
Post-Surgical Prognosis of Patients with Pineoblastoma: A Systematic Review and Individual Patient Data Analysis with Trends over Time

 , , ,
, , , 

Simple Summary
Abstract
1. Introduction
2. Methods
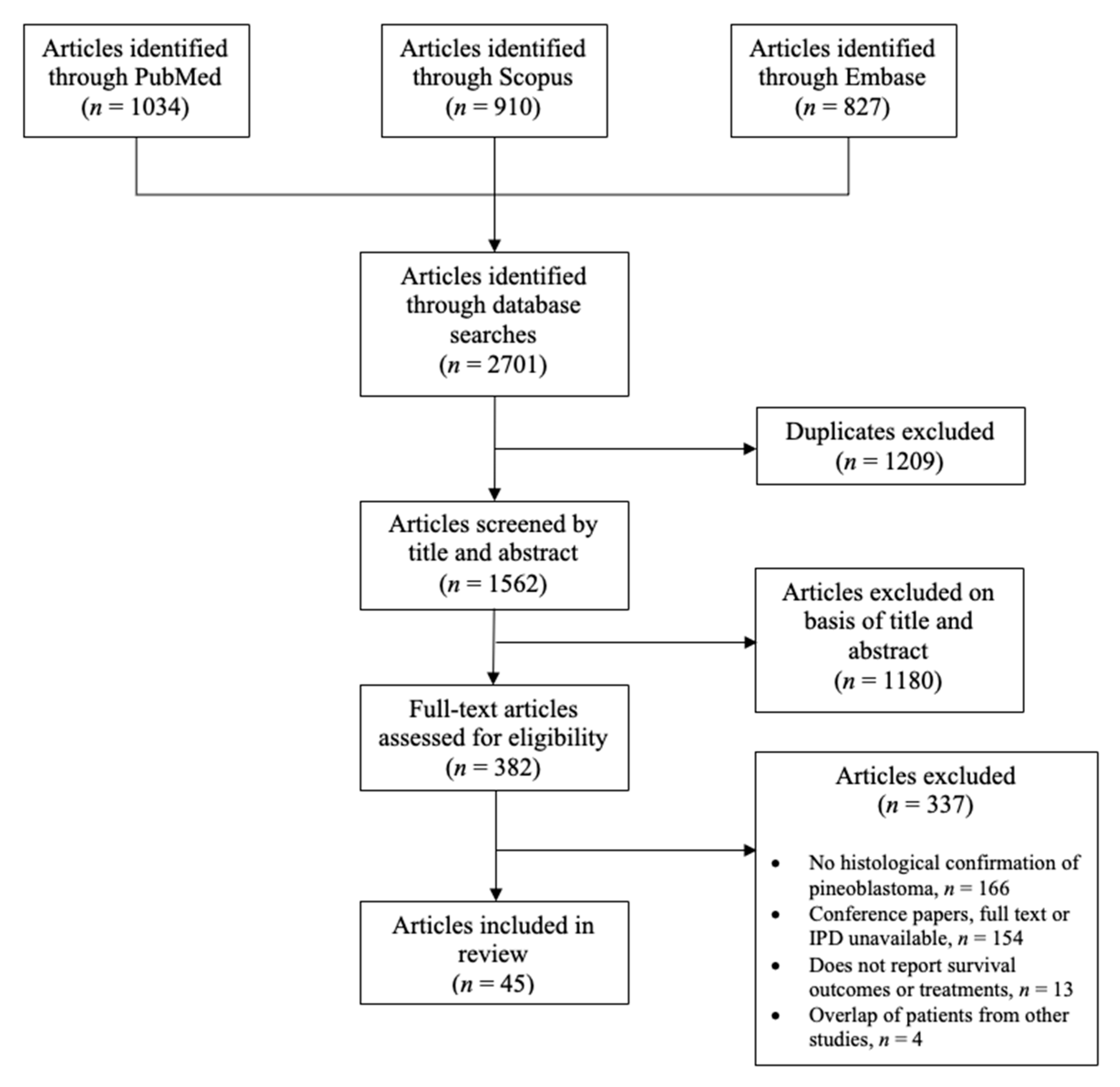
2.1. Design and Search
2.2. Data Extraction
2.3. Statistical Analysis
3. Results
3.1. Included Studies
3.2. Individual Patient Data
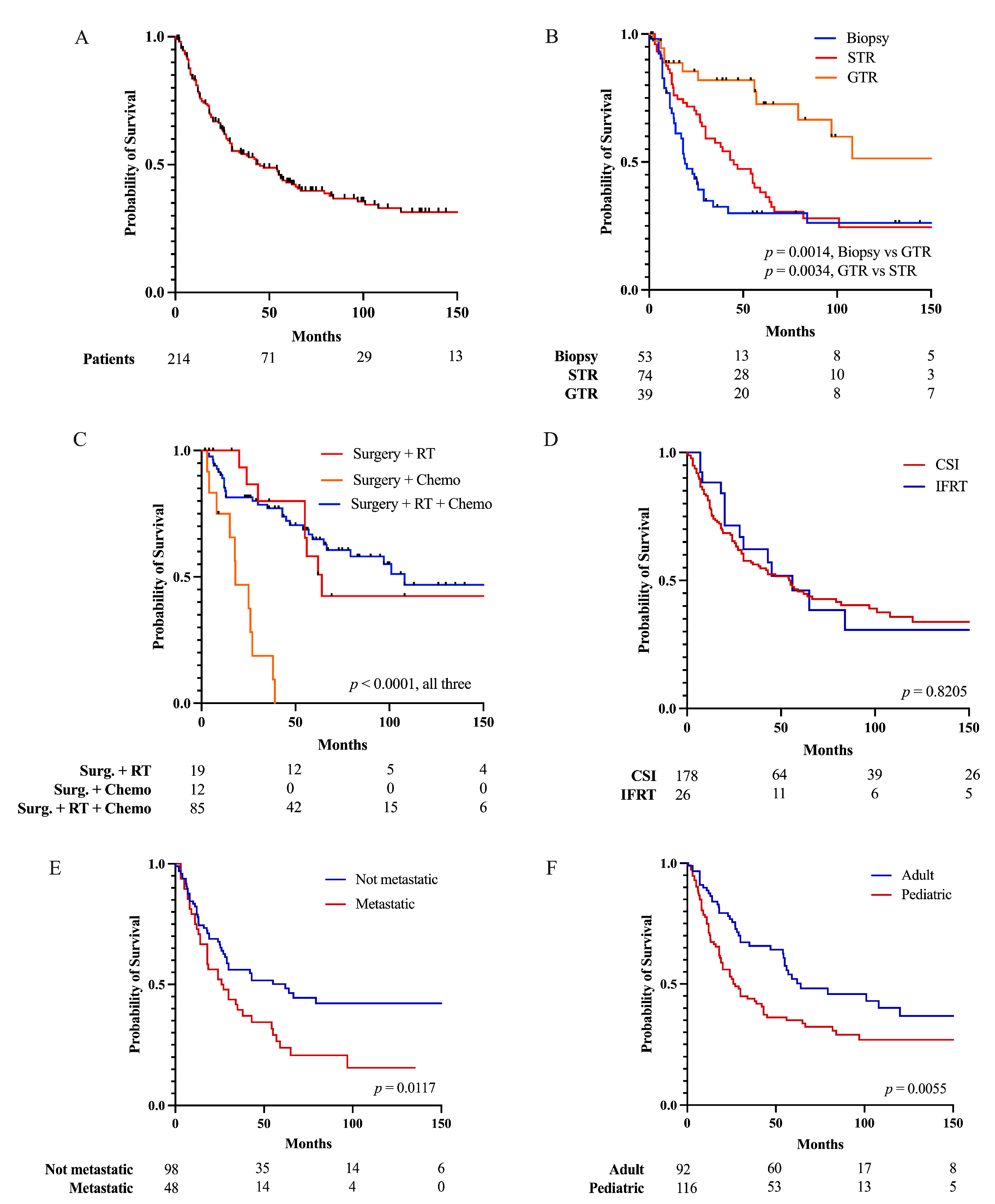
3.3. Factors Affecting Patient Survival
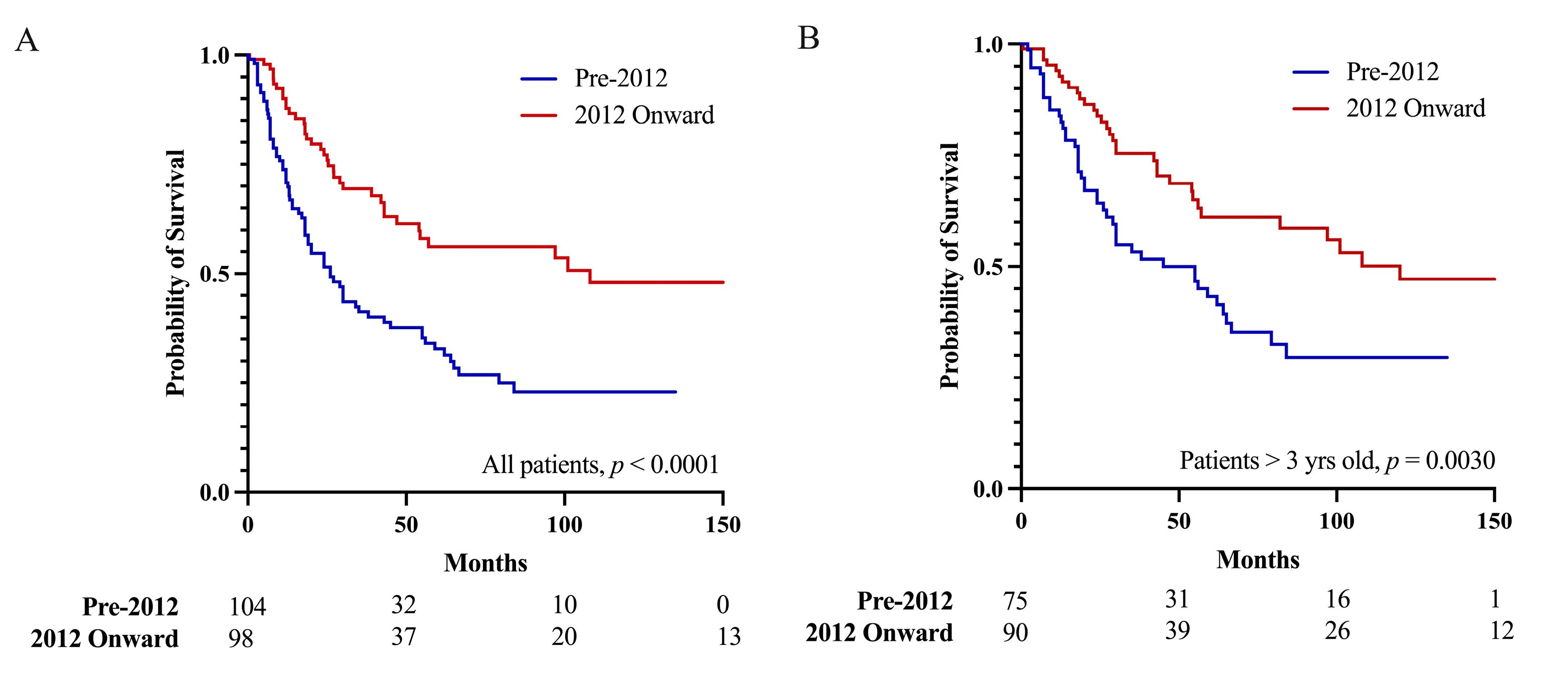
3.4. Pineoblastoma Prognosis since 2012
4. Discussion
Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2015–2019. Neuro-Oncology 2022, 24, v1–v95. [Google Scholar] [CrossRef]
- Greppin, K.; Cioffi, G.; Waite, K.A.; Ostrom, Q.T.; Landi, D.; Takaoka, K.; Kruchko, C.; Barnholtz-Sloan, J.S. Epidemiology of pineoblastoma in the United States, 2000–2017. Neuro-Oncol. Pract. 2022, 9, 149–157. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Jooma, R.; Kendall, B.E. Diagnosis and management of pineal tumors. J. Neurosurg. 1983, 58, 654–665. [Google Scholar] [CrossRef]
- Borit, A.; Blackwood, W.; Mair, W.G. The separation of pineocytoma from pineoblastoma. Cancer 1980, 45, 1408–1418. [Google Scholar] [CrossRef]
- Mynarek, M.; Pizer, B.; Dufour, C.; van Vuurden, D.; Garami, M.; Massimino, M.; Fangusaro, J.; Davidson, T.; Gil-da-Costa, M.J.; Sterba, J.; et al. Evaluation of age-dependent treatment strategies for children and young adults with pineoblastoma: Analysis of pooled European Society for Paediatric Oncology (SIOP-E) and US Head Start data. Neuro-Oncology 2016, 19, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Farnia, B.; Allen, P.K.; Brown, P.D.; Khatua, S.; Levine, N.B.; Li, J.; Penas-Prado, M.; Mahajan, A.; Ghia, A.J. Clinical outcomes and patterns of failure in pineoblastoma: A 30-year, single-institution retrospective review. World Neurosurg. 2014, 82, 1232–1241. [Google Scholar] [CrossRef]
