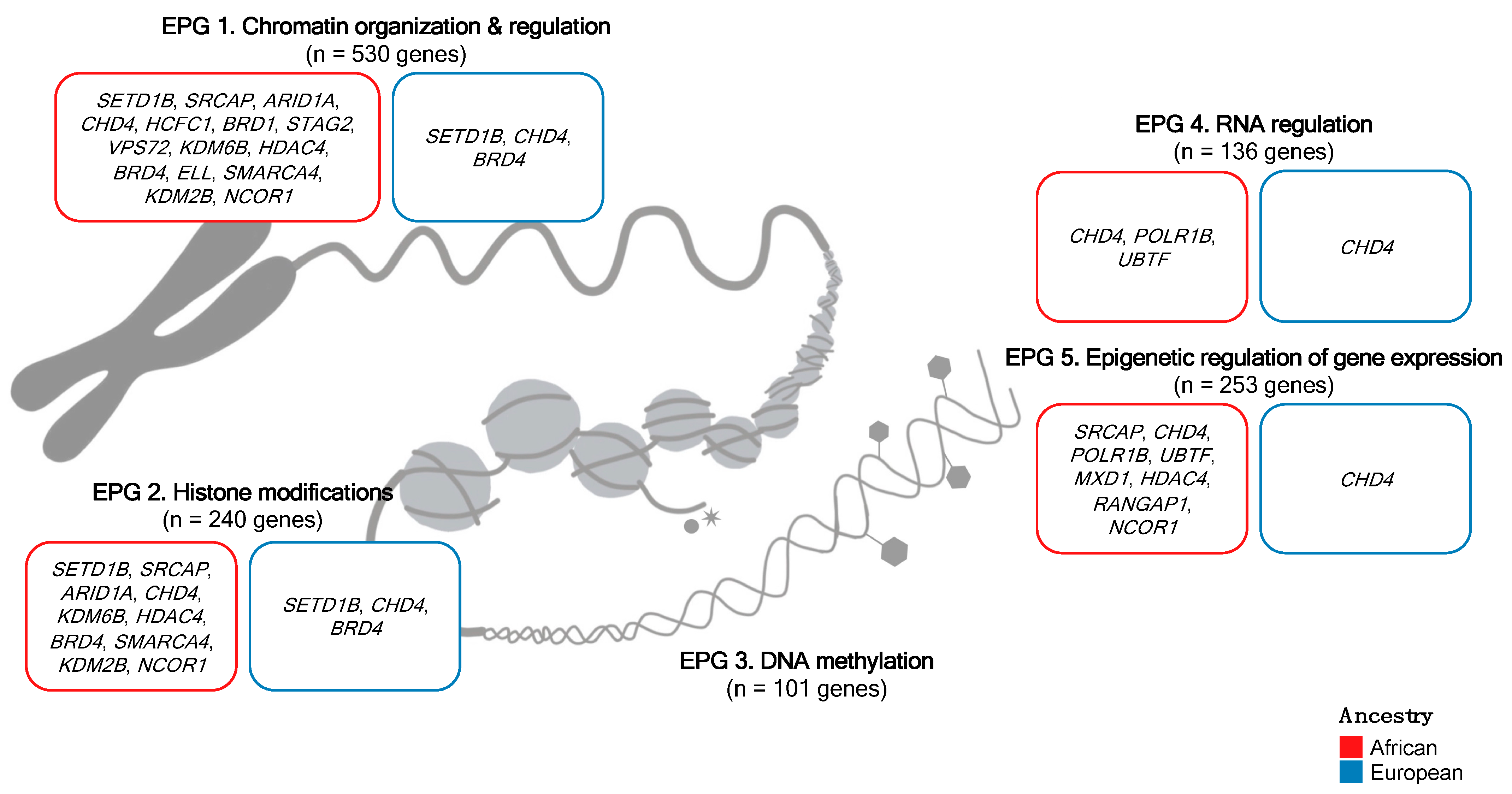
2.2. Epigenetic Process Group Classification
2.3. Tumor Mutational Burden (TMB), Damaging Variant Detection, and Mutational Frequency Analysis
2.4. Integrative Analysis of Epigenetic Machinery-Driven Prostate Cancer Subtypes
2.5. Statistical Analyses
3. Results
3.1. Tumor Mutational Landscape
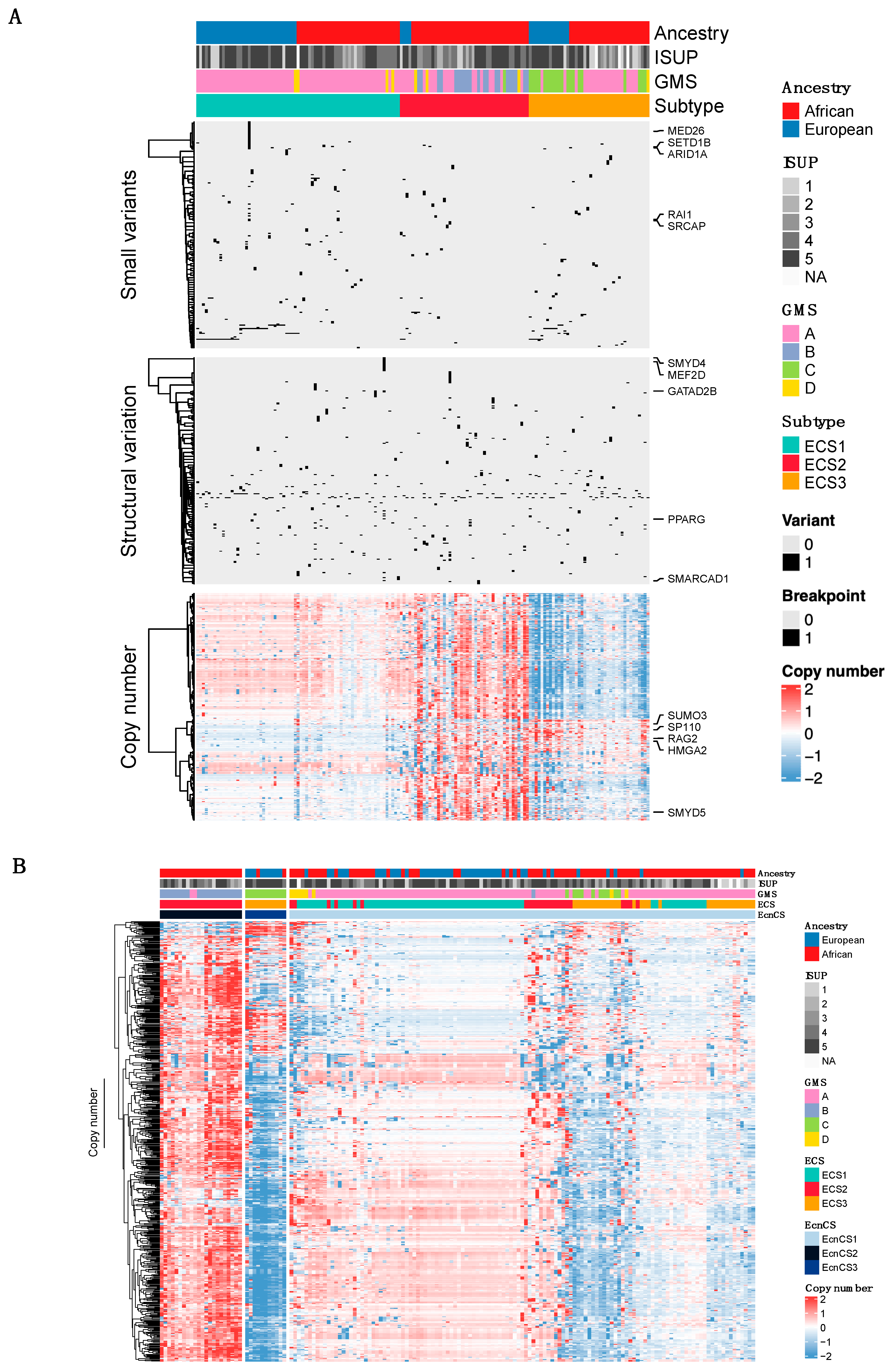
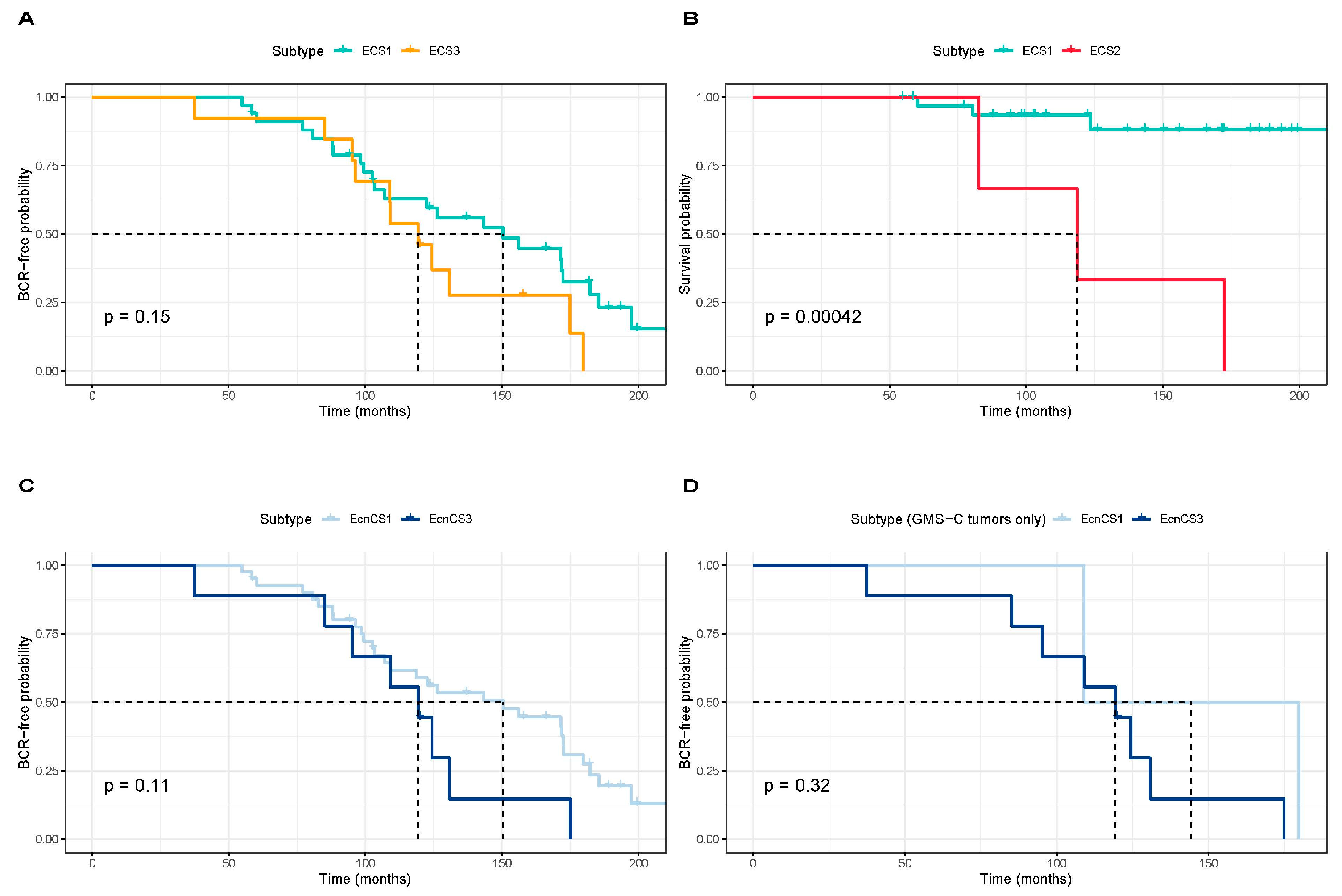
3.2. Integrative Clustering Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fahrner, J.A.; Bjornsson, H.T. Mendelian disorders of the epigenetic machinery: Postnatal malleability and therapeutic prospects. Hum. Mol. Genet. 2019, 28, 2–254. [Google Scholar] [CrossRef]
- Nilsson, E.E.; Sadler-Riggleman, I.; Skinner, M.K. Environmentally induced epigenetic transgenerational inheritance of disease. Environ. Epigenet. 2018, 4, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.H.; Morton, R.A.; Epstein, J.I.; Brooks, J.D.; Campbell, P.A.; Bova, G.S.; Hsieh, W.S.; Isaacs, W.B.; Nelson, W.G. Cytidine methylation of regulatory sequences near the pi-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 11733–11737. [Google Scholar] [CrossRef] [PubMed]
- Martignano, F.; Gurioli, G.; Salvi, S.; Calistri, D.; Costantini, M.; Gunelli, R.; de Giorgi, U.; Foca, F.; Casadio, V. GSTP1 methylation and protein expression in prostate cancer: Diagnostic implications. Dis. Markers 2016, 2016, 4358292. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Carroll, J.S.; Brown, M. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Mol. Cell 2005, 19, 631–642. [Google Scholar] [CrossRef]
- Wissmann, M.; Yin, N.; Müller, J.M.; Greschik, H.; Günther, T.; Metzger, E.; Schüle, R.; Fodor, B.D.; Jenuwein, T.; Vogler, C.; et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat. Cell Biol. 2007, 9, 347–353. [Google Scholar] [CrossRef]
- Yegnasubramanian, S.; de Marzo, A.M.; Nelson, W.G. Prostate cancer epigenetics: From basic mechanisms to clinical implications. Cold Spring Harb. Perspect. Med. 2019, 9, a030445. [Google Scholar] [CrossRef]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef]
- Jones, S.; Li, M.; Williams Parsons, D.; Zhang, X.; Wesseling, J.; Kristel, P.; Schmidt, M.K.; Markowitz, S.; Yan, H.; Bigner, D.; et al. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Hum. Mutat. 2012, 33, 100–103. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, L.; Yao, W.; Chen, K.; Xu, H.; Ye, Z. Integrated analysis of genetic abnormalities of the histone lysine methyltransferases in prostate cancer. Med. Sci. Monit. 2019, 25, 193. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The molecular taxonomy of primary prostate cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed]
