

Residues from Homologous Transmembrane Helices 4 and 10 Are Critical for P-Glycoprotein (ABCB1)-Mediated Drug Transport
 , ,
, ,  ,
,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Antibodies
2.2. Cell Line and Culture Conditions
2.3. BacMam Baculovirus Transduction of HeLa-S3 Cells and Cell Surface Expression of the Mutants
2.4. Transport of Fluorescent Substrates
2.5. Cytotoxicity Assays
2.6. Preparation of Membrane Vesicles of High Five Insect Cells
2.7. SDS-PAGE and Western Blotting
2.8. ATPase Activity
2.9. Molecular Modeling and Electrostatic Surface Potential
2.10. Molecular Dynamics (MD) Simulations
2.11. Statistical Analysis
3. Results
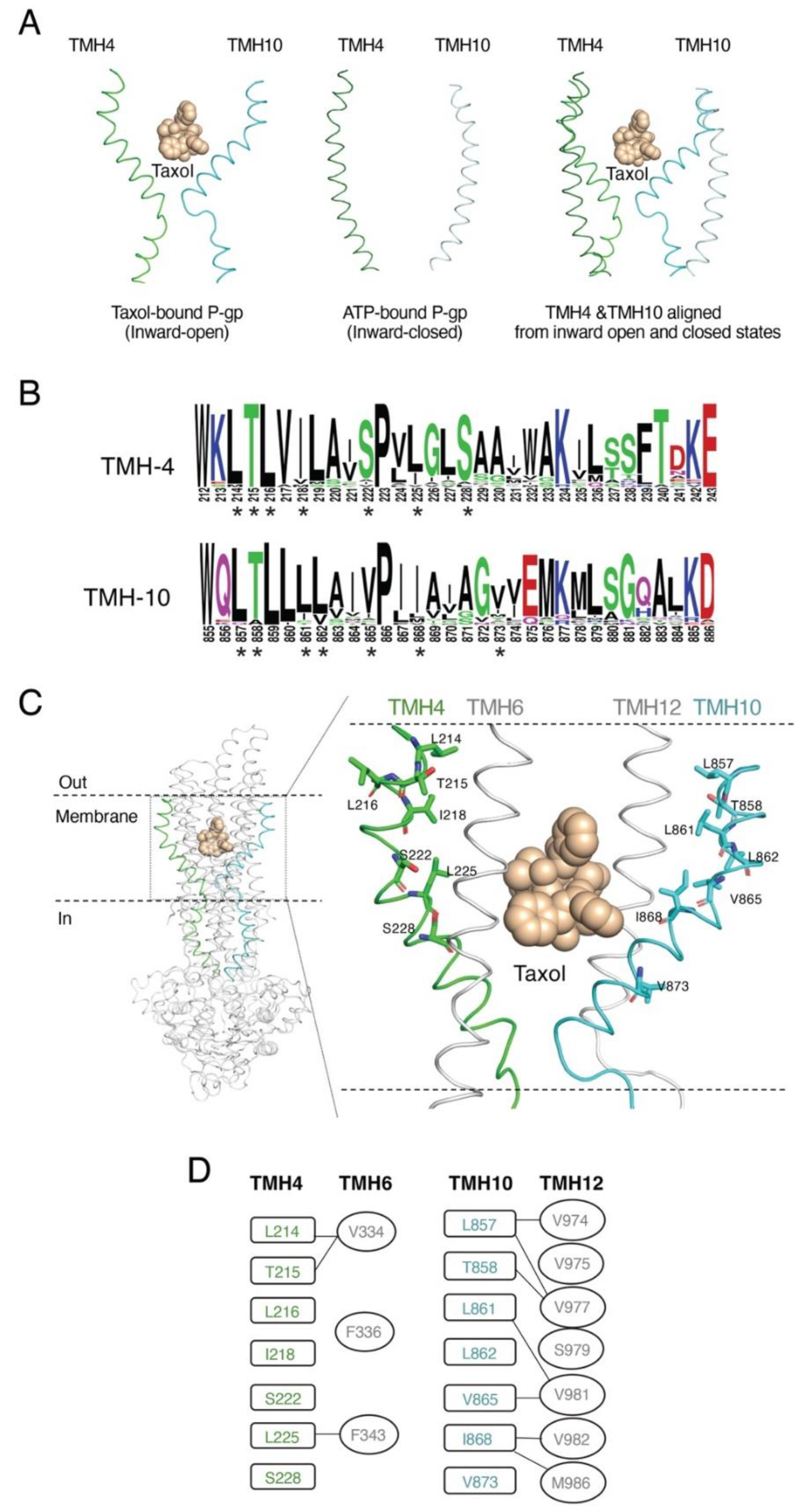
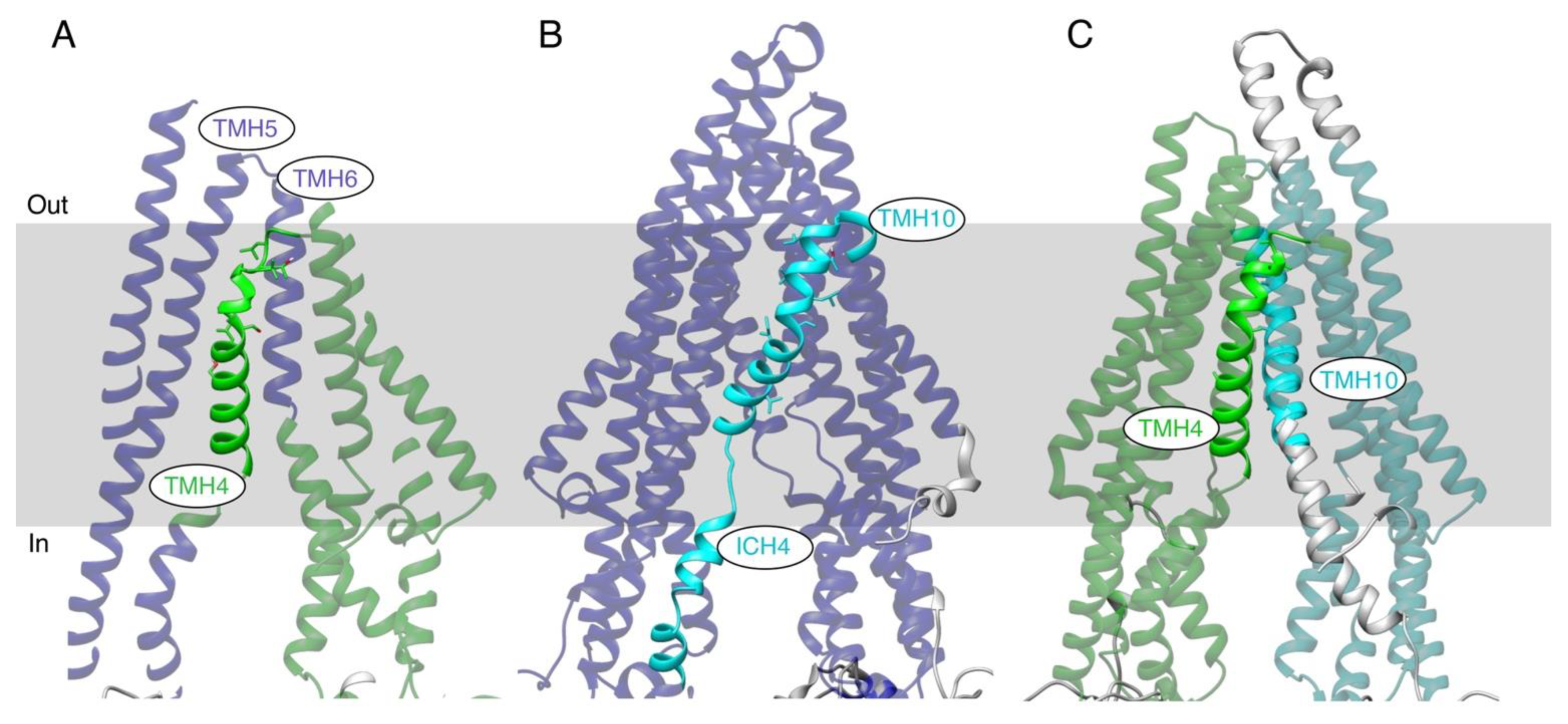
3.1. Rationale for Studying Homologous Transmembrane Helices TMH4 and TMH10 and the Selection of Residues to Generate Mutants
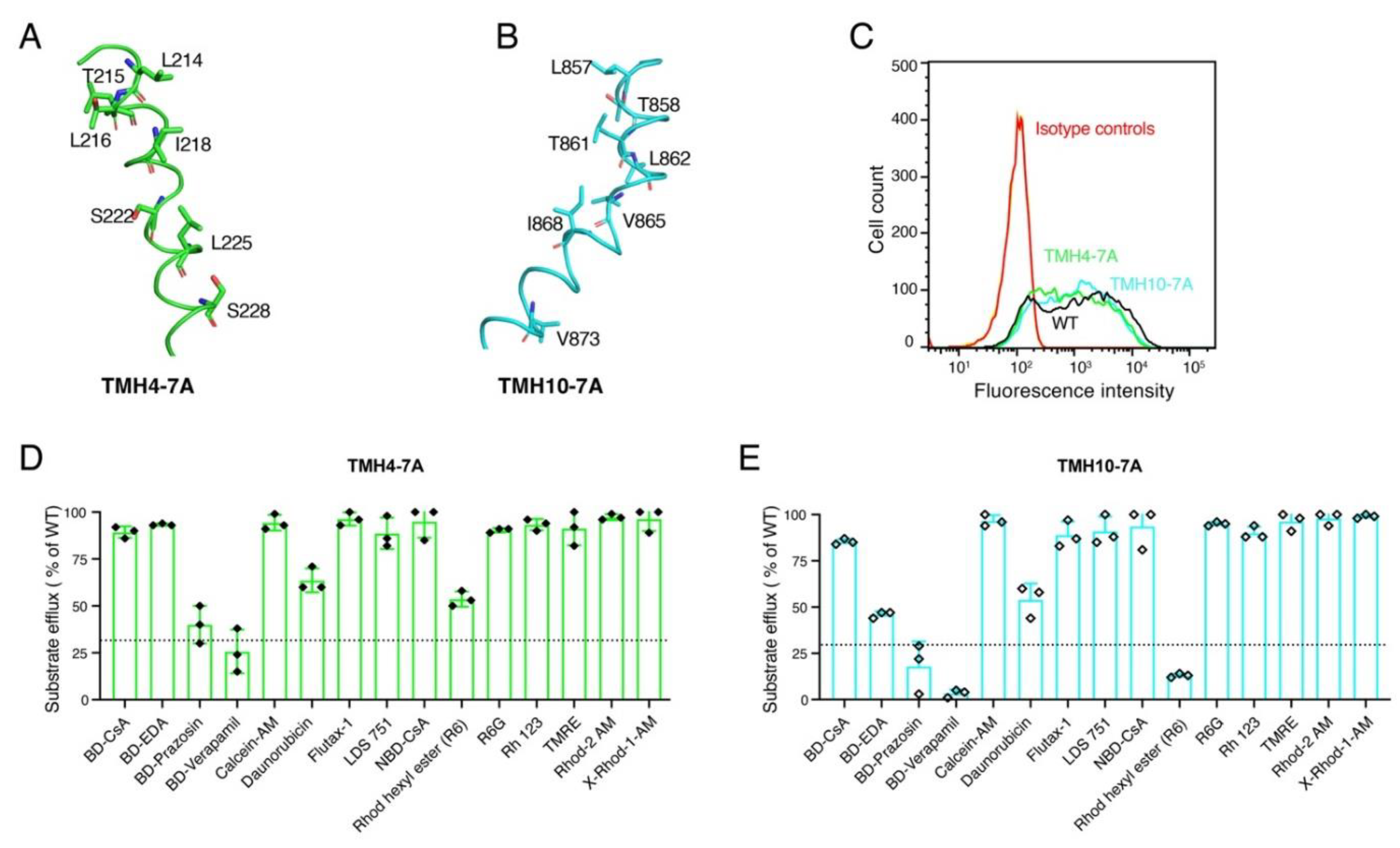
3.2. Both TMH4-7A and TMH10-7A Mutants Have a Similar Level of Expression as WT P-gp
3.3. Seven Alanine Substitutions in TMHs 4 and 10 Do Not Have Any Significant Effect on the Transport Function
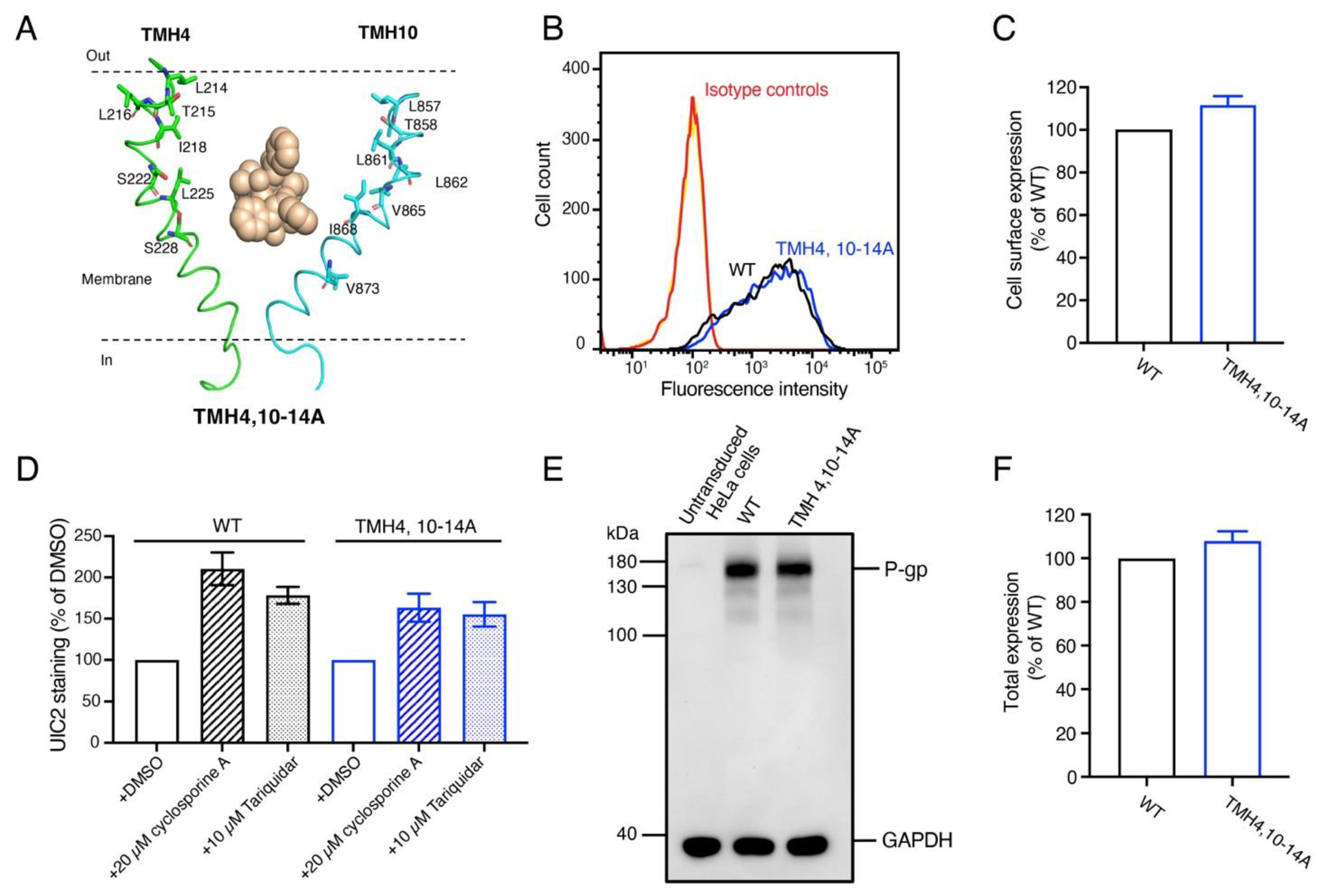
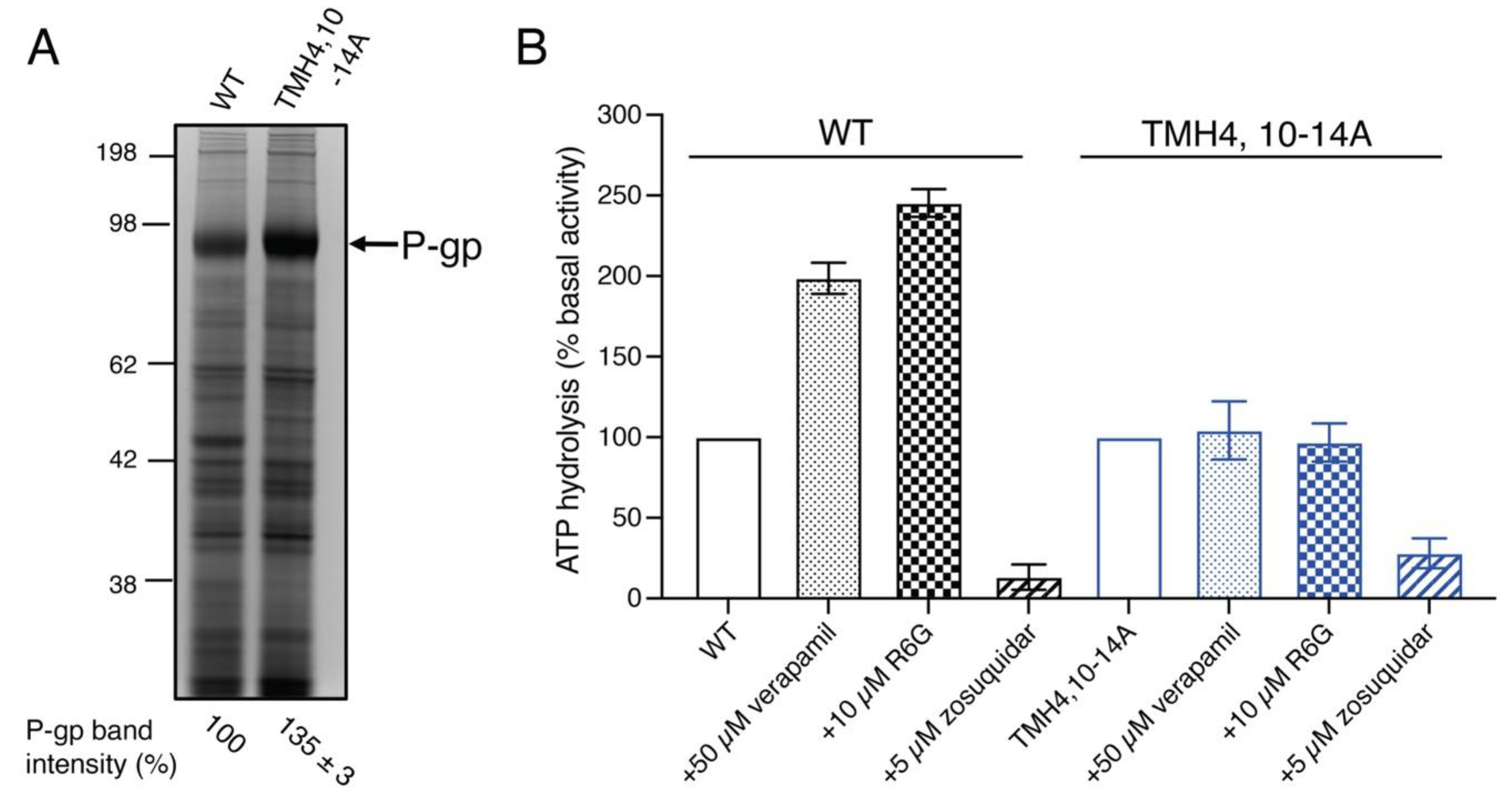
3.4. The Expression and Overall Conformation of the TMH4,10-14A Mutant Is the Same as WT P-gp
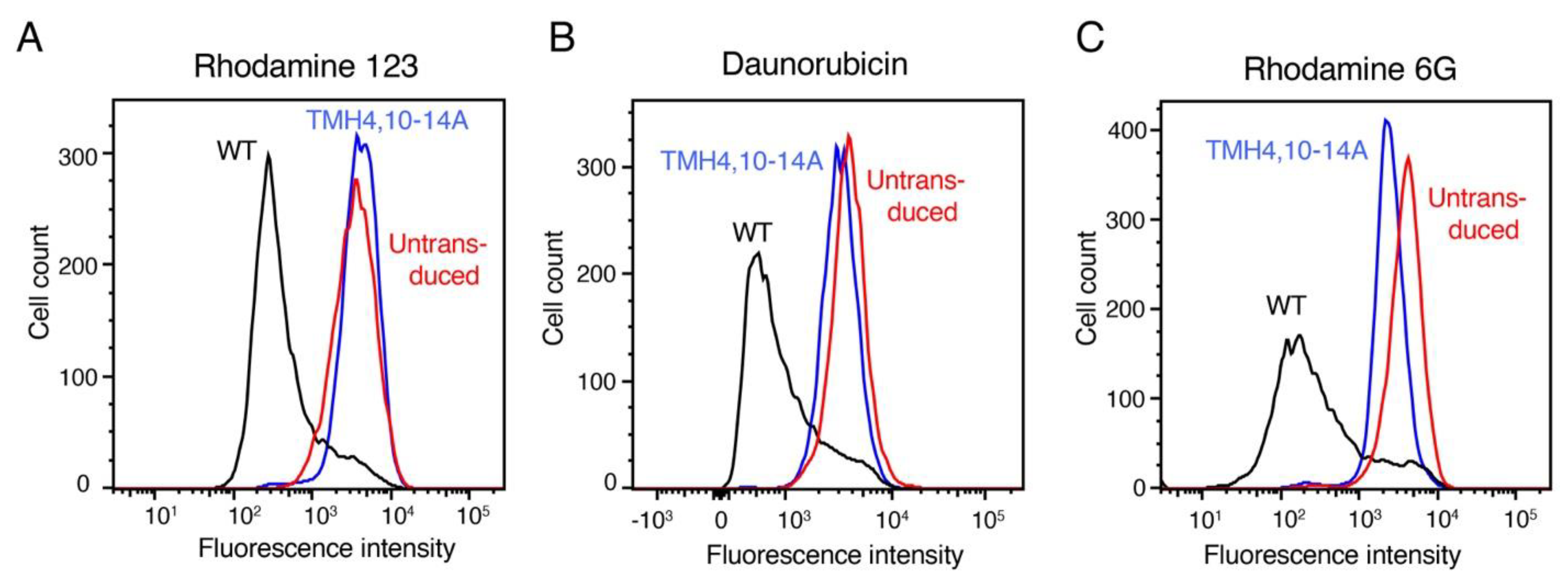
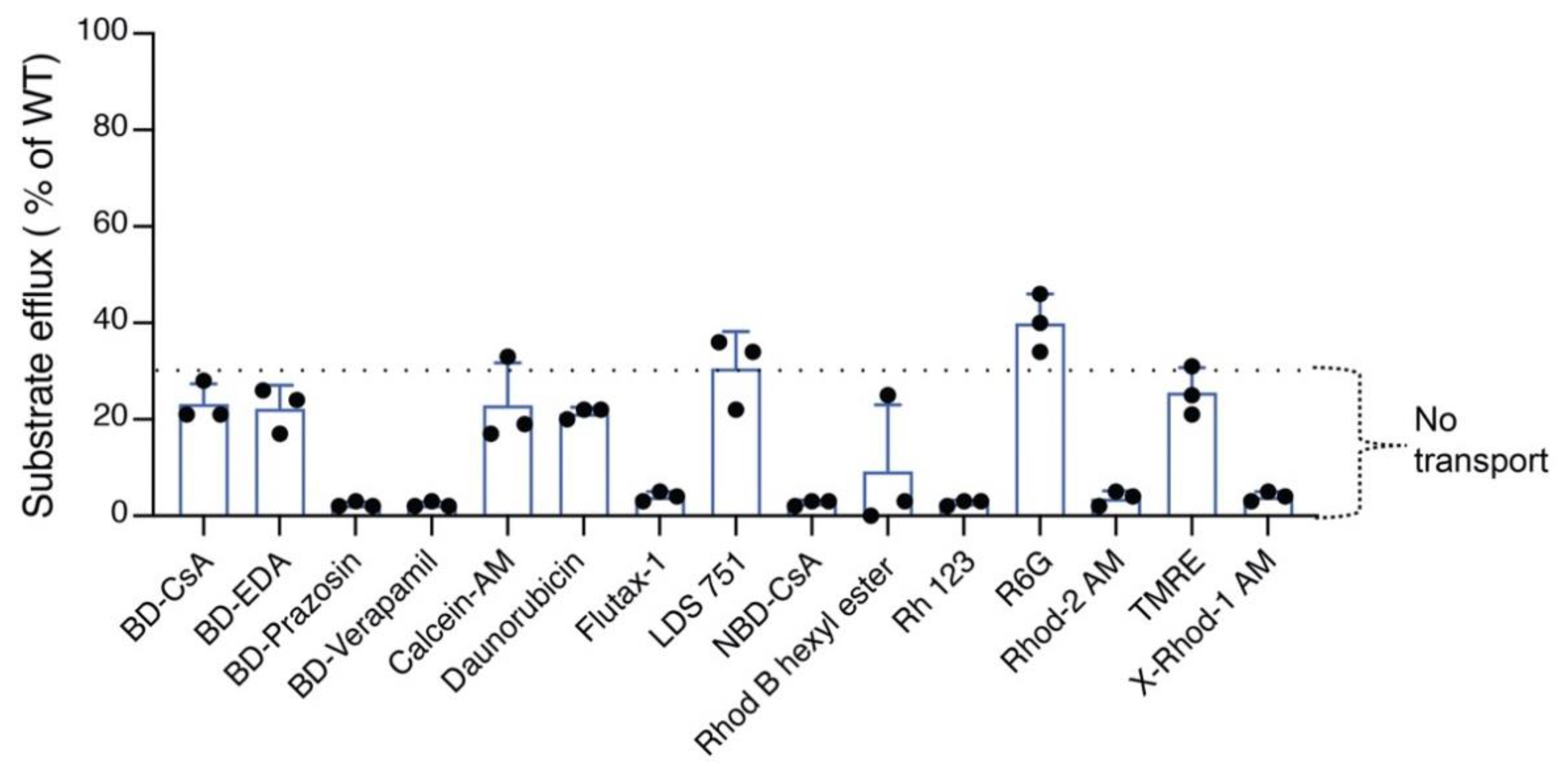
3.5. TMH4,10-14A Mutant Failed to Transport All the Tested Substrates
3.6. Although the TMH4,10-14A Mutant Exhibits Basal ATPase Activity the Same as That of WT P-gp, Substrates Fail to Stimulate It
3.7. Mutations in TMH4,10-14A Do Not Significantly Affect the Electrostatic Surface Potential in the Drug-Binding Cavity
3.8. MD Simulation of TMHs 4 and 10 of the TMH4,10-14A Mutant and WT P-gp
3.9. Omitting Three Alanine Substitutions in TMH 4 in the TMH4,10-14A Mutant Recovers the Transport of a Few Substrates
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Lusvarghi, S.; Robey, R.W.; Gottesman, M.M.; Ambudkar, S.V. Multidrug transporters: Recent insights from cryo-electron microscopy-derived atomic structures and animal models. F1000Research 2020, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Amiri-Kordestani, L.; Basseville, A.; Kurdziel, K.; Fojo, A.T.; Bates, S.E. Targeting MDR in breast and lung cancer: Discriminating its potential importance from the failure of drug resistance reversal studies. Drug Resist. Updates 2012, 15, 50–61. [Google Scholar] [CrossRef]
- Goldstein, L.J.; Galski, H.; Fojo, A.; Willingham, M.; Lai, S.L.; Gazdar, A.; Pirker, R.; Green, A.; Crist, W.; Brodeur, G.M.; et al. Expression of a multidrug resistance gene in human cancers. J. Natl. Cancer Inst. 1989, 81, 116–124. [Google Scholar] [CrossRef]
- Patel, N.R.; Rathi, A.; Mongayt, D.; Torchilin, V.P. Reversal of multidrug resistance by co-delivery of tariquidar (XR9576) and paclitaxel using long-circulating liposomes. Int. J. Pharm. 2011, 416, 296–299. [Google Scholar] [CrossRef]
- Dash, R.P.; Jayachandra Babu, R.; Srinivas, N.R. Therapeutic Potential and Utility of Elacridar with Respect to P-glycoprotein Inhibition: An Insight from the Published In Vitro, Preclinical and Clinical Studies. Eur. J. Drug Metab. Pharm. 2017, 42, 915–933. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, N.R. Understanding the role of tariquidar, a potent Pgp inhibitor, in combination trials with cytotoxic drugs: What is missing? Cancer Chemother. Pharm. 2016, 78, 1097–1098. [Google Scholar] [CrossRef] [PubMed]