- Jakacki, R.I.; Zeltzer, P.M.; Boyett, J.M.; Albright, A.L.; Allen, J.C.; Geyer, J.R.; Rorke, L.B.; Stanley, P.; Stevens, K.R.; Wisoff, J.; et al. Survival and prognostic factors following radiation and/or chemotherapy for primitive neuroectodermal tumors of the pineal region in infants and children: A report of the Childrens Cancer Group. J. Clin. Oncol. 1995, 13, 1377–1383. [Google Scholar] [CrossRef]
- Raleigh, D.R.; Solomon, D.A.; Lloyd, S.A.; Lazar, A.; Garcia, M.A.; Sneed, P.K.; Clarke, J.L.; McDermott, M.W.; Berger, M.S.; Tihan, T.; et al. Histopathologic review of pineal parenchymal tumors identifies novel morphologic subtypes and prognostic factors for outcome. Neuro-Oncology 2017, 19, 78–88. [Google Scholar] [CrossRef]
- Tate, M.; Sughrue, M.E.; Rutkowski, M.J.; Kane, A.J.; Aranda, D.; McClinton, L.; McClinton, L.; Barani, I.J.; Parsa, A.T. The long-term postsurgical prognosis of patients with pineoblastoma. Cancer 2012, 118, 173–179. [Google Scholar] [CrossRef]
- Fauchon, F.; Jouvet, A.; Paquis, P.; Saint-Pierre, G.; Mottolese, C.; Ben Hassel, M.; Chauveinc, L.; Sichez, J.P.; Philippon, J.; Schlienger, M.; et al. Parenchymal pineal tumors: A clinicopathological study of 76 cases. Int. J. Radiat. Oncol. Biol. Phys. 2000, 46, 959–968. [Google Scholar] [CrossRef]
- Jin, M.C.; Prolo, L.M.; Wu, A.; Azad, T.D.; Shi, S.; Rodrigues, A.J.; Soltys, S.G.; Pollom, E.L.; Li, G.; Hiniker, S.M.; et al. Patterns of Care and Age-Specific Impact of Extent of Resection and Adjuvant Radiotherapy in Pediatric Pineoblastoma. Neurosurgery 2020, 86, E426–E435. [Google Scholar] [CrossRef]
- Kerezoudis, P.; Yolcu, Y.U.; Laack, N.N.; Ruff, M.W.; Khatua, S.; Daniels, D.J.; Burns, T.C.; Kizilbash, S.H. Survival and associated predictors for patients with pineoblastoma or pineal parenchymal tumors of intermediate differentiation older than 3 years: Insights from the National Cancer Database. Neurooncol. Adv. 2022, 4, vdac057. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.Y.; Gilbert, A.; Cachia, D.; Mandel, J.; Fuller, G.N.; Penas-Prado, M.; de Groot, J.; Kamiya-Matsuoka, C. Pineal parenchymal tumor of intermediate differentiation: A single-institution experience. Neurooncol. Pract. 2020, 7, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Yang, Z.; Zhang, X.; Lin, D.; Xu, X.; Lu, X.; Chen, S.; Lin, J. Prognosis of Pediatric Patients with Pineoblastoma: A SEER Analysis 1990–2013. World Neurosurg. 2018, 118, e871–e879. [Google Scholar] [CrossRef] [PubMed]
- Stewart, L.A.; Clarke, M.; Rovers, M.; Riley, R.D.; Simmonds, M.; Stewart, G.; Tierney, J.F.; for the PRISMA-IPD Development Group. Preferred Reporting Items for a Systematic Review and Meta-Analysis of Individual Participant Data: The PRISMA-IPD Statement. JAMA 2015, 313, 1657–1665. [Google Scholar] [CrossRef]
- Page, M.J.; Moher, D.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E. PRISMA 2020 explanation and elaboration: Updated guidance and exemplars for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]
- Nandoliya, K.R.; Sadagopan, N.S.; Thirunavu, V.; Houskamp, E.J.; Karras, C.L.; Chaliparambil, R.K.; Jamshidi, P.; Raleigh, D.R.; Lukas, R.V.; Magill, S.T. Post-Surgical Prognosis of Patients with Pineoblastoma: A Systematic Review and Individual Patient Data Analysis with Trends over Time. PROSPERO 2023 CRD42023424651. Available online: https://www.crd.york.ac.uk/prospero/display_record.php?ID=CRD42023424651 (accessed on 18 May 2023).
- Ouzzani, M.; Hammady, H.; Fedorowicz, Z.; Elmagarmid, A. Rayyan—A web and mobile app for systematic reviews. Syst. Rev. 2016, 5, 210. [Google Scholar] [CrossRef] [PubMed]
- Guyatt, G.H.; Oxman, A.D.; Vist, G.E.; Kunz, R.; Falck-Ytter, Y.; Alonso-Coello, P.; Schünemann, H.J. GRADE: An emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008, 336, 924–926. [Google Scholar] [CrossRef]
- Sterne, J.A.; Hernán, M.A.; Reeves, B.C.; Savović, J.; Berkman, N.D.; Viswanathan, M.; Henry, D.; Altman, D.G.; Ansari, M.T.; Boutron, I. ROBINS-I: A tool for assessing risk of bias in non-randomised studies of interventions. BMJ 2016, 355, i4919. [Google Scholar] [CrossRef] [PubMed]
- Abbassy, M.; Aref, K.; Farhoud, A.; Hekal, A. The supracerebellar infratentorial approach in pineal region tumors: Technique and outcome in an underprivileged setting. Alex. J. Med. 2018, 54, 725–729. [Google Scholar] [CrossRef]
- Ai, P.; Peng, X.; Jiang, Y.; Zhang, H.; Wang, S.; Wei, Y. Complete regression of adult pineoblastoma following radiotherapy: A case report and review of the literature. Oncol. Lett. 2015, 10, 2329–2332. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Alsultan, A.; Alharbi, M.; Al-Dandan, S.; Bayoumi, Y.; Alharbi, T.; Alsudairy, R.; Alomari, A.; Aljamaan, K.; Musleh, O.; Alharbi, Q.; et al. High-dose chemotherapy with autologous stem cell rescue in saudi children less than 3 years of age with embryonal brain tumors. J. Pediatr. Hematol. Oncol. 2015, 37, 204–208. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ashley, D.M.; Longee, D.; Tien, R.; Fuchs, H.; Graham, M.L.; Kurtzberg, J.; Casey, J.; Olson, J.; Meier, L.; Ferrell, L.; et al. Treatment of patients with pineoblastoma with high dose cyclophosphamide. Med. Pediatr. Oncol. 1996, 26, 387–392. [Google Scholar] [CrossRef]
- Bernstock, J.D.; Alva, E.; Cohen, J.L.; Lobbous, M.; Chagoya, G.; Elsayed, G.A.; Orr, B.A.; Rozzelle, C.; Rocque, B.; Blount, J.; et al. Treatment of pediatric high-grade central nervous system tumors with high-dose methotrexate in combination with multiagent chemotherapy: A single-institution experience. Pediatr. Blood Cancer 2020, 67, e28119. [Google Scholar] [CrossRef]
- Biswas, A.; Mallick, S.; Purkait, S.; Gandhi, A.; Sarkar, C.; Singh, M.; Julka, P.K.; Rath, G.K. Treatment outcome and patterns of failure in patients of pinealoblastoma: Review of literature and clinical experience from a regional cancer centre in north India. Childs Nerv. Syst. 2015, 31, 1291–1304. [Google Scholar] [CrossRef]
- Broniscer, A.; Nicolaides, T.P.; Dunkel, I.J.; Gardner, S.L.; Johnson, J., Jr.; Allen, J.C.; Sposto, R.; Finlay, J.L. High-dose chemotherapy with autologous stem-cell rescue in the treatment of patients with recurrent non-cerebellar primitive neuroectodermal tumors. Pediatr. Blood Cancer 2004, 42, 261–267. [Google Scholar] [CrossRef]
- Cai, Y.; Xiong, Z.; Xin, C.; Chen, J.; Liu, K. Endoscope-Assisted Microsurgery in Pediatric Cases With Pineal Region Tumors: A Study of 18 Cases Series. Front. Surg. 2021, 8, 641196. [Google Scholar] [CrossRef]
- Chang, S.M.; Lillis-Hearne, P.K.; Larson, D.A.; Wara, W.M.; Bollen, A.W.; Prados, M.D. Pineoblastoma in adults. Neurosurgery 1995, 37, 381–391; discussion 390–391. [Google Scholar] [CrossRef]
- Charafe-Jauffret, E.; Lehmann, G.; Fauchon, F.; Michiels, J.F.; Paquis, P.; Maraninchi, D.; Hassoun, J. Vertebral metastases from pineoblastoma. Arch. Pathol. Lab. Med. 2001, 125, 939–943. [Google Scholar] [CrossRef]
- Choque-Velasquez, J.; Resendiz-Nieves, J.C.; Jahromi, B.R.; Colasanti, R.; Tynninen, O.; Collan, J.; Niemelä, M.; Hernesniemi, J. Pineoblastomas: A long-term follow up study of three cases in Helsinki Neurosurgery. Interdiscip. Neurosurg. Adv. Tech. Case Manag. 2019, 18, 100477. [Google Scholar] [CrossRef]
- Cuccia, F.; Mortellaro, G.; Cespuglio, D.; Valenti, V.; Gregorio, G.D.E.; Quartuccio, E.; Blasi, L.; Francaviglia, N.; Gallo, C.; Casto, A.L.O.; et al. A case report of adult pineoblastoma occurring in a pregnant woman. Anticancer Res. 2019, 39, 2627–2631. [Google Scholar] [CrossRef]
- Cuccia, V.; Rodríguez, F.; Palma, F.; Zuccaro, G. Pinealoblastomas in children. Childs Nerv. Syst. 2006, 22, 577–585. [Google Scholar] [CrossRef]
- Duffner, P.K.; Cohen, M.E.; Sanford, R.A.; Horowitz, M.E.; Krischer, J.P.; Burger, P.C.; Friedman, H.S.; Kun, L.E. Lack of efficacy of postoperative chemotherapy and delayed radiation in very young children with pineoblastoma. Pediatric Oncology Group. Med. Pediatr. Oncol. 1995, 25, 38–44. [Google Scholar] [CrossRef]
- Elshahoubi, A.; Khattab, E.; Halalsheh, H.; Khaleifeh, K.; Bouffet, E.; Amayiri, N. Feasibility of high-dose chemotherapy protocols to treat infants with malignant central nervous system tumors: Experience from a middle-income country. Pediatr. Blood Cancer 2019, 66, e27464. [Google Scholar] [CrossRef]
- Friedrich, C.; Müller, K.; Von Hoff, K.; Kwiecien, R.; Pietsch, T.; Warmuth-Metz, M.; Gerber, N.U.; Hau, P.; Kuehl, J.; Kortmann, R.D.; et al. Adults with CNS primitive neuroectodermal tumors/pineoblastomas: Results of multimodal treatment according to the pediatric HIT 2000 protocol. J. Neuro-Oncol. 2014, 116, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Gaito, S.; Malagoli, M.; Depenni, R.; Pavesi, G.; Bruni, A. Pineoblastoma in Adults: A Rare Case Successfully Treated with Multimodal Approach Including Craniospinal Irradiation Using Helical Tomotherapy. Cureus 2019, 11, e5852. [Google Scholar] [CrossRef] [PubMed]
- Gener, M.A.; Conger, A.R.; Van Gompel, J.; Ariai, M.S.; Jentoft, M.; Meyer, F.B.; Cardinal, J.S.; Bonnin, J.M.; Cohen-Gadol, A.A. Clinical, Pathological, and Surgical Outcomes for Adult Pineoblastomas. World Neurosurg. 2015, 84, 1816–1824. [Google Scholar] [CrossRef] [PubMed]
- Ghim, T.T.; Davis, P.; Seo, J.J.; Crocker, I.; O’Brien, M.; Krawiecki, N. Response to neoadjuvant chemotherapy in children with pineoblastoma. Cancer 1993, 72, 1795–1800. [Google Scholar] [CrossRef]
- Gilheeney, S.W.; Saad, A.; Chi, S.; Turner, C.; Ullrich, N.J.; Goumnerova, L.; Scott, R.M.; Marcus, K.; Lehman, L.; De Girolami, U.; et al. Outcome of pediatric pineoblastoma after surgery, radiation and chemotherapy. J. Neurooncol. 2008, 89, 89–95. [Google Scholar] [CrossRef]
- Golbin, D.; Nikitin, K.V.; Konovalov, A.N.; Pitskhelauri, D.I.; Shishkina, L.V.; Golanov, A.V.; Cherekaev, V.A.; Kobiakov, G.L.; Absalyamova, O.V.; Lasunin, N.; et al. Intraosseous Metastasizing of Pineoblastoma into the Anterior Skull Base, Calvarial Bones, and Vertebrae. Cureus 2015, 7, e437. [Google Scholar] [CrossRef] [PubMed]
- Görgün, Ö.; Koç, B.; Kebudi, R.; Wolff, J.E.; Kebudi, A.; Darendeliler, E. Clinical characteristics, late effects and outcomes in pineoblastomas in children: A single center experience. Turk. J. Pediatr. 2021, 63, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Kondziolka, D.; Hadjipanayis, C.G.; Flickinger, J.C.; Lunsford, L.D. The role of radiosurgery for the treatment of pineal parenchymal tumors. Neurosurgery 2002, 51, 880–889. [Google Scholar] [CrossRef] [PubMed]
- Herrick, M.K.; Rubinstein, L.J. The cytological differentiating potential of pineal parenchymal neoplasms (true pinealomas). A clinicopathological study of 28 tumours. Brain 1979, 102, 289–320. [Google Scholar] [CrossRef] [PubMed]
- Hinkes, B.G.; von Hoff, K.; Deinlein, F.; Warmuth-Metz, M.; Soerensen, N.; Timmermann, B.; Mittler, U.; Urban, C.; Bode, U.; Pietsch, T.; et al. Childhood pineoblastoma: Experiences from the prospective multicenter trials HIT-SKK87, HIT-SKK92 and HIT91. J. Neuro-Oncol. 2007, 81, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Kanno, H.; Sato, K.; Oikawa, M.; Ozaki, Y.; Nakamura, H.; Terasaka, S.; Kobayashi, H.; Houkin, K.; Hatanaka, K.; et al. Clinicopathologic study of pineal parenchymal tumors of intermediate differentiation. World Neurosurg. 2014, 81, 783–789. [Google Scholar] [CrossRef]
- Kang, Y.-M.; Lin, S.-C.; Lee, Y.-Y.; Chang, F.-C.; Liang, M.-L.; Chen, H.-H.; Liu, Y.-M.; Wong, T.-T.; Chen, Y.-W. Treatment outcomes for pediatric pineoblastoma: A single institute experience in Taiwan. Ther. Radiol. Oncol. 2018, 2, 19–27. [Google Scholar] [CrossRef][Green Version]
- Kellie, S.J.; Kovnar, E.H.; Kun, L.E.; Horowitz, M.E.; Heideman, R.L.; Douglass, E.C.; Langston, J.W.; Sanford, R.A.; Jenkins Iii, J.J.; Fairclough, D.L.; et al. Neuraxis dissemination in pediatric brain tumors: Response to preirradiation chemotherapy. Cancer 1992, 69, 1061–1066. [Google Scholar] [CrossRef]
- Kumar, N.; Srinivasa, G.Y.; Madan, R.; Salunke, P. Role of radiotherapy in residual pineal parenchymal tumors. Clin. Neurol. Neurosurg. 2018, 166, 91–98. [Google Scholar] [CrossRef]
- Lesnick, J.E.; Chayt, K.J.; Bruce, D.A. Familial pineoblastoma. Report of two cases. J. Neurosurg. 1985, 62, 930–932. [Google Scholar] [CrossRef] [PubMed]
- Linggood, R.M.; Chapman, P.H. Pineal tumors. J. Neurooncol. 1992, 12, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Maarouf, M.; El Majdoub, F.; Bührle, C.; Voges, J.; Lehrke, R.; Kocher, M.; Hunsche, S.; Treuer, H.; Sturm, V. Pineal parenchymal tumors. Management with interstitial iodine-125 radiosurgery. Strahlenther. Onkol. 2010, 186, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.L.; Chong, E.G.; Lee, J.; Mirshahidi, S.; Mirshahidi, H. A rare case of extremely delayed osseous metastasis of pineoblastoma. Rare Tumors 2021, 13, 2036361320975752. [Google Scholar] [CrossRef] [PubMed]
- Nozza, P.; Casciana, M.L.; Rossi, A.; Cama, A.; Milanaccio, C.; Raso, A.; Ravegnani, M.; Morreale, G.; Garre, M.L. Post-chemotherapy maturation of a pineoblastoma. Acta Neuropathol. 2010, 119, 651–653. [Google Scholar] [CrossRef]
- Panosyan, E.H.; Ikeda, A.K.; Chang, V.Y.; Laks, D.R.; Reeb, C.L.; Bowles, L.V.; Lasky, J.L., 3rd; Moore, T.B. High-dose chemotherