
Emerging Role of Glioma Stem Cells in Mechanisms of Therapy Resistance



{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Cancer Stem Cells in Glioblastoma
2.1. Tumor Initiation and Cell of Origin
2.1.1. NSCs, Committed Precurser Cells, and the Subventricular Zone
2.1.2. Cell of Origin in IDH-Mutant and H3K27-Altered Gliomas
3. Tumor Heterogeneity
3.1. Intertumor Heterogeneity and GBM Classification and Subtypes
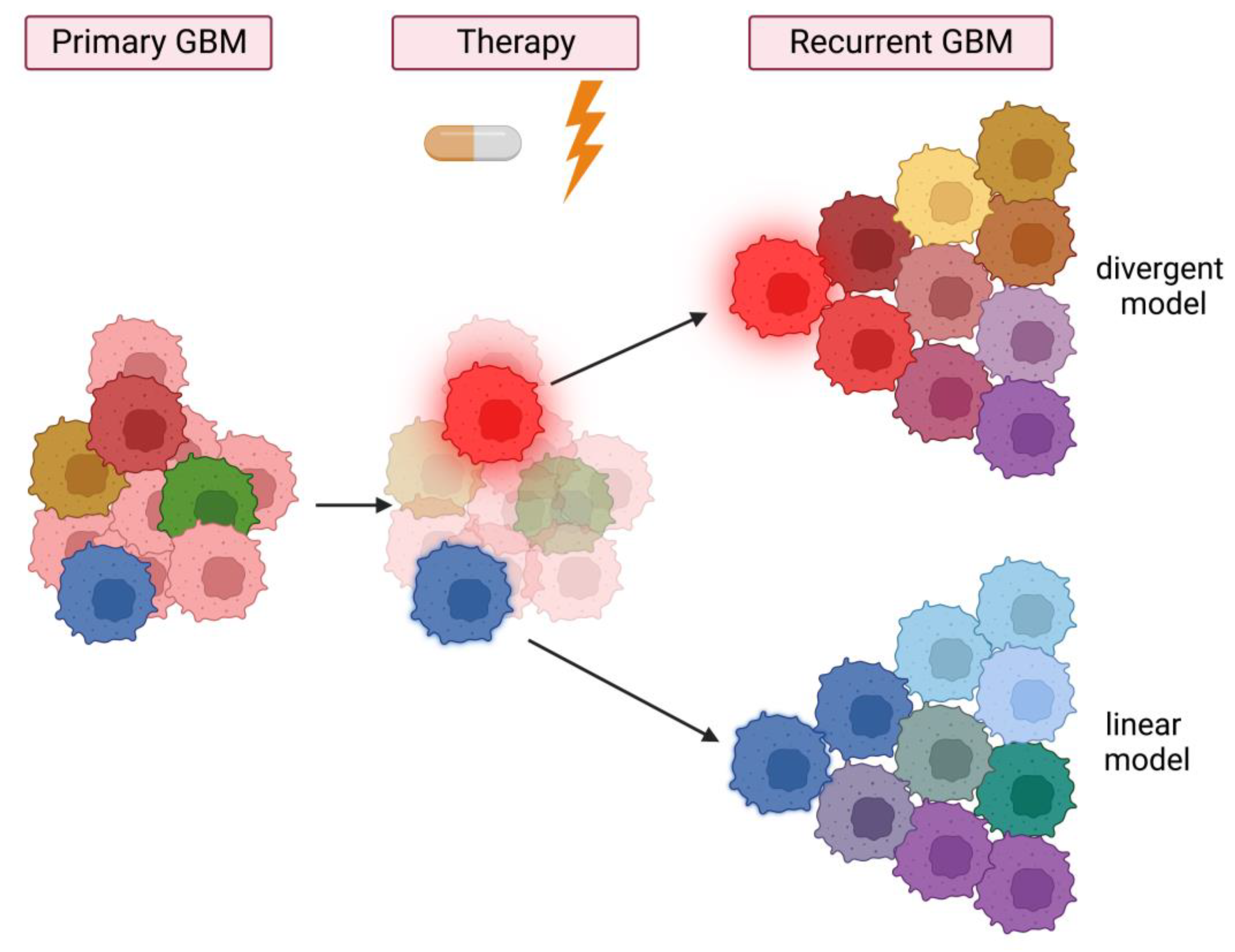
3.2. Intratumor Heterogeneity
4. Plasticity
5. Therapy Resistance
5.1. DNA Repair Systems
5.2. DNA Damage Response (DDR)
5.3. Immunosuppression by Dynamic Crosstalk with Cells of the Immune System
5.4. Chemoresistance in GSCs
6. Targeting Therapy Resistance in GSCs
6.1. Targeting Self-Renewal and Resistance in GSCs
6.2. GSCs in the Context of Immunomodulatory Approaches
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, A.M.; Taylor, J.W.; Wiencke, J.K.; Wrensch, M.R. Genetic and molecular epidemiology of adult diffuse glioma. Nat. Rev. Neurol. 2019, 15, 405–417. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro Oncol. 2020, 22, iv1–iv96. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef]
- Sell, S. Stem cell origin of cancer and differentiation therapy. Crit. Rev. Oncol. Hematol. 2004, 51, 1–28. [Google Scholar] [CrossRef]
- McCulloch, E.A.; Till, J.E. The radiation sensitivity of normal mouse bone marrow cells, determined by quantitative marrow transplantation into irradiated mice. Radiat. Res. 1960, 13, 115–125. [Google Scholar] [CrossRef]
- Becker, A.J.; Mc, C.E.; Till, J.E. Cytological demonstration of the clonal nature of spleen colonies derived from transplanted mouse marrow cells. Nature 1963, 197, 452–454. [Google Scholar] [CrossRef]
- Beck, B.; Blanpain, C. Unravelling cancer stem cell potential. Nat. Rev. Cancer 2013, 13, 727–738. [Google Scholar] [CrossRef]
- Furth, J.; Kahn, M.C.; Breedis, C. The Transmission of Leukemia of Mice with a Single Cell. Am. Journal. Cancer 1937, 31, 276–282. [Google Scholar]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, H.J.; Blair, A.; Zapf, R.W. Characterization of a hierarchy in human acute myeloid leukemia progenitor cells. Blood 1996, 87, 4754–4761. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Todaro, M.; Alea, M.P.; Di Stefano, A.B.; Cammareri, P.; Vermeulen, L.; Iovino, F.; Tripodo, C.; Russo, A.; Gulotta, G.; Medema, J.P.; et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell 2007, 1, 389–402. [Google Scholar] [CrossRef]
- Tomizawa, Y.; Wu, T.T.; Wang, K.K. Epithelial mesenchymal transition and cancer stem cells in esophageal adenocarcinoma originating from Barrett’s esophagus. Oncol. Lett. 2012, 3, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Chan, K.W.; Hu, L.; Lee, T.K.; Wo, J.Y.; Ng, I.O.; Zheng, B.J.; Guan, X.Y. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 2007, 132, 2542–2556. [Google Scholar] [CrossRef]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef]
- Alvero, A.B.; Chen, R.; Fu, H.H.; Montagna, M.; Schwartz, P.E.; Rutherford, T.; Silasi, D.A.; Steffensen, K.D.; Waldstrom, M.; Visintin, I.; et al. Molecular phenotyping of human ovarian cancer stem cells unravels the mechanisms for repair and chemoresistance. Cell Cycle 2009, 8, 158–166. [Google Scholar] [CrossRef]
- Fioriti, D.; Mischitelli, M.; Di Monaco, F.; Di Silverio, F.; Petrangeli, E.; Russo, G.; Giordano, A.; Pietropaolo, V. Cancer stem cells in prostate adenocarcinoma: A target for new anticancer strategies. J. Cell. Physiol. 2008, 216, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Takaishi, S.; Okumura, T.; Wang, T.C. Gastric cancer stem cells. J. Clin. Oncol. 2008, 26, 2876–2882. [Google Scholar] [CrossRef] [PubMed]
- Todaro, M.; Iovino, F.; Eterno, V.; Cammareri, P.; Gambara, G.; Espina, V.; Gulotta, G.; Dieli, F.; Giordano, S.; De Maria, R.; et al. Tumorigenic and metastatic activity of human thyroid cancer stem cells. Cancer Res. 2010, 70, 8874–8885. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Ailles, L.E.; Weissman, I.L. Cancer stem cells in solid tumors. Curr. Opin. Biotechnol. 2007, 18, 460–466. [Google Scholar] [CrossRef]
- Magee, J.A.; Piskounova, E.; Morrison, S.J. Cancer stem cells: Impact, heterogeneity, and uncertainty. Cancer Cell 2012, 21, 283–296. [Google Scholar] [CrossRef]
- Meyer, M.; Reimand, J.; Lan, X.; Head, R.; Zhu, X.; Kushida, M.; Bayani, J.; Pressey, J.C.; Lionel, A.C.; Clarke, I.D.; et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc. Natl. Acad. Sci. USA 2015, 112, 851–856. [Google Scholar] [CrossRef]
- Kim, H.; Zheng, S.; Amini, S.S.; Virk, S.M.; Mikkelsen, T.; Brat, D.J.; Grimsby, J.; Sougnez, C.; Muller, F.; Hu, J.; et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015, 25, 316–327. [Google Scholar] [CrossRef]
- Kumar, A.; Boyle, E.A.; Tokita, M.; Mikheev, A.M.; Sanger, M.C.; Girard, E.; Silber, J.R.; Gonzalez-Cuyar, L.F.; Hiatt, J.B.; Adey, A.; et al. Deep sequencing of multiple regions of glial tumors reveals spatial heterogeneity for mutations in clinically relevant genes. Genome Biol. 2014, 15, 530. [Google Scholar] [CrossRef]