- Morano, A.; Angrisano, T.; Russo, G.; Landi, R.; Pezone, A.; Bartollino, S.; Zuchegna, C.; Babbio, F.; Bonapace, I.M.; Allen, B.; et al. Targeted DNA methylation by homology-directed repair in mammalian cells. Transcription reshapes methylation on the repaired gene. Nucleic Acids Res. 2014, 42, 804–821. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.; Landi, R.; Pezone, A.; Morano, A.; Zuchegna, C.; Romano, A.; Muller, M.T.; Gottesman, M.E.; Porcellini, A.; Avvedimento, E.V. DNA damage and repair modify DNA methylation and chromatin domain of the targeted locus: Mechanism of allele methylation polymorphism. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.; Pezone, A.; Porcellini, A.; Muller, M.T.; Masternak, M.M. Non-homologous end joining induced alterations in DNA methylation: A source of permanent epigenetic change. Oncotarget 2017, 8, 40359. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, L.; Kucherlapati, M.; Hadjipanayis, A.; Pantazi, A.; Bristow, C.A.; Lee, E.A.; Mahadeshwar, H.S.; Tang, J.; Zhang, J.; et al. Global impact of somatic structural variation on the DNA methylome of human cancers. Genome Biol. 2019, 20, 1–24. [Google Scholar] [CrossRef]
- Sun, W.; Bunn, P.; Jin, C.; Little, P.; Zhabotynsky, V.; Perou, C.M.; Hayes, D.N.; Chen, M.; Lin, D.Y. The association between copy number aberration, DNA methylation and gene expression in tumor samples. Nucleic Acids Res. 2018, 46, 3009–3018. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Gheybi, K.; Jiang, J.; Mutambirwa, S.B.A.; Soh, P.X.Y.; Kote-Jarai, Z.; Jaratlerdsiri, W.; Eeles, R.A.; Bornman, M.S.R.; Hayes, V.M. Evaluating germline testing panels in southern African males with advanced prostate cancer. J. Natl. Compr. Cancer Netw. 2023, 21, 289–296. [Google Scholar] [CrossRef]
- Conti, D.V.; Darst, B.F.; Moss, L.C.; Saunders, E.J.; Sheng, X.; Chou, A.; Schumacher, F.R.; Olama, A.A.A.; Benlloch, S.; Dadaev, T.; et al. Trans-ancestry genome-wide association meta-analysis of prostate cancer identifies new susceptibility loci and informs genetic risk prediction. Nat. Genet. 2021, 53, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Lowder, D.; Rizwan, K.; McColl, C.; Paparella, A.; Ittmann, M.; Mitsiades, N.; Kaochar, S. Racial disparities in prostate cancer: A complex interplay between socioeconomic inequities and genomics. Cancer Lett. 2022, 531, 71–82. [Google Scholar] [CrossRef]