- Ambudkar, S.V.; Dey, S.; Hrycyna, C.A.; Ramachandra, M.; Pastan, I.; Gottesman, M.M. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 361–398. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Pastan, I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu. Rev. Biochem. 1993, 62, 385–427. [Google Scholar] [CrossRef]
- Chufan, E.E.; Kapoor, K.; Sim, H.M.; Singh, S.; Talele, T.T.; Durell, S.R.; Ambudkar, S.V. Multiple transport-active binding sites are available for a single substrate on human P-glycoprotein (ABCB1). PLoS ONE 2013, 8, e82463. [Google Scholar] [CrossRef]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753–756. [Google Scholar] [CrossRef]
- Vahedi, S.; Chufan, E.E.; Ambudkar, S.V. Global alteration of the drug-binding pocket of human P-glycoprotein (ABCB1) by substitution of fifteen conserved residues reveals a negative correlation between substrate size and transport efficiency. Biochem. Pharmacol. 2017, 143, 53–64. [Google Scholar] [CrossRef]
- Nosol, K.; Romane, K.; Irobalieva, R.N.; Alam, A.; Kowal, J.; Fujita, N.; Locher, K.P. Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. Proc. Natl. Acad. Sci. USA 2020, 117, 26245–26253. [Google Scholar] [CrossRef]
- Xiong, J.; Feng, J.; Yuan, D.; Zhou, J.; Miao, W. Tracing the structural evolution of eukaryotic ATP binding cassette transporter superfamily. Sci. Rep. 2015, 5, 16724. [Google Scholar] [CrossRef]
- Sajid, A.; Lusvarghi, S.; Chufan, E.E.; Ambudkar, S.V. Evidence for the critical role of transmembrane helices 1 and 7 in substrate transport by human P-glycoprotein (ABCB1). PLoS ONE 2018, 13, e0204693. [Google Scholar] [CrossRef] [PubMed]
- Sajid, A.; Lusvarghi, S.; Murakami, M.; Chufan, E.E.; Abel, B.; Gottesman, M.M.; Durell, S.R.; Ambudkar, S.V. Reversing the direction of drug transport mediated by the human multidrug transporter P-glycoprotein. Proc. Natl. Acad. Sci. USA 2020, 117, 29609–29617. [Google Scholar] [CrossRef]
- Kodan, A.; Yamaguchi, T.; Nakatsu, T.; Sakiyama, K.; Hipolito, C.J.; Fujioka, A.; Hirokane, R.; Ikeguchi, K.; Watanabe, B.; Hiratake, J.; et al. Structural basis for gating mechanisms of a eukaryotic P-glycoprotein homolog. Proc. Natl. Acad. Sci. USA 2014, 111, 4049–4054. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Chen, J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. [Google Scholar] [CrossRef]
- Ward, A.B.; Szewczyk, P.; Grimard, V.; Lee, C.W.; Martinez, L.; Doshi, R.; Caya, A.; Villaluz, M.; Pardon, E.; Cregger, C.; et al. Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc. Natl. Acad. Sci. USA 2013, 110, 13386–13391. [Google Scholar] [CrossRef]
- Li, J.; Jaimes, K.F.; Aller, S.G. Refined structures of mouse P-glycoprotein. Protein Sci. 2014, 23, 34–46. [Google Scholar] [CrossRef]
- Szewczyk, P.; Tao, H.; McGrath, A.P.; Villaluz, M.; Rees, S.D.; Lee, S.C.; Doshi, R.; Urbatsch, I.L.; Zhang, Q.; Chang, G. Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta. Crystallogr. D Biol. Crystallogr. 2015, 71, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Esser, L.; Zhou, F.; Pluchino, K.M.; Shiloach, J.; Ma, J.; Tang, W.K.; Gutierrez, C.; Zhang, A.; Shukla, S.; Madigan, J.P.; et al. Structures of the Multidrug Transporter P-glycoprotein Reveal Asymmetric ATP Binding and the Mechanism of Polyspecificity. J. Biol. Chem. 2017, 292, 446–461. [Google Scholar] [CrossRef]
- Le, C.A.; Harvey, D.S.; Aller, S.G. Structural definition of polyspecific compensatory ligand recognition by P-glycoprotein. IUCrJ 2020, 7, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Nosol, K.; Bang-Sørensen, R.; Irobalieva, R.N.; Erramilli, S.K.; Stieger, B.; Kossiakoff, A.A.; Locher, K.P. Structures of ABCB4 provide insight into phosphatidylcholine translocation. Proc. Natl. Acad. Sci. USA 2021, 118, e2106702118. [Google Scholar] [CrossRef]
- Schleker, E.S.M.; Buschmann, S.; Xie, H.; Welsch, S.; Michel, H.; Reinhart, C. Structural and functional investigation of ABC transporter STE6-2p from Pichia pastoris reveals unexpected interaction with sterol molecules. Proc. Natl. Acad. Sci. USA 2022, 119, e2202822119. [Google Scholar] [CrossRef]
- Shukla, S.; Schwartz, C.; Kapoor, K.; Kouanda, A.; Ambudkar, S.V. Use of baculovirus BacMam vectors for expression of ABC drug transporters in mammalian cells. Drug Metab. Dispos 2012, 40, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Mechetner, E.B.; Roninson, I.B. Efficient inhibition of P-glycoprotein-mediated multidrug resistance with a monoclonal antibody. Proc. Natl. Acad. Sci. USA 1992, 89, 5824–5828. [Google Scholar] [CrossRef]
- Mechetner, E.B.; Schott, B.; Morse, B.S.; Stein, W.D.; Druley, T.; Davis, K.A.; Tsuruo, T.; Roninson, I.B. P-glycoprotein function involves conformational transitions detectable by differential immunoreactivity. Proc. Natl. Acad. Sci. USA 1997, 94, 12908–12913. [Google Scholar] [CrossRef]
- Alam, A.; Küng, R.; Kowal, J.; McLeod, R.A.; Tremp, N.; Broude, E.V.; Roninson, I.B.; Stahlberg, H.; Locher, K.P. Structure of a zosuquidar and UIC2-bound human-mouse chimeric ABCB1. Proc. Natl. Acad. Sci. USA 2018, 115, E1973–E1982. [Google Scholar] [CrossRef]
- Vahedi, S.; Lusvarghi, S.; Pluchino, K.; Shafrir, Y.; Durell, S.R.; Gottesman, M.M.; Ambudkar, S.V. Mapping discontinuous epitopes for MRK-16, UIC2 and 4E3 antibodies to extracellular loops 1 and 4 of human P-glycoprotein. Sci. Rep. 2018, 8, 12716. [Google Scholar] [CrossRef]
- Kerr, K.M.; Sauna, Z.E.; Ambudkar, S.V. Correlation between steady-state ATP hydrolysis and vanadate-induced ADP trapping in Human P-glycoprotein. Evidence for ADP release as the rate-limiting step in the catalytic cycle and its modulation by substrates. J. Biol. Chem. 2001, 276, 8657–8664. [Google Scholar] [CrossRef] [PubMed]
- Pluchino, K.M.; Hall, M.D.; Moen, J.K.; Chufan, E.E.; Fetsch, P.A.; Shukla, S.; Gill, D.R.; Hyde, S.C.; Xia, D.; Ambudkar, S.V.; et al. Human-Mouse Chimeras with Normal Expression and Function Reveal That Major Domain Swapping Is Tolerated by P-Glycoprotein (ABCB1). Biochemistry 2016, 55, 1010–1023. [Google Scholar] [CrossRef] [PubMed]
- Nandigama, K.; Lusvarghi, S.; Shukla, S.; Ambudkar, S.V. Large-scale purification of functional human P-glycoprotein (ABCB1). Protein Expr. Purif. 2019, 159, 60–68. [Google Scholar] [CrossRef]
- Schaffner, W.; Weissmann, C. A rapid, sensitive, and specific method for the determination of protein in dilute solution. Anal. Biochem. 1973, 56, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Ambudkar, S.V. Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells. Methods Enzym. 1998, 292, 504–514. [Google Scholar]
- Ramachandra, M.; Ambudkar, S.V.; Chen, D.; Hrycyna, C.A.; Dey, S.; Gottesman, M.M.; Pastan, I. Human P-glycoprotein exhibits reduced affinity for substrates during a catalytic transition state. Biochemistry 1998, 37, 5010–5019. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Moritsugu, K.; Koike, R.; Yamada, K.; Kato, H.; Kidera, A. Motion Tree Delineates Hierarchical Structure of Protein Dynamics Observed in Molecular Dynamics Simulation. PLoS ONE 2015, 10, e0131583. [Google Scholar] [CrossRef]
- Mora Lagares, L.; Pérez-Castillo, Y.; Minovski, N.; Novič, M. Structure-Function Relationships in the Human P-Glycoprotein (ABCB1): Insights from Molecular Dynamics Simulations. Int. J. Mol. Sci. 2021, 23, 362. [Google Scholar] [CrossRef]
- Chufan, E.E.; Sim, H.M.; Ambudkar, S.V. Molecular basis of the polyspecificity of P-glycoprotein (ABCB1): Recent biochemical and structural studies. Adv. Cancer Res. 2015, 125, 71–96. [Google Scholar] [CrossRef]
- Ritchie, T.K.; Kwon, H.; Atkins, W.M. Conformational analysis of human ATP-binding cassette transporter ABCB1 in lipid nanodiscs and inhibition by the antibodies MRK16 and UIC2. J. Biol. Chem. 2011, 286, 39489–39496. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrates | TMH4,10-14A | TMH4,10-11A | TMH4-7A | TMH10-7A |
|---|---|---|---|---|
| BD-CsA | 33 ± 3 | 3 ± 0.6 | 89 ± 3 | 85 ± 2 |
| BD-EDA | 22 ± 5 | 66 ± 6 | 93 ± 67 | 46 ± 2 |
| BD-Prazosin | 2 ± 0.6 | 54 ± 4 | 40 ± 10 | 18 ± 13 |
| BD-Verapamil | ND | 3 ± 0.6 | 26 ± 12 | 3 ± 2 |
| Calcein-AM | 23 ± 9 | 58 ± 9 | 94 ± 4 | 97 ± 3 |
| Daunorubicin | 21 ± 1 | 34 ± 2 | 64 ± 6 | 54 ± 9 |
| Flutax-1 | ND | 4 ± 2 | 98 ± 4 | 92 ± 7 |
| LDS 751 | 31 ± 8 | 43 ± 6 | 89 ± 8 | 91 ± 8 |
| NBD-CsA | 3 ± 1 | 4 ± 1 | 95 ± 9 | 94 ± 11 |
| R6 | 13 ± 11 | 6 ± 2 | 54 ± 6 | 13 ± 1 |
| R6G | 43 ± 3 | 78 ± 3 | 90 ± 1 | 95 ± 1 |
| Rh123 | 3 ± 0.58 | 28 ± 5 | 93 ± 3 | 90 ± 3 |
| Rhod-2 AM | ND | 5 ± 2 | 96 ± 6 | 96 ± 5 |
| TMRE | 26 ± 5 | 85 ± 1 | 100 ± 0 | 98 ± 3 |
| X-Rhod-1 AM | 4 ± 1 | 4 ± 1 | 96 ± 6 | 99 ± 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, H.; Ware, M.J.; Sajid, A.; Lusvarghi, S.; Durell, S.R.; Ambudkar, S.V. Residues from Homologous Transmembrane Helices 4 and 10 Are Critical for P-Glycoprotein (ABCB1)-Mediated Drug Transport. Cancers 2023, 15, 3459. https://doi.org/10.3390/cancers15133459
Rahman H, Ware MJ, Sajid A, Lusvarghi S, Durell SR, Ambudkar SV. Residues from Homologous Transmembrane Helices 4 and 10 Are Critical for P-Glycoprotein (ABCB1)-Mediated Drug Transport. Cancers. 2023; 15(13):3459. https://doi.org/10.3390/cancers15133459
Chicago/Turabian StyleRahman, Hadiar, Mark J. Ware, Andaleeb Sajid, Sabrina Lusvarghi, Stewart R. Durell, and Suresh V. Ambudkar. 2023. "Residues from Homologous Transmembrane Helices 4 and 10 Are Critical for P-Glycoprotein (ABCB1)-Mediated Drug Transport" Cancers 15, no. 13: 3459. https://doi.org/10.3390/cancers15133459
APA StyleRahman, H., Ware, M. J., Sajid, A., Lusvarghi, S., Durell, S. R., & Ambudkar, S. V. (2023). Residues from Homologous Transmembrane Helices 4 and 10 Are Critical for P-Glycoprotein (ABCB1)-Mediated Drug Transport. Cancers, 15(13), 3459. https://doi.org/10.3390/cancers15133459