- Lee, J.K.; Wang, J.; Sa, J.K.; Ladewig, E.; Lee, H.O.; Lee, I.H.; Kang, H.J.; Rosenbloom, D.S.; Camara, P.G.; Liu, Z.; et al. Spatiotemporal genomic architecture informs precision oncology in glioblastoma. Nat. Genet. 2017, 49, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavare, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.I.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.J.; et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e46. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Snuderl, M.; Fazlollahi, L.; Le, L.P.; Nitta, M.; Zhelyazkova, B.H.; Davidson, C.J.; Akhavanfard, S.; Cahill, D.P.; Aldape, K.D.; Betensky, R.A.; et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 2011, 20, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, I.H.; Cho, H.J.; Park, C.K.; Jung, Y.S.; Kim, Y.; Nam, S.H.; Kim, B.S.; Johnson, M.D.; Kong, D.S.; et al. Spatiotemporal Evolution of the Primary Glioblastoma Genome. Cancer Cell 2015, 28, 318–328. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Reinartz, R.; Wang, S.; Kebir, S.; Silver, D.J.; Wieland, A.; Zheng, T.; Kupper, M.; Rauschenbach, L.; Fimmers, R.; Shepherd, T.M.; et al. Functional Subclone Profiling for Prediction of Treatment-Induced Intratumor Population Shifts and Discovery of Rational Drug Combinations in Human Glioblastoma. Clin. Cancer Res. 2017, 23, 562–574. [Google Scholar] [CrossRef]
- Qin, S.; Jiang, J.; Lu, Y.; Nice, E.C.; Huang, C.; Zhang, J.; He, W. Emerging role of tumor cell plasticity in modifying therapeutic response. Signal. Transduct. Target. Ther. 2020, 5, 228. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e821. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, R.; Wu, M.; Johnson, K.; Kim, H.; Celebre, A.; Shahzad, U.; Graham, M.S.; Kessler, J.A.; Chuang, J.H.; Karamchandani, J.; et al. BMP signaling mediates glioma stem cell quiescence and confers treatment resistance in glioblastoma. Sci. Rep. 2019, 9, 14569. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Jorg, D.J.; Cavalli, F.M.G.; Richards, L.M.; Nguyen, L.V.; Vanner, R.J.; Guilhamon, P.; Lee, L.; Kushida, M.M.; Pellacani, D.; et al. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 2017, 549, 227–232. [Google Scholar] [CrossRef]
- Ding, B.S.; James, D.; Iyer, R.; Falciatori, I.; Hambardzumyan, D.; Wang, S.; Butler, J.M.; Rabbany, S.Y.; Hormigo, A. Prominin 1/CD133 endothelium sustains growth of proneural glioma. PLoS ONE 2013, 8, e62150. [Google Scholar] [CrossRef]
- Beier, D.; Hau, P.; Proescholdt, M.; Lohmeier, A.; Wischhusen, J.; Oefner, P.J.; Aigner, L.; Brawanski, A.; Bogdahn, U.; Beier, C.P. CD133(+) and CD133(-) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007, 67, 4010–4015. [Google Scholar] [CrossRef]
- Joo, K.M.; Kim, S.Y.; Jin, X.; Song, S.Y.; Kong, D.S.; Lee, J.I.; Jeon, J.W.; Kim, M.H.; Kang, B.G.; Jung, Y.; et al. Clinical and biological implications of CD133-positive and CD133-negative cells in glioblastomas. Lab. Investig. 2008, 88, 808–815. [Google Scholar] [CrossRef]
- Chen, R.; Nishimura, M.C.; Bumbaca, S.M.; Kharbanda, S.; Forrest, W.F.; Kasman, I.M.; Greve, J.M.; Soriano, R.H.; Gilmour, L.L.; Rivers, C.S.; et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell 2010, 17, 362–375. [Google Scholar] [CrossRef]
- Wang, J.; Sakariassen, P.O.; Tsinkalovsky, O.; Immervoll, H.; Boe, S.O.; Svendsen, A.; Prestegarden, L.; Rosland, G.; Thorsen, F.; Stuhr, L.; et al. CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. Int. J. Cancer 2008, 122, 761–768. [Google Scholar] [CrossRef]
- Jijiwa, M.; Demir, H.; Gupta, S.; Leung, C.; Joshi, K.; Orozco, N.; Huang, T.; Yildiz, V.O.; Shibahara, I.; de Jesus, J.A.; et al. CD44v6 regulates growth of brain tumor stem cells partially through the AKT-mediated pathway. PLoS ONE 2011, 6, e24217. [Google Scholar] [CrossRef]
- Anido, J.; Saez-Borderias, A.; Gonzalez-Junca, A.; Rodon, L.; Folch, G.; Carmona, M.A.; Prieto-Sanchez, R.M.; Barba, I.; Martinez-Saez, E.; Prudkin, L.; et al. TGF-beta Receptor Inhibitors Target the CD44(high)/Id1(high) Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell 2010, 18, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Son, M.J.; Woolard, K.; Nam, D.H.; Lee, J.; Fine, H.A. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell 2009, 4, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.C.; Roy, N.S.; Keyoung, H.M.; Goodman, R.R.; McKhann, G., 2nd; Jiang, L.; Kang, J.; Nedergaard, M.; Goldman, S.A. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat. Med. 2003, 9, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Ogden, A.T.; Waziri, A.E.; Lochhead, R.A.; Fusco, D.; Lopez, K.; Ellis, J.A.; Kang, J.; Assanah, M.; McKhann, G.M.; Sisti, M.B.; et al. Identification of A2B5+CD133- tumor-initiating cells in adult human gliomas. Neurosurgery 2008, 62, 505–514, discussion 514–505. [Google Scholar] [CrossRef]
- He, J.; Liu, Y.; Zhu, T.; Zhu, J.; Dimeco, F.; Vescovi, A.L.; Heth, J.A.; Muraszko, K.M.; Fan, X.; Lubman, D.M. CD90 is identified as a candidate marker for cancer stem cells in primary high-grade gliomas using tissue microarrays. Mol. Cell. Proteomics 2012, 11, M111.010744. [Google Scholar] [CrossRef]
- Lathia, J.D.; Gallagher, J.; Heddleston, J.M.; Wang, J.; Eyler, C.E.; Macswords, J.; Wu, Q.; Vasanji, A.; McLendon, R.E.; Hjelmeland, A.B.; et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell 2010, 6, 421–432. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; Li, Z.; Sathornsumetee, S.; Wang, H.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 2008, 68, 6043–6048. [Google Scholar] [CrossRef]
- Hemmati, H.D.; Nakano, I.; Lazareff, J.A.; Masterman-Smith, M.; Geschwind, D.H.; Bronner-Fraser, M.; Kornblum, H.I. Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 15178–15183. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef]
- Tunici, P.; Bissola, L.; Lualdi, E.; Pollo, B.; Cajola, L.; Broggi, G.; Sozzi, G.; Finocchiaro, G. Genetic alterations and in vivo tumorigenicity of neurospheres derived from an adult glioblastoma. Mol. Cancer 2004, 3, 25. [Google Scholar] [CrossRef]
- Castellan, M.; Guarnieri, A.; Fujimura, A.; Zanconato, F.; Battilana, G.; Panciera, T.; Sladitschek, H.L.; Contessotto, P.; Citron, A.; Grilli, A.; et al. Single-cell analyses reveal YAP/TAZ as regulators of stemness and cell plasticity in Glioblastoma. Nat. Cancer 2021, 2, 174–188. [Google Scholar] [CrossRef]
- Ligon, K.L.; Huillard, E.; Mehta, S.; Kesari, S.; Liu, H.; Alberta, J.A.; Bachoo, R.M.; Kane, M.; Louis, D.N.; Depinho, R.A.; et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron 2007, 53, 503–517. [Google Scholar] [CrossRef]
- Suva, M.L.; Rheinbay, E.; Gillespie, S.M.; Patel, A.P.; Wakimoto, H.; Rabkin, S.D.; Riggi, N.; Chi, A.S.; Cahill, D.P.; Nahed, B.V.; et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014, 157, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes. Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, T.; Kageyama, R. Regulation of temporal properties of neural stem cells and transition timing of neurogenesis and gliogenesis during mammalian neocortical development. Semin. Cell Dev. Biol. 2019, 95, 4–11. [Google Scholar] [CrossRef]
- Eriksson, P.S.; Perfilieva, E.; Bjork-Eriksson, T.; Alborn, A.M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998, 4, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Deng, W.; Gage, F.H. Mechanisms and functional implications of adult neurogenesis. Cell 2008, 132, 645–660. [Google Scholar] [CrossRef]
- Nishiyama, A.; Komitova, M.; Suzuki, R.; Zhu, X. Polydendrocytes (NG2 cells): Multifunctional cells with lineage plasticity. Nat. Rev. Neurosci. 2009, 10, 9–22. [Google Scholar] [CrossRef]
- Kempermann, G. Neuronal stem cells and adult neurogenesis. Ernst Schering Res. Found. Workshop 2002, 35, 17–28. [Google Scholar] [CrossRef]
- Bedard, A.; Cossette, M.; Levesque, M.; Parent, A. Proliferating cells can differentiate into neurons in the striatum of normal adult monkey. Neurosci. Lett. 2002, 328, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Arsenijevic, Y.; Villemure, J.G.; Brunet, J.F.; Bloch, J.J.; Deglon, N.; Kostic, C.; Zurn, A.; Aebischer, P. Isolation of multipotent neural precursors residing in the cortex of the adult human brain. Exp. Neurol. 2001, 170, 48–62. [Google Scholar] [CrossRef]
- Reynolds, B.A.; Weiss, S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 1992, 255, 1707–1710. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Buylla, A.; Seri, B.; Doetsch, F. Identification of neural stem cells in the adult vertebrate brain. Brain Res. Bull. 2002, 57, 751–758. [Google Scholar] [CrossRef]
- Sanai, N.; Alvarez-Buylla, A.; Berger, M.S. Neural stem cells and the origin of gliomas. N. Engl. J. Med. 2005, 353, 811–822. [Google Scholar] [CrossRef]
- Waker, C.A.; Lober, R.M. Brain Tumors of Glial Origin. Adv. Exp. Med. Biol. 2019, 1190, 281–297. [Google Scholar]
- Chesler, D.A.; Berger, M.S.; Quinones-Hinojosa, A. The potential origin of glioblastoma initiating cells. Front. Biosci. (Schol. Ed.) 2012, 4, 190–205. [Google Scholar] [CrossRef]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, R.J.; Rich, J.N. Making a tumour’s bed: Glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 2007, 7, 733–736. [Google Scholar] [CrossRef]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Wang, Q.; Wang, Y.A.; Hua, S.; Sauve, C.G.; Ong, D.; Lan, Z.D.; Chang, Q.; Ho, Y.W.; Monasterio, M.M.; et al. Epigenetic Activation of WNT5A Drives Glioblastoma Stem Cell Differentiation and Invasive Growth. Cell 2016, 167, 1281–1295.e1218. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Uhrbom, L. On the origin of glioma. Ups. J. Med. Sci. 2012, 117, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Coskun, V.; Liang, A.; Yu, J.; Cheng, L.; Ge, W.; Shi, Z.; Zhang, K.; Li, C.; Cui, Y.; et al. Single-cell transcriptome analyses reveal signals to activate dormant neural stem cells. Cell 2015, 161, 1175–1186. [Google Scholar] [CrossRef]
- Xie, X.P.; Laks, D.R.; Sun, D.; Poran, A.; Laughney, A.M.; Wang, Z.; Sam, J.; Belenguer, G.; Farinas, I.; Elemento, O.; et al. High-resolution mouse subventricular zone stem-cell niche transcriptome reveals features of lineage, anatomy, and aging. Proc. Natl. Acad. Sci. USA 2020, 117, 31448–31458. [Google Scholar] [CrossRef] [PubMed]
- Vanner, R.J.; Remke, M.; Gallo, M.; Selvadurai, H.J.; Coutinho, F.; Lee, L.; Kushida, M.; Head, R.; Morrissy, S.; Zhu, X.; et al. Quiescent sox2(+) cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell 2014, 26, 33–47. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.Y.; Kim, W.K.; Lee, J.K.; Park, J.; et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.S.; Romero-Garcia, R.; Hart, M.G.; Suckling, J. Genetic, cellular, and connectomic characterization of the brain regions commonly plagued by glioma. Brain 2020, 143, 3294–3307. [Google Scholar] [CrossRef]
- Holland, E.C.; Celestino, J.; Dai, C.; Schaefer, L.; Sawaya, R.E.; Fuller, G.N. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat. Genet. 2000, 25, 55–57. [Google Scholar] [CrossRef]
- Alcantara Llaguno, S.; Sun, D.; Pedraza, A.M.; Vera, E.; Wang, Z.; Burns, D.K.; Parada, L.F. Cell-of-origin susceptibility to glioblastoma formation declines with neural lineage restriction. Nat. Neurosci. 2019, 22, 545–555. [Google Scholar] [CrossRef]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 2017, 355, eaai8478. [Google Scholar] [CrossRef]
- Tirosh, I.; Venteicher, A.S.; Hebert, C.; Escalante, L.E.; Patel, A.P.; Yizhak, K.; Fisher, J.M.; Rodman, C.; Mount, C.; Filbin, M.G.; et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 2016, 539, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Filbin, M.G.; Tirosh, I.; Hovestadt, V.; Shaw, M.L.; Escalante, L.E.; Mathewson, N.D.; Neftel, C.; Frank, N.; Pelton, K.; Hebert, C.M.; et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 2018, 360, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Hide, T.; Takezaki, T.; Nakatani, Y.; Nakamura, H.; Kuratsu, J.; Kondo, T. Combination of a ptgs2 inhibitor and an epidermal growth factor receptor-signaling inhibitor prevents tumorigenesis of oligodendrocyte lineage-derived glioma-initiating cells. Stem Cells 2011, 29, 590–599. [Google Scholar] [CrossRef]
- Alcantara Llaguno, S.R.; Wang, Z.; Sun, D.; Chen, J.; Xu, J.; Kim, E.; Hatanpaa, K.J.; Raisanen, J.M.; Burns, D.K.; Johnson, J.E.; et al. Adult Lineage-Restricted CNS Progenitors Specify Distinct Glioblastoma Subtypes. Cancer Cell 2015, 28, 429–440. [Google Scholar] [CrossRef]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Richards, L.M.; Whitley, O.K.N.; MacLeod, G.; Cavalli, F.M.G.; Coutinho, F.J.; Jaramillo, J.E.; Svergun, N.; Riverin, M.; Croucher, D.C.; Kushida, M.; et al. Gradient of Developmental and Injury Response transcriptional states defines functional vulnerabilities underpinning glioblastoma heterogeneity. Nat. Cancer 2021, 2, 157–173. [Google Scholar] [CrossRef]
- Garofano, L.; Migliozzi, S.; Oh, Y.T.; D’Angelo, F.; Najac, R.D.; Ko, A.; Frangaj, B.; Caruso, F.P.; Yu, K.; Yuan, J.; et al. Pathway-based classification of glioblastoma uncovers a mitochondrial subtype with therapeutic vulnerabilities. Nat. Cancer 2021, 2, 141–156. [Google Scholar] [CrossRef]
- Zhang, P.; Xia, Q.; Liu, L.; Li, S.; Dong, L. Current Opinion on Molecular Characterization for GBM Classification in Guiding Clinical Diagnosis, Prognosis, and Therapy. Front. Mol. Biosci. 2020, 7, 562798. [Google Scholar] [CrossRef] [PubMed]
- Ferris, S.P.; Hofmann, J.W.; Solomon, D.A.; Perry, A. Characterization of gliomas: From morphology to molecules. Virchows Arch. 2017, 471, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Francis, J.M.; Zhang, C.Z.; Maire, C.L.; Jung, J.; Manzo, V.E.; Adalsteinsson, V.A.; Homer, H.; Haidar, S.; Blumenstiel, B.; Pedamallu, C.S.; et al. EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Discov. 2014, 4, 956–971. [Google Scholar] [CrossRef]
- Szerlip, N.J.; Pedraza, A.; Chakravarty, D.; Azim, M.; McGuire, J.; Fang, Y.; Ozawa, T.; Holland, E.C.; Huse, J.T.; Jhanwar, S.; et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc. Natl. Acad. Sci. USA 2012, 109, 3041–3046. [Google Scholar] [CrossRef] [PubMed]
- Bhaduri, A.; Di Lullo, E.; Jung, D.; Muller, S.; Crouch, E.E.; Espinosa, C.S.; Ozawa, T.; Alvarado, B.; Spatazza, J.; Cadwell, C.R.; et al. Outer Radial Glia-like Cancer Stem Cells Contribute to Heterogeneity of Glioblastoma. Cell Stem Cell 2020, 26, 48–63.e46. [Google Scholar] [CrossRef]
- Jin, X.; Kim, L.J.Y.; Wu, Q.; Wallace, L.C.; Prager, B.C.; Sanvoranart, T.; Gimple, R.C.; Wang, X.; Mack, S.C.; Miller, T.E.; et al. Targeting glioma stem cells through combined BMI1 and EZH2 inhibition. Nat. Med. 2017, 23, 1352–1361. [Google Scholar] [CrossRef]
- Wang, L.B.; Karpova, A.; Gritsenko, M.A.; Kyle, J.E.; Cao, S.; Li, Y.; Rykunov, D.; Colaprico, A.; Rothstein, J.H.; Hong, R.; et al. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 2021, 39, 509–528.e520. [Google Scholar] [CrossRef]
- Segerman, A.; Niklasson, M.; Haglund, C.; Bergstrom, T.; Jarvius, M.; Xie, Y.; Westermark, A.; Sonmez, D.; Hermansson, A.; Kastemar, M.; et al. Clonal Variation in Drug and Radiation Response among Glioma-Initiating Cells Is Linked to Proneural-Mesenchymal Transition. Cell Rep. 2016, 17, 2994–3009. [Google Scholar] [CrossRef]
- Wang, L.; Babikir, H.; Muller, S.; Yagnik, G.; Shamardani, K.; Catalan, F.; Kohanbash, G.; Alvarado, B.; Di Lullo, E.; Kriegstein, A.; et al. The Phenotypes of Proliferating Glioblastoma Cells Reside on a Single Axis of Variation. Cancer Discov. 2019, 9, 1708–1719. [Google Scholar] [CrossRef]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Minata, M.; Audia, A.; Shi, J.; Lu, S.; Bernstock, J.; Pavlyukov, M.S.; Das, A.; Kim, S.H.; Shin, Y.J.; Lee, Y.; et al. Phenotypic Plasticity of Invasive Edge Glioma Stem-like Cells in Response to Ionizing Radiation. Cell Rep. 2019, 26, 1893–1905.e1897. [Google Scholar] [CrossRef] [PubMed]
- Almairac, F.; Turchi, L.; Sakakini, N.; Debruyne, D.N.; Elkeurti, S.; Gjernes, E.; Polo, B.; Bianchini, L.; Fontaine, D.; Paquis, P.; et al. ERK-Mediated Loss of miR-199a-3p and Induction of EGR1 Act as a “Toggle Switch” of GBM Cell Dedifferentiation into NANOG- and OCT4-Positive Cells. Cancer Res. 2020, 80, 3236–3250. [Google Scholar] [CrossRef]
- Brescia, P.; Ortensi, B.; Fornasari, L.; Levi, D.; Broggi, G.; Pelicci, G. CD133 is essential for glioblastoma stem cell maintenance. Stem Cells 2013, 31, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.V.; Filiz, G.; Daniel, P.M.; Hollande, F.; Dworkin, S.; Amiridis, S.; Kountouri, N.; Ng, W.; Morokoff, A.P.; Mantamadiotis, T. Expression of CD133 and CD44 in glioblastoma stem cells correlates with cell proliferation, phenotype stability and intra-tumor heterogeneity. PLoS ONE 2017, 12, e0172791. [Google Scholar] [CrossRef]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef] [PubMed]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef]
- Auffinger, B.; Tobias, A.L.; Han, Y.; Lee, G.; Guo, D.; Dey, M.; Lesniak, M.S.; Ahmed, A.U. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014, 21, 1119–1131. [Google Scholar] [CrossRef]
- Kaina, B.; Christmann, M. DNA repair in personalized brain cancer therapy with temozolomide and nitrosoureas. DNA Repair. (Amst) 2019, 78, 128–141. [Google Scholar] [CrossRef]
- Qazi, M.A.; Salim, S.K.; Brown, K.R.; Mikolajewicz, N.; Savage, N.; Han, H.; Subapanditha, M.K.; Bakhshinyan, D.; Nixon, A.; Vora, P.; et al. Characterization of the minimal residual disease state reveals distinct evolutionary trajectories of human glioblastoma. Cell Rep. 2022, 40, 111420. [Google Scholar] [CrossRef]
- Touat, M.; Li, Y.Y.; Boynton, A.N.; Spurr, L.F.; Iorgulescu, J.B.; Bohrson, C.L.; Cortes-Ciriano, I.; Birzu, C.; Geduldig, J.E.; Pelton, K.; et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 2020, 580, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Oren, Y.; Tsabar, M.; Cuoco, M.S.; Amir-Zilberstein, L.; Cabanos, H.F.; Hutter, J.C.; Hu, B.; Thakore, P.I.; Tabaka, M.; Fulco, C.P.; et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature 2021, 596, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, A.M.; Halle, B.; Cedile, O.; Burton, M.; Baun, C.; Thisgaard, H.; Anand, A.; Hubert, C.; Thomassen, M.; Michaelsen, S.R.; et al. Surgical resection of glioblastomas induces pleiotrophin-mediated self-renewal of glioblastoma stem cells in recurrent tumors. Neuro Oncol. 2022, 24, 1074–1087. [Google Scholar] [CrossRef]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Eramo, A.; Ricci-Vitiani, L.; Zeuner, A.; Pallini, R.; Lotti, F.; Sette, G.; Pilozzi, E.; Larocca, L.M.; Peschle, C.; De Maria, R. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006, 13, 1238–1241. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Beier, D.; Schulz, J.B.; Beier, C.P. Chemoresistance of glioblastoma cancer stem cells--much more complex than expected. Mol. Cancer 2011, 10, 128. [Google Scholar] [CrossRef]
- Sorensen, M.D.; Fosmark, S.; Hellwege, S.; Beier, D.; Kristensen, B.W.; Beier, C.P. Chemoresistance and chemotherapy targeting stem-like cells in malignant glioma. Adv. Exp. Med. Biol. 2015, 853, 111–138. [Google Scholar]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef]
- Beier, D.; Rohrl, S.; Pillai, D.R.; Schwarz, S.; Kunz-Schughart, L.A.; Leukel, P.; Proescholdt, M.; Brawanski, A.; Bogdahn, U.; Trampe-Kieslich, A.; et al. Temozolomide preferentially depletes cancer stem cells in glioblastoma. Cancer Res. 2008, 68, 5706–5715. [Google Scholar] [CrossRef]
- Bleau, A.M.; Hambardzumyan, D.; Ozawa, T.; Fomchenko, E.I.; Huse, J.T.; Brennan, C.W.; Holland, E.C. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell 2009, 4, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.; Smith, R.; Cahill, D.P.; Stephens, P.; Stevens, C.; Teague, J.; Greenman, C.; Edkins, S.; Bignell, G.; Davies, H.; et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 2006, 66, 3987–3991. [Google Scholar] [CrossRef]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef]
- Indraccolo, S.; Lombardi, G.; Fassan, M.; Pasqualini, L.; Giunco, S.; Marcato, R.; Gasparini, A.; Candiotto, C.; Nalio, S.; Fiduccia, P.; et al. Genetic, Epigenetic, and Immunologic Profiling of MMR-Deficient Relapsed Glioblastoma. Clin. Cancer Res. 2019, 25, 1828–1837. [Google Scholar] [CrossRef] [PubMed]
- van Thuijl, H.F.; Mazor, T.; Johnson, B.E.; Fouse, S.D.; Aihara, K.; Hong, C.; Malmstrom, A.; Hallbeck, M.; Heimans, J.J.; Kloezeman, J.J.; et al. Evolution of DNA repair defects during malignant progression of low-grade gliomas after temozolomide treatment. Acta Neuropathol. 2015, 129, 597–607. [Google Scholar] [CrossRef]
- Yu, D.; Khan, O.F.; Suva, M.L.; Dong, B.; Panek, W.K.; Xiao, T.; Wu, M.; Han, Y.; Ahmed, A.U.; Balyasnikova, I.V.; et al. Multiplexed RNAi therapy against brain tumor-initiating cells via lipopolymeric nanoparticle infusion delays glioblastoma progression. Proc. Natl. Acad. Sci. USA 2017, 114, E6147–E6156. [Google Scholar] [CrossRef] [PubMed]
- Sarkaria, J.N.; Kitange, G.J.; James, C.D.; Plummer, R.; Calvert, H.; Weller, M.; Wick, W. Mechanisms of chemoresistance to alkylating agents in malignant glioma. Clin. Cancer Res. 2008, 14, 2900–2908. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- McCord, M.; Bartom, E.; Burdett, K.; Baran, A.; Eckerdt, F.D.; Balyasnikova, I.V.; McCortney, K.; Sears, T.; Cheng, S.Y.; Sarkaria, J.N.; et al. Modeling Therapy-Driven Evolution of Glioblastoma with Patient-Derived Xenografts. Cancers 2022, 14, 5494. [Google Scholar] [CrossRef]
- Ferri, A.; Stagni, V.; Barila, D. Targeting the DNA Damage Response to Overcome Cancer Drug Resistance in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 4910. [Google Scholar] [CrossRef]
- MacLeod, G.; Bozek, D.A.; Rajakulendran, N.; Monteiro, V.; Ahmadi, M.; Steinhart, Z.; Kushida, M.M.; Yu, H.; Coutinho, F.J.; Cavalli, F.M.G.; et al. Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell Rep. 2019, 27, 971–986.e979. [Google Scholar] [CrossRef] [PubMed]
- Tentori, L.; Ricci-Vitiani, L.; Muzi, A.; Ciccarone, F.; Pelacchi, F.; Calabrese, R.; Runci, D.; Pallini, R.; Caiafa, P.; Graziani, G. Pharmacological inhibition of poly(ADP-ribose) polymerase-1 modulates resistance of human glioblastoma stem cells to temozolomide. BMC Cancer 2014, 14, 151. [Google Scholar] [CrossRef] [PubMed]
- Ropolo, M.; Daga, A.; Griffero, F.; Foresta, M.; Casartelli, G.; Zunino, A.; Poggi, A.; Cappelli, E.; Zona, G.; Spaziante, R.; et al. Comparative analysis of DNA repair in stem and nonstem glioma cell cultures. Mol. Cancer Res. 2009, 7, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wakeman, T.P.; Lathia, J.D.; Hjelmeland, A.B.; Wang, X.F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch promotes radioresistance of glioma stem cells. Stem Cells 2010, 28, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Venere, M.; Hamerlik, P.; Wu, Q.; Rasmussen, R.D.; Song, L.A.; Vasanji, A.; Tenley, N.; Flavahan, W.A.; Hjelmeland, A.B.; Bartek, J.; et al. Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma-initiating cells. Cell Death Differ. 2014, 21, 258–269. [Google Scholar] [CrossRef]
- Kim, S.H.; Joshi, K.; Ezhilarasan, R.; Myers, T.R.; Siu, J.; Gu, C.; Nakano-Okuno, M.; Taylor, D.; Minata, M.; Sulman, E.P.; et al. EZH2 protects glioma stem cells from radiation-induced cell death in a MELK/FOXM1-dependent manner. Stem Cell Reports 2015, 4, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Erasimus, H.; Gobin, M.; Niclou, S.; Van Dyck, E. DNA repair mechanisms and their clinical impact in glioblastoma. Mutat. Res. Rev. Mutat. Res. 2016, 769, 19–35. [Google Scholar] [CrossRef]
- Nathansen, J.; Meyer, F.; Muller, L.; Schmitz, M.; Borgmann, K.; Dubrovska, A. Beyond the Double-Strand Breaks: The Role of DNA Repair Proteins in Cancer Stem-Cell Regulation. Cancers 2021, 13, 4818. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Lim, M.; Weller, M.; Idbaih, A.; Steinbach, J.; Finocchiaro, G.; Raval, R.R.; Ansstas, G.; Baehring, J.; Taylor, J.W.; Honnorat, J.; et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol. 2022, 24, 1935–1949. [Google Scholar] [CrossRef]
- Omuro, A.; Reardon, D.A.; Sampson, J.H.; Baehring, J.; Sahebjam, S.; Cloughesy, T.F.; Chalamandaris, A.G.; Potter, V.; Butowski, N.; Lim, M. Nivolumab plus radiotherapy with or without temozolomide in newly diagnosed glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neurooncol Adv. 2022, 4, vdac025. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bahr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef]
- Lee, A.H.; Sun, L.; Mochizuki, A.Y.; Reynoso, J.G.; Orpilla, J.; Chow, F.; Kienzler, J.C.; Everson, R.G.; Nathanson, D.A.; Bensinger, S.J.; et al. Neoadjuvant PD-1 blockade induces T cell and cDC1 activation but fails to overcome the immunosuppressive tumor associated macrophages in recurrent glioblastoma. Nat. Commun. 2021, 12, 6938. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, L.; Zhang, H.; Zhang, Y.; Ju, H.; Wang, X.; Ren, H.; Zhu, X.; Dong, Y. The immunosuppressive microenvironment and immunotherapy in human glioblastoma. Front. Immunol. 2022, 13, 1003651. [Google Scholar] [CrossRef]
- Arrieta, V.A.; Dmello, C.; McGrail, D.J.; Brat, D.J.; Lee-Chang, C.; Heimberger, A.B.; Chand, D.; Stupp, R.; Sonabend, A.M. Immune checkpoint blockade in glioblastoma: From tumor heterogeneity to personalized treatment. J. Clin. Investig. 2023, 133. [Google Scholar] [CrossRef]
- Alvarado, A.G.; Thiagarajan, P.S.; Mulkearns-Hubert, E.E.; Silver, D.J.; Hale, J.S.; Alban, T.J.; Turaga, S.M.; Jarrar, A.; Reizes, O.; Longworth, M.S.; et al. Glioblastoma Cancer Stem Cells Evade Innate Immune Suppression of Self-Renewal through Reduced TLR4 Expression. Cell Stem Cell 2017, 20, 450–461.e454. [Google Scholar] [CrossRef]
- Otvos, B.; Silver, D.J.; Mulkearns-Hubert, E.E.; Alvarado, A.G.; Turaga, S.M.; Sorensen, M.D.; Rayman, P.; Flavahan, W.A.; Hale, J.S.; Stoltz, K.; et al. Cancer Stem Cell-Secreted Macrophage Migration Inhibitory Factor Stimulates Myeloid Derived Suppressor Cell Function and Facilitates Glioblastoma Immune Evasion. Stem Cells 2016, 34, 2026–2039. [Google Scholar] [CrossRef]
- Prosniak, M.; Harshyne, L.A.; Andrews, D.W.; Kenyon, L.C.; Bedelbaeva, K.; Apanasovich, T.V.; Heber-Katz, E.; Curtis, M.T.; Cotzia, P.; Hooper, D.C. Glioma grade is associated with the accumulation and activity of cells bearing M2 monocyte markers. Clin. Cancer Res. 2013, 19, 3776–3786. [Google Scholar] [CrossRef]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef]
- Wu, A.; Wei, J.; Kong, L.Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010, 12, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Marisetty, A.; Schrand, B.; Gabrusiewicz, K.; Hashimoto, Y.; Ott, M.; Grami, Z.; Kong, L.Y.; Ling, X.; Caruso, H.; et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J. Clin. Investig. 2019, 129, 137–149. [Google Scholar] [CrossRef]
- Ricklefs, F.L.; Alayo, Q.; Krenzlin, H.; Mahmoud, A.B.; Speranza, M.C.; Nakashima, H.; Hayes, J.L.; Lee, K.; Balaj, L.; Passaro, C.; et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci. Adv. 2018, 4, eaar2766. [Google Scholar] [CrossRef]
- Domenis, R.; Cesselli, D.; Toffoletto, B.; Bourkoula, E.; Caponnetto, F.; Manini, I.; Beltrami, A.P.; Ius, T.; Skrap, M.; Di Loreto, C.; et al. Systemic T Cells Immunosuppression of Glioma Stem Cell-Derived Exosomes Is Mediated by Monocytic Myeloid-Derived Suppressor Cells. PLoS ONE 2017, 12, e0169932. [Google Scholar] [CrossRef]
- Shi, Y.; Ping, Y.F.; Zhou, W.; He, Z.C.; Chen, C.; Bian, B.S.; Zhang, L.; Chen, L.; Lan, X.; Zhang, X.C.; et al. Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat. Commun. 2017, 8, 15080. [Google Scholar] [CrossRef]
- Ye, X.Z.; Xu, S.L.; Xin, Y.H.; Yu, S.C.; Ping, Y.F.; Chen, L.; Xiao, H.L.; Wang, B.; Yi, L.; Wang, Q.L.; et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-beta1 signaling pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef]
- Ravi, V.M.; Will, P.; Kueckelhaus, J.; Sun, N.; Joseph, K.; Salie, H.; Vollmer, L.; Kuliesiute, U.; von Ehr, J.; Benotmane, J.K.; et al. Spatially resolved multi-omics deciphers bidirectional tumor-host interdependence in glioblastoma. Cancer Cell 2022, 40, 639–655.e613. [Google Scholar] [CrossRef] [PubMed]
- Goenka, A.; Tiek, D.; Song, X.; Huang, T.; Hu, B.; Cheng, S.Y. The Many Facets of Therapy Resistance and Tumor Recurrence in Glioblastoma. Cells 2021, 10, 484. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Lee, H.; Kim, D.; Kim, B.; Kang, J.; Kim, H.Y.; Youn, H.; Youn, B. Targeting Glioblastoma Stem Cells to Overcome Chemoresistance: An Overview of Current Therapeutic Strategies. Biomedicines 2022, 10, 1308. [Google Scholar] [CrossRef] [PubMed]
- Warren, K.E.; Gururangan, S.; Geyer, J.R.; McLendon, R.E.; Poussaint, T.Y.; Wallace, D.; Balis, F.M.; Berg, S.L.; Packer, R.J.; Goldman, S.; et al. A phase II study of O6-benzylguanine and temozolomide in pediatric patients with recurrent or progressive high-grade gliomas and brainstem gliomas: A Pediatric Brain Tumor Consortium study. J. Neurooncol 2012, 106, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.A.; Jiang, S.X.; Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Rich, J.N.; Gururangan, S.; Friedman, A.H.; Bigner, D.D.; Sampson, J.H.; et al. Phase II trial of temozolomide plus o6-benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma. J. Clin. Oncol. 2009, 27, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Troike, K.; Fodor, R.; Lathia, J.D. Unexplored Functions of Sex Hormones in Glioblastoma Cancer Stem Cells. Endocrinology 2022, 163, bqac002. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, C.; Hallett, R.; Vora, P.; Manoranjan, B.; Mahendram, S.; Qazi, M.A.; McFarlane, N.; Subapanditha, M.; Nolte, S.M.; Singh, M.; et al. Pyrvinium Targets CD133 in Human Glioblastoma Brain Tumor-Initiating Cells. Clin. Cancer Res. 2015, 21, 5324–5337. [Google Scholar] [CrossRef] [PubMed]
- Emlet, D.R.; Gupta, P.; Holgado-Madruga, M.; Del Vecchio, C.A.; Mitra, S.S.; Han, S.Y.; Li, G.; Jensen, K.C.; Vogel, H.; Xu, L.W.; et al. Targeting a glioblastoma cancer stem-cell population defined by EGF receptor variant III. Cancer Res. 2014, 74, 1238–1249. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Gaedicke, S.; Machein, M.; Mittler, G.; Braun, F.; Hettich, M.; Firat, E.; Klingner, K.; Schuler, J.; Wider, D.; et al. Effective Eradication of Glioblastoma Stem Cells by Local Application of an AC133/CD133-Specific T-cell-Engaging Antibody and CD8 T Cells. Cancer Res. 2015, 75, 2166–2176. [Google Scholar] [CrossRef]
- Datta, S.; Sears, T.; Cortopassi, G.; Woolard, K.; Angelastro, J.M. Repurposing FDA approved drugs inhibiting mitochondrial function for targeting glioma-stem like cells. Biomed. Pharmacother. 2021, 133, 111058. [Google Scholar] [CrossRef]
- Moon, B.S.; Cai, M.; Lee, G.; Zhao, T.; Song, X.; Giannotta, S.L.; Attenello, F.J.; Yu, M.; Lu, W. Epigenetic modulator inhibition overcomes temozolomide chemoresistance and antagonizes tumor recurrence of glioblastoma. J. Clin. Investig. 2020, 130, 5782–5799. [Google Scholar] [CrossRef]
- Huang, T.; Kim, C.K.; Alvarez, A.A.; Pangeni, R.P.; Wan, X.; Song, X.; Shi, T.; Yang, Y.; Sastry, N.; Horbinski, C.M.; et al. MST4 Phosphorylation of ATG4B Regulates Autophagic Activity, Tumorigenicity, and Radioresistance in Glioblastoma. Cancer Cell 2017, 32, 840–855.e848. [Google Scholar] [CrossRef]
- Huang, T.; Yang, Y.; Song, X.; Wan, X.; Wu, B.; Sastry, N.; Horbinski, C.M.; Zeng, C.; Tiek, D.; Goenka, A.; et al. PRMT6 methylation of RCC1 regulates mitosis, tumorigenicity, and radiation response of glioblastoma stem cells. Mol. Cell 2021, 81, 1276–1291.e1279. [Google Scholar] [CrossRef]
- Eckerdt, F.D.; Bell, J.B.; Gonzalez, C.; Oh, M.S.; Perez, R.E.; Mazewski, C.; Fischietti, M.; Goldman, S.; Nakano, I.; Platanias, L.C. Combined PI3Kalpha-mTOR Targeting of Glioma Stem Cells. Sci. Rep. 2020, 10, 21873. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.B.; Eckerdt, F.; Dhruv, H.D.; Finlay, D.; Peng, S.; Kim, S.; Kroczynska, B.; Beauchamp, E.M.; Alley, K.; Clymer, J.; et al. Differential Response of Glioma Stem Cells to Arsenic Trioxide Therapy Is Regulated by MNK1 and mRNA Translation. Mol. Cancer Res. 2018, 16, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, A.; Eckerdt, F.; Bell, J.; Nakano, I.; Giles, F.J.; Cheng, S.Y.; Lulla, R.R.; Goldman, S.; Platanias, L.C. Targeting of glioblastoma cell lines and glioma stem cells by combined PIM kinase and PI3K-p110alpha inhibition. Oncotarget 2016, 7, 33192–33201. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Wu, Q.; Hitomi, M.; Rahim, N.; Kim, Y.; Sloan, A.E.; Weil, R.J.; Nakano, I.; Sarkaria, J.N.; Stringer, B.W.; et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat. Neurosci. 2013, 16, 1373–1382. [Google Scholar] [CrossRef]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef]
- Chedeville, A.L.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Radiotherapy Resistance. Cancers 2021, 13, 542. [Google Scholar] [CrossRef]
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma stem cells: Lessons from the tumor hierarchy in a lethal cancer. Genes. Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef]
- Wang, X.; Yang, K.; Wu, Q.; Kim, L.J.Y.; Morton, A.R.; Gimple, R.C.; Prager, B.C.; Shi, Y.; Zhou, W.; Bhargava, S.; et al. Targeting pyrimidine synthesis accentuates molecular therapy response in glioblastoma stem cells. Sci. Transl. Med. 2019, 11, eaau4972. [Google Scholar] [CrossRef]
- Dong, Z.; Zhang, G.; Qu, M.; Gimple, R.C.; Wu, Q.; Qiu, Z.; Prager, B.C.; Wang, X.; Kim, L.J.Y.; Morton, A.R.; et al. Targeting Glioblastoma Stem Cells through Disruption of the Circadian Clock. Cancer Discov. 2019, 9, 1556–1573. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.; Sprowls, S.A.; Arora, S.; Shakya, S.; Silver, D.J.; Goins, C.M.; Wallace, L.; Roversi, G.; Schafer, R.; Kay, K.; et al. WDR5 represents a therapeutically exploitable target for cancer stem cells in glioblastoma. Genes. Dev. 2023, 37, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Hao, Y.; Shao, C.; Wu, Q.; Prager, B.C.; Gimple, R.C.; Sulli, G.; Kim, L.J.; Zhang, G.; Qiu, Z.; et al. ADAR1-mediated RNA editing links ganglioside catabolism to glioblastoma stem cell maintenance. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef] [PubMed]
- Lv, D.; Gimple, R.C.; Zhong, C.; Wu, Q.; Yang, K.; Prager, B.C.; Godugu, B.; Qiu, Z.; Zhao, L.; Zhang, G.; et al. PDGF signaling inhibits mitophagy in glioblastoma stem cells through N(6)-methyladenosine. Dev. Cell 2022, 57, 1466–1481.e1466. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.; Xie, G.S.; Ma, L.; Li, P.; Li, L.; Richard, H.T. USP36 promotes tumorigenesis and drug sensitivity of glioblastoma by deubiquitinating and stabilizing ALKBH5. Neuro Oncol. 2022, 25, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Dixit, D.; Prager, B.C.; Gimple, R.C.; Poh, H.X.; Wang, Y.; Wu, Q.; Qiu, Z.; Kidwell, R.L.; Kim, L.J.Y.; Xie, Q.; et al. The RNA m6A Reader YTHDF2 Maintains Oncogene Expression and Is a Targetable Dependency in Glioblastoma Stem Cells. Cancer Discov. 2021, 11, 480–499. [Google Scholar] [CrossRef]
- Qiu, Z.; Zhao, L.; Shen, J.Z.; Liang, Z.; Wu, Q.; Yang, K.; Min, L.; Gimple, R.C.; Yang, Q.; Bhargava, S.; et al. Transcription Elongation Machinery Is a Druggable Dependency and Potentiates Immunotherapy in Glioblastoma Stem Cells. Cancer Discov. 2022, 12, 502–521. [Google Scholar] [CrossRef]
- Hendriks, L.E.L.; Henon, C.; Auclin, E.; Mezquita, L.; Ferrara, R.; Audigier-Valette, C.; Mazieres, J.; Lefebvre, C.; Rabeau, A.; Le Moulec, S.; et al. Outcome of Patients with Non-Small Cell Lung Cancer and Brain Metastases Treated with Checkpoint Inhibitors. J. Thorac. Oncol. 2019, 14, 1244–1254. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Forsyth, P.A.; Algazi, A.; Hamid, O.; Hodi, F.S.; Moschos, S.J.; Khushalani, N.I.; Lewis, K.; Lao, C.D.; Postow, M.A.; et al. Combined Nivolumab and Ipilimumab in Melanoma Metastatic to the Brain. N. Engl. J. Med. 2018, 379, 722–730. [Google Scholar] [CrossRef]
- Li, F.; Lv, B.; Liu, Y.; Hua, T.; Han, J.; Sun, C.; Xu, L.; Zhang, Z.; Feng, Z.; Cai, Y.; et al. Blocking the CD47-SIRPalpha axis by delivery of anti-CD47 antibody induces antitumor effects in glioma and glioma stem cells. Oncoimmunology 2018, 7, e1391973. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e1110. [Google Scholar] [CrossRef] [PubMed]
- Choe, J.H.; Watchmaker, P.B.; Simic, M.S.; Gilbert, R.D.; Li, A.W.; Krasnow, N.A.; Downey, K.M.; Yu, W.; Carrera, D.A.; Celli, A.; et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci. Transl. Med. 2021, 13, eabe7378. [Google Scholar] [CrossRef]
- Vora, P.; Venugopal, C.; Salim, S.K.; Tatari, N.; Bakhshinyan, D.; Singh, M.; Seyfrid, M.; Upreti, D.; Rentas, S.; Wong, N.; et al. The Rational Development of CD133-Targeting Immunotherapies for Glioblastoma. Cell Stem Cell 2020, 26, 832–844.e836. [Google Scholar] [CrossRef]
- Chen, C.; Jing, W.; Chen, Y.; Wang, G.; Abdalla, M.; Gao, L.; Han, M.; Shi, C.; Li, A.; Sun, P.; et al. Intracavity generation of glioma stem cell-specific CAR macrophages primes locoregional immunity for postoperative glioblastoma therapy. Sci. Transl. Med. 2022, 14, eabn1128. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Prager, B.C.; Gimple, R.C.; Aguilar, B.; Alizadeh, D.; Tang, H.; Lv, D.; Starr, R.; Brito, A.; Wu, Q.; et al. CRISPR Screening of CAR T Cells and Cancer Stem Cells Reveals Critical Dependencies for Cell-Based Therapies. Cancer Discov. 2021, 11, 1192–1211. [Google Scholar] [CrossRef] [PubMed]
- Lindo, L.; Wilkinson, L.H.; Hay, K.A. Befriending the Hostile Tumor Microenvironment in CAR T-Cell Therapy. Front. Immunol. 2020, 11, 618387. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Mackall, C.L. CAR T cell therapy: Inroads to response and resistance. Nat. Rev. Immunol. 2019, 19, 73–74. [Google Scholar] [CrossRef]
- Lee-Chang, C.; Miska, J.; Hou, D.; Rashidi, A.; Zhang, P.; Burga, R.A.; Jusue-Torres, I.; Xiao, T.; Arrieta, V.A.; Zhang, D.Y.; et al. Activation of 4-1BBL+ B cells with CD40 agonism and IFNgamma elicits potent immunity against glioblastoma. J. Exp. Med. 2021, 218, e20200913. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eckerdt, F.; Platanias, L.C. Emerging Role of Glioma Stem Cells in Mechanisms of Therapy Resistance. Cancers 2023, 15, 3458. https://doi.org/10.3390/cancers15133458
Eckerdt F, Platanias LC. Emerging Role of Glioma Stem Cells in Mechanisms of Therapy Resistance. Cancers. 2023; 15(13):3458. https://doi.org/10.3390/cancers15133458
Chicago/Turabian StyleEckerdt, Frank, and Leonidas C. Platanias. 2023. "Emerging Role of Glioma Stem Cells in Mechanisms of Therapy Resistance" Cancers 15, no. 13: 3458. https://doi.org/10.3390/cancers15133458
APA StyleEckerdt, F., & Platanias, L. C. (2023). Emerging Role of Glioma Stem Cells in Mechanisms of Therapy Resistance. Cancers, 15(13), 3458. https://doi.org/10.3390/cancers15133458