- Popejoy, A.B.; Fullerton, S.M. Genomics is failing on diversity. Nature 2016, 538, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Hayes, V.M.; Gong, T.; Mutambirwa, S.B.A.; Jaratlerdsiri, W.; Bornman, M.S.R. African inclusion in prostate cancer genomic studies provides the first glimpses into addressing health disparities through tailored clinical care. Clin. Transl. Med. 2023, 13, e1142. [Google Scholar] [CrossRef] [PubMed]
- Enokida, H.; Shiina, H.; Urakami, S.; Igawa, M.; Ogishima, T.; Pookot, D.; Li, L.C.; Tabatabai, Z.L.; Kawahara, M.; Nakagawa, M.; et al. Ethnic group-related differences in CpG hypermethylation of the GSTP1 gene promoter among African-American, Caucasian and Asian patients with prostate cancer. Int. J. Cancer 2005, 116, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Kwabi-Addo, B.; Wang, S.; Chung, W.; Jelinek, J.; Patierno, S.R.; Wang, B.D.; Andrawis, R.; Lee, N.H.; Apprey, V.; Issa, J.P.; et al. Identification of differentially methylated genes in normal prostate tissues from African American and Caucasian men. Clin. Cancer Res. 2010, 16, 3539–3547. [Google Scholar] [CrossRef]
- Devaney, J.M.; Wang, S.; Furbert-Harris, P.; Apprey, V.; Ittmann, M.; Wang, B.D.; Olender, J.; Lee, N.H.; Kwabi-Addo, B. Genome-wide differentially methylated genes in prostate cancer tissues from African-American and Caucasian men. Epigenetics 2015, 10, 319–328. [Google Scholar] [CrossRef]
- Rubicz, R.; Zhao, S.; Geybels, M.; Wright, J.L.; Kolb, S.; Klotzle, B.; Bibikova, M.; Lance, R.; Ostrander, E.A.; Feng, Z.; et al. DNA methylation profiles in African American prostate cancer patients in relation to disease progression. Genomics 2016, 111, 10–16. [Google Scholar] [CrossRef]
- Woodson, K.; Hayes, R.; Wideroff, L.; Villaruz, L.; Tangrea, J. Hypermethylation of GSTP1, CD44, and E-cadherin genes in prostate cancer among US Blacks and Whites. Prostate 2003, 55, 199–205. [Google Scholar] [CrossRef]
- Tang, D.; Kryvenko, O.N.; Mitrache, N.; Do, K.C.; Jankowski, M.; Chitale, D.A.; Trudeau, S.; Rundle, A.; Belinsky, S.A.; Rybicki, B.A. Methylation of the RARB gene increases prostate cancer risk in black Americans. J. Urol. 2013, 190, 317–324. [Google Scholar] [CrossRef]
- Belinky, F.; Nativ, N.; Stelzer, G.; Zimmerman, S.; Stein, T.I.; Safran, M.; Lancet, D. PathCards: Multi-source consolidation of human biological pathways. Database 2015, 2015, bav006. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.M.; Walsh, L.A.; Chan, T.A. Driver mutations of cancer epigenomes. Protein Cell 2014, 5, 265–296. [Google Scholar] [CrossRef]
- Guo, M.; Peng, Y.; Gao, A.; Du, C.; Herman, J.G. Epigenetic heterogeneity in cancer. Biomark. Res. 2019, 7, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, J.A.; Bjornsson, H.T. Mendelian disorders of the epigenetic machinery: Tipping the balance of chromatin states. Annu. Rev. Genom. Hum. Genet. 2014, 15, 269–293. [Google Scholar] [CrossRef]
- Jaratlerdsiri, W.; Jiang, J.; Gong, T.; Patrick, S.M.; Willet, C.; Chew, T.; Lyons, R.J.; Haynes, A.M.; Pasqualim, G.; Louw, M.; et al. African-specific molecular taxonomy of prostate cancer. Nature 2022, 609, 552–559. [Google Scholar] [CrossRef]
- Raj, A.; Stephens, M.; Pritchard, J.K. fastSTRUCTURE: Variational inference of population structure in large SNP data sets. Genetics 2014, 197, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef]
- Kinsella, R.J.; Kähäri, A.; Haider, S.; Zamora, J.; Proctor, G.; Spudich, G.; Almeida-King, J.; Staines, D.; Derwent, P.; Kerhornou, A.; et al. Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database 2011, 2011, bar030. [Google Scholar] [CrossRef]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2016, 11, 1–9. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef]
- Oscanoa, J.; Sivapalan, L.; Gadaleta, E.; Dayem Ullah, A.Z.; Lemoine, N.R.; Chelala, C. SNPnexus: A web server for functional annotation of human genome sequence variation (2020 update). Nucleic Acids Res. 2020, 48, W185–W192. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Xu, H.; Yang, Y.; Zhu, J.; Liu, P.; Lu, Y. DrGaP: A powerful tool for identifying driver genes and pathways in cancer sequencing studies. Am. J. Hum. Genet. 2013, 93, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Pleasance, E.D. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Lu, X.; Meng, J.; Zhou, Y.; Jiang, L.; Yan, F. MOVICS: An R package for multi-omics integration and visualization in cancer subtyping. Bioinformatics 2020, 36, 5539–5541. [Google Scholar] [CrossRef]
- Monti, S.; Tamayo, P.; Mesirov, J.; Golub, T. Consensus clustering: A resampling-based method for class discovery and visualization of gene expression microarray data. Mach. Learn. 2003, 52, 91–118. [Google Scholar] [CrossRef]
- Mo, Q.; Shen, R.; Guo, C.; Vannucci, M.; Chan, K.S.; Hilsenbeck, S.G. A fully Bayesian latent variable model for integrative clustering analysis of multi-type omics data. Biostatistics 2018, 19, 71–86. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Kolde, R. Pheatmap: Pretty Heatmaps, R Package Version 1.0.12; 2019. Available online: https://cran.r-project.org/package=pheatmap (accessed on 28 June 2023).
- Jaratlerdsiri, W.; Chan, E.K.F.; Gong, T.; Petersen, D.C.; Kalsbeek, A.M.F.; Venter, P.A.; Stricker, P.D.; Bornman, M.S.R.; Hayes, V.M. Whole-genome sequencing reveals elevated tumor mutational burden and initiating driver mutations in African men with treatment-naïve, high-risk prostate cancer. Cancer Res. 2018, 78, 6736–6746. [Google Scholar] [CrossRef]
- Fagan, R.J.; Dingwall, A.K. COMPASS ascending: Emerging clues regarding the roles of MLL3/KMT2C and MLL2/KMT2D proteins in cancer. Cancer Lett. 2019, 458, 56–65. [Google Scholar] [CrossRef]
- Grypari, I.M.; Logotheti, S.; Zolota, V.; Troncoso, P.; Efstathiou, E.; Bravou, V.; Melachrinou, M.; Logothetis, C.; Tzelepi, V.; Zhang, H. The protein arginine methyltransferases (PRMTs) PRMT1 and CARM1 as candidate epigenetic drivers in prostate cancer progression. Medicine 2021, 100, e27094. [Google Scholar] [CrossRef]
- Fan, H.T.; Shi, Y.Y.; Lin, Y.; Yang, X.P. EHMT2 promotes the development of prostate cancer by inhibiting PI3K/AKT/mTOR pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 7808–7815. [Google Scholar] [PubMed]
- Burleson, M.; Deng, J.J.; Qin, T.; Duong, T.M.; Yan, Y.; Gu, X.; Das, D.; Easley, A.; Liss, M.A.; Renee Yew, P.; et al. GLI3 is stabilized by SPOP mutations and promotes castration resistance via functional cooperation with androgen receptor in prostate cancer. Mol. Cancer Res. 2022, 20, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Burdelski, C.; Ruge, O.M.; Melling, N.; Koop, C.; Simon, R.; Steurer, S.; Sauter, G.; Kluth, M.; Hube-Magg, C.; Minner, S.; et al. HDAC1 overexpression independently predicts biochemical recurrence and is associated with rapid tumor cell proliferation and genomic instability in prostate cancer. Exp. Mol. Pathol. 2015, 98, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Shi, X.; Tian, F.; Fang, Y.; Wu, J.B.; Mrdenovic, S.; Nian, X.; Ji, J.; Xu, H.; Kong, C.; et al. KDM6B is an androgen regulated gene and plays oncogenic roles by demethylating H3K27me3 at cyclin D1 promoter in prostate cancer. Cell Death Dis. 2021, 12, 2. [Google Scholar] [CrossRef]
- Xie, H.; Zhao, J.; Wan, J.; Zhao, J.; Wang, Q.; Yang, X.; Yang, W.; Lin, P.; Yu, X. Long non-coding RNA AC245100.4 promotes prostate cancer tumorigenesis via the microRNA-145-5p/RBBP5 axis. Oncol. Rep. 2021, 45, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.T.; Lin, T.P.; Campbell, M.; Pan, C.C.; Lee, S.H.; Lee, H.C.; Yang, M.H.; Kung, H.J.; Chang, P.C. REST is a crucial regulator for acquiring EMT-like and stemness phenotypes in hormone-refractory prostate cancer. Sci. Rep. 2017, 7, 42795. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.M.; Qiao, Q.D.; Xie, H.F.; Wei, J.X. Breast cancer metastasis suppressor 1 (BRMS1) suppresses prostate cancer progression by inducing apoptosis and regulating invasion. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 68–75. [Google Scholar]
- Zhu, S.; Xu, Y.; Song, M.; Chen, G.; Wang, H.; Zhao, Y.; Wang, Z.; Li, F. PRDM16 is associated with evasion of apoptosis by prostatic cancer cells according to RNA interference screening. Mol. Med. Rep. 2016, 14, 3357–3361. [Google Scholar] [CrossRef]
- Goll, M.G.; Bestor, T.H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef]
- Tao, Q.; Huang, H.; Geiman, T.M.; Lim, C.Y.; Fu, L.; Qiu, G.H.; Robertson, K.D. Defective de novo methylation of viral and cellular DNA sequences in ICF syndrome cells. Hum. Mol. Genet. 2002, 11, 2091–2102. [Google Scholar] [CrossRef]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef]
- Singal, R.; Das, P.; Manoharan, M.; Reis, I.; Schlesselman, J. Polymorphisms in the DNA methyltransferase 3b gene and prostate cancer risk. Oncol. Rep. 2005, 14, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Ranieri, G.; Muzi, P.; Marampon, F.; Mancini, A.; Di Pasquale, B.; Di Clemente, L.; Dolo, V.; D’Alessandro, A.M.; Festuccia, C. Increased levels of DNA methyltransferases are associated with the tumorigenic capacity of prostate cancer cells. Oncol. Rep. 2013, 29, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Reddy Parine, N.; Alanazi, I.O.; Shaik, J.P.; Aldhaian, S.; Aljebreen, A.M.; Alharbi, O.; Almadi, M.A.; Azzam, N.A.; Alanazi, M.; Yang, Q. TDG gene polymorphisms and their possible association with colorectal cancer: A case control study. J. Oncol. 2019, 2019, 7091815. [Google Scholar] [CrossRef]
- Ruczinski, I.; Jorgensen, T.J.; Shugart, Y.Y.; Schaad, Y.B.; Kessing, B.; Hoffman-Bolton, J.; Helzlsouer, K.J.; Kao, W.H.L.; Wheless, L.; Francis, L.; et al. A population-based study of DNA repair gene variants in relation to non-melanoma skin cancer as a marker of a cancer-prone phenotype. Carcinogenesis 2012, 33, 1692–1698. [Google Scholar] [CrossRef]
- Xu, X.; Yu, T.; Shi, J.; Chen, X.; Zhang, W.; Lin, T.; Liu, Z.; Wang, Y.; Zeng, Z.; Wang, C.; et al. Thymine DNA glycosylase is a positive regulator of Wnt signaling in colorectal cancer. J. Biol. Chem. 2014, 289, 8881–8890. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Li, S.; Zhang, K.; Zhao, R.; Cui, J.; Zhou, W.; Liu, Y.; Zhang, L.; Cheng, Y. N 6-methyladenosine demethylase ALKBH5 suppresses malignancy of esophageal cancer by regulating microRNA biogenesis and RAI1 expression. Oncogene 2021, 40, 5600–5612. [Google Scholar] [CrossRef]
- Meeks, J.J.; Shilatifard, A. Multiple roles for the MLL/COMPASS family in the epigenetic regulation of gene expression and in cancer. Annu. Rev. Cancer Biol. 2017, 1, 425–446. [Google Scholar] [CrossRef]
- Choi, Y.J.; Oh, H.R.; Choi, M.R.; Gwak, M.; An, C.H.; Chung, Y.J.; Yoo, N.J.; Lee, S.H. Frameshift mutation of a histone methylation-related gene SETD1B and its regional heterogeneity in gastric and colorectal cancers with high microsatellite instability. Hum. Pathol. 2014, 45, 1674–1681. [Google Scholar] [CrossRef] [PubMed]
- Fowler, T.; Ghatak, P.; Price, D.H.; Conaway, R.; Conaway, J.; Chiang, C.M.; Bradner, J.E.; Shilatifard, A.; Roy, A.L. Regulation of MYC expression and differential JQ1 sensitivity in cancer cells. PLoS ONE 2014, 9, e87003. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Peng, S.; Li, Z.; Zhang, R.; Xiao, Y.; Chen, X.; Zhu, J.; Li, B.; Lv, X. Identification of therapeutic targets and prognostic biomarkers among genes from the mediator complex family in the hepatocellular carcinoma tumour-immune microenvironment. Comput. Math. Methods Med. 2022, 2022, 2021613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, Y.; Shi, Z.; Liu, Z.; Chen, H.; Wang, X.; Cheng, Y.; Xi, L.; Li, X.; Zhang, C.; et al. Integrated analysis of genes encoding ATP-dependent chromatin remodellers identifies CHD7 as a potential target for colorectal cancer therapy. Clin. Transl. Med. 2022, 12, e953. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Liu, H.; Zhao, J.; Ma, X.; Qi, W. POLR1B is upregulated and promotes cell proliferation in non-small cell lung cancer. Oncol. Lett. 2020, 19, 671–680. [Google Scholar] [CrossRef]
- Chen, T.; Tu, Y.; Lv, D.; Lin, K.; Tang, H.; Huang, W. Vacuolar protein sorting-associated protein 72 homolog (VPS72) binding to lysine acetyltransferase 5 (KAT5) promotes the proliferation, invasion and migration of hepatocellular carcinoma through regulating phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) signaling pathway. Bioengineered 2022, 13, 9197–9210. [Google Scholar]
- Zhang, J.; Zhang, J.; Liu, W.; Ge, R.; Gao, T.; Tian, Q.; Mu, X.; Zhao, L.; Li, X. UBTF facilitates melanoma progression via modulating MEK1/2-ERK1/2 signalling pathways by promoting GIT1 transcription. Cancer Cell Int. 2021, 21, 543. [Google Scholar] [CrossRef]
- Lin, W.; Dai, W.; Xu, X.; Yu, Q.; Zhang, B.; Li, J.; Li, H. Downregulation of DPF3 promotes the proliferation and motility of breast cancer cells through activating JAK2/STAT3 signaling. Biochem. Biophys. Res. Commun. 2019, 514, 639–644. [Google Scholar] [CrossRef]
- Chen, D.; Li, T.; Wang, C.; Lei, G.; Wang, R.; Wang, Z.; Yu, L.; Yan, J.; Zhang, P.; Wang, X.; et al. High-level SETD1B gene expression is associated with unfavorable prognosis in hepatocellular carcinoma. Mol. Med. Rep. 2019, 19, 1587–1594. [Google Scholar] [CrossRef]
- Hu, Z.; Zhou, J.; Jiang, J.; Yuan, J.; Zhang, Y.; Wei, X.; Loo, N.; Wang, Y.; Pan, Y.; Zhang, T.; et al. Genomic characterization of genes encoding histone acetylation modulator proteins identifies therapeutic targets for cancer treatment. Nat. Commun. 2019, 10, 733. [Google Scholar] [CrossRef]
- Moradi Marjaneh, R.; Hassanian, S.M.; Ghobadi, N.; Ferns, G.A.; Karimi, A.; Jazayeri, M.H.; Nasiri, M.; Avan, A.; Khazaei, M. Targeting the death receptor signaling pathway as a potential therapeutic target in the treatment of colorectal cancer. J. Cell. Physiol. 2018, 233, 6538–6549. [Google Scholar] [CrossRef] [PubMed]
- Grzeskowiak, C.L.; Kundu, S.T.; Mo, X.; Ivanov, A.A.; Zagorodna, O.; Lu, H.; Chapple, R.H.; Tsang, Y.H.; Moreno, D.; Mosqueda, M.; et al. In vivo screening identifies GATAD2B as a metastasis driver in KRAS-driven lung cancer. Nat. Commun. 2018, 9, 2732. [Google Scholar] [CrossRef] [PubMed]
- Korakiti, A.M.; Moutafi, M.; Zografos, E.; Dimopoulos, M.A.; Zagouri, F. The genomic profile of pregnancy-associated breast cancer: A systematic review. Front. Oncol. 2020, 10, 1773. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Sentürk, N.; Song, C.; Grummt, I. lncRNA PAPAS tethered to the rDNA enhancer recruits hypophosphorylated CHD4/NuRD to repress rRNA synthesis at elevated temperatures. Genes Dev. 2018, 32, 836–848. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Africans (n = 109) | Europeans (n = 56) | |||||
|---|---|---|---|---|---|---|
| Total Genes | Total Damaging Variants | Samples that Contain Damaging Variants | Total Damaging Variants | Samples that Contain Damaging Variants | p-Value * | |
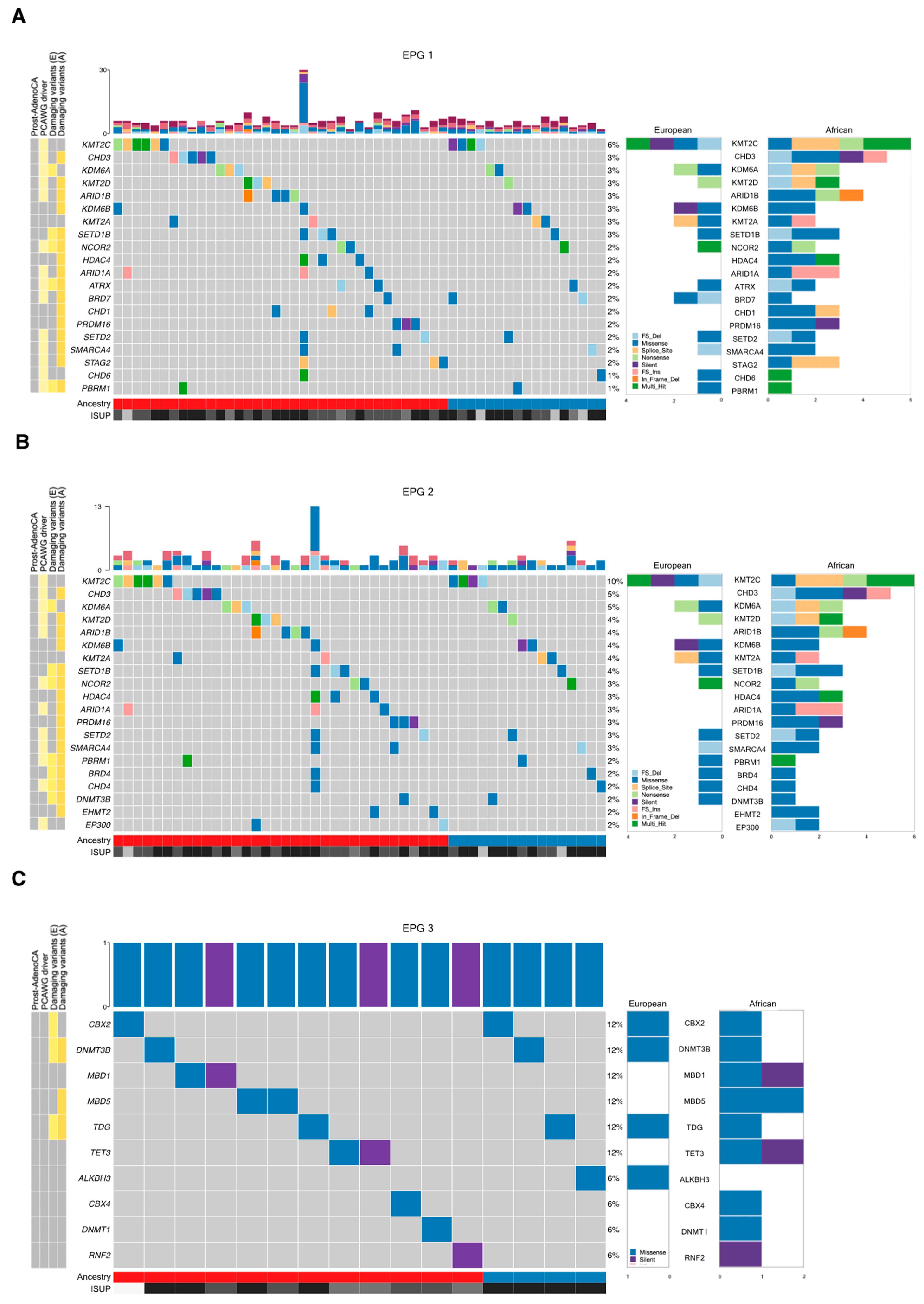
| EPG 1 | 530 | 71 | 37 | 26 | 22 | 0.4988 |
| EPG 2 | 240 | 35 | 21 | 13 | 12 | 0.8375 |
| EPG 3 | 101 | 5 | 5 | 3 | 3 | 1 |
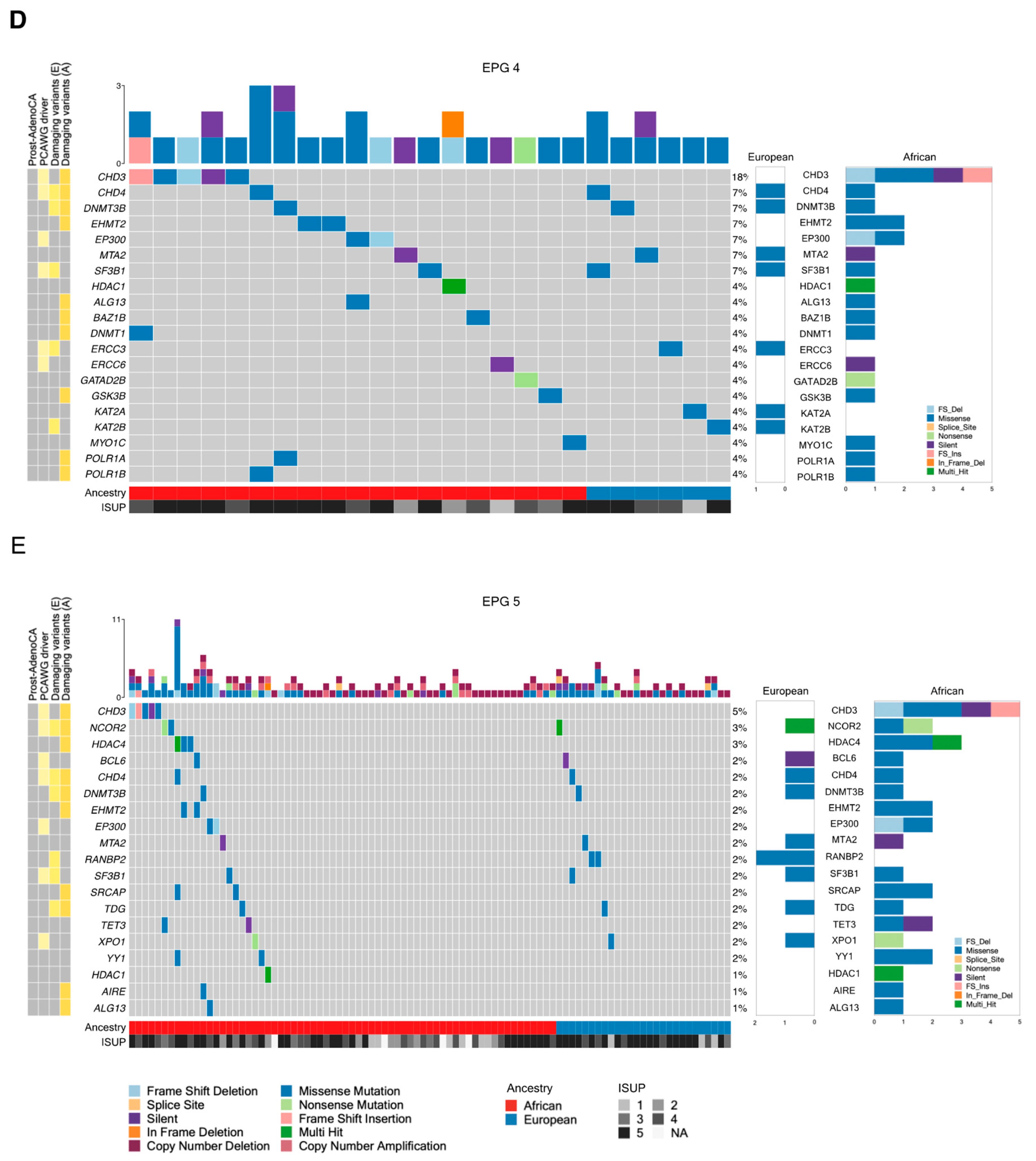
| EPG 4 | 136 | 12 | 9 | 5 | 4 | 1 |
| EPG 5 | 253 | 31 | 23 | 15 | 13 | 0.8426 |
| Africans | Africans and Europeans (Shared) | Europeans | |
|---|---|---|---|
| (Unique) | (Unique) | ||
| EPG 1 | AIRE, ARID1A 2, ARID1B 2, BAZ1B, BRD1, BRD7, BRWD1 2, CARM1, CHD1 1,2, CHD3 2, CXXC1, DNMT1, EHMT1, EHMT2, ELL, GLI1, HCFC1, HDAC4 1,2, HJURP, KAT8, KDM2B, KDM6B 1, KMT2D 2, MBD5, MED12, MEN1, NCOR1, NUP188, NUP93, PAX3, PELP1, POLR1A, POLR3A, PRDM16, PRMT6, PSIP1 2, RBBP5, REST, RNF20, RTF1, SATB2, SETD2 2, SMARCA1 2, SMARCA4 1, SRCAP, SSRP1, STAG2 1,2, TADA2B 1,2, TAF1, TAF6L, TPR, VPS72, WAPL, WHSC1L1 1 | ATRX 2, BRD4, CHD4, DNMT3B, NCOR2 2, PADI1, PBRM1 1, SETD1B 2 | AR, CBX2, CLOCK, ELOA, ERCC3, HNRNPA2B1, JAK2, KAT2B, KAT6A, KDM1B, KDM6A, KMT2B 3, KMT5C, PRDM5, RANBP2, RB1, RNF40, SIRT3 |
| EPG 2 | AIRE, ARID1A 2, ARID1B 2, CARM1 2, CHD3 2, DNMT1, EHMT1, EHMT2 2, HDAC4 1,2, KDM2B, KDM6B 1,2, KMT2D 2, MBD5, NCOR1, PRDM16 2, PRMT6, RBBP5 2, REST 2, SETBP1, SETD2 2, SMARCA4 1,2, SRCAP, WHSC1L1 1 | BRD4, CHD4, DNMT3B, NCOR2 2, PBRM1 1, SETD1B 2 | JAK2, KDM1B, KDM6A 4, KMT2B 3, KMT5C, PRDM5, SETD5 |
| EPG 3 | DNMT1, MBD5 | DNMT3B, TDG | CBX2 |
| EPG 4 | ALG13, BAZ1B, CHD3 2, DNMT1, EHMT2, GSK3B, POLR1A, POLR1B, TDRD7, UBTF | CHD4, DNMT3B | ERCC3, KAT2B, SF3B1 |
| EPG 5 | AIRE, ALG13, BAZ1B, CHD3 2, DNMT1, EHMT2, FOXO3, GSK3B, HDAC4 1,2, MXD1, NCOR1, POLR1A, POLR1B, RANGAP1, REST, SRCAP, UBTF, XRCC6 | CHD4, DNMT3B, NCOR2, TDG, TP53 1,2,3,4 | ERCC3, HDAC9, KAT2B, RANBP2, RXRB, SF3B1, SIRT3, STAT3 |
| Africans | Africans and Europeans | Europeans | |
|---|---|---|---|
| (Unique) | (Shared) | (Unique) | |
| EPG 1 | ARID1A 1,2, ARID1B 1,2, ATRX 1,2, BRWD1 1,2, CHD1 2, CHD3 1,2, CHD7, DPF3, ELP2, EP300 1, GATAD2B, GLI3, HDAC1, HDAC3 1, HDAC4 2, KMT2A, KMT2D 1,2, NCOR2 1,2, NUP35, PSIP1 1,2, SETD1B 2, SETD2 1,2, SMARCA1 1,2, STAG2 1,2, TADA2B 2, XPO1 1 | KMT2C 1 | — |
| EPG 2 | ARID1A 1,2, ARID1B 1,2, ARID5B 1, BRMS1, CARM1 2, CHD3 1,2, EHMT2 2, EP300 1, GATAD2B, HDAC1, HDAC3 1, HDAC4 2, KDM6B 2, KMT2A, KMT2D 1,2, NCOR2 1,2, PRDM16 2, RBBP5 2, REST 2, SETD1B 2, SETD2 1,2, SMARCA4 1,2 | KDM6A 1,3, KMT2C 1 | — |
| EPG 3 | — | — | — |
| EPG 4 | CHD3 1,2, HDAC1 | — | — |
| EPG 5 | CHD31,2, HDAC1, HDAC4 2 | TP53 1,2,3 | — |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Craddock, J.; Jiang, J.; Patrick, S.M.; Mutambirwa, S.B.A.; Stricker, P.D.; Bornman, M.S.R.; Jaratlerdsiri, W.; Hayes, V.M. Alterations in the Epigenetic Machinery Associated with Prostate Cancer Health Disparities. Cancers 2023, 15, 3462. https://doi.org/10.3390/cancers15133462
Craddock J, Jiang J, Patrick SM, Mutambirwa SBA, Stricker PD, Bornman MSR, Jaratlerdsiri W, Hayes VM. Alterations in the Epigenetic Machinery Associated with Prostate Cancer Health Disparities. Cancers. 2023; 15(13):3462. https://doi.org/10.3390/cancers15133462
Chicago/Turabian StyleCraddock, Jenna, Jue Jiang, Sean M. Patrick, Shingai B. A. Mutambirwa, Phillip D. Stricker, M. S. Riana Bornman, Weerachai Jaratlerdsiri, and Vanessa M. Hayes. 2023. "Alterations in the Epigenetic Machinery Associated with Prostate Cancer Health Disparities" Cancers 15, no. 13: 3462. https://doi.org/10.3390/cancers15133462
APA StyleCraddock, J., Jiang, J., Patrick, S. M., Mutambirwa, S. B. A., Stricker, P. D., Bornman, M. S. R., Jaratlerdsiri, W., & Hayes, V. M. (2023). Alterations in the Epigenetic Machinery Associated with Prostate Cancer Health Disparities. Cancers, 15(13), 3462. https://doi.org/10.3390/cancers15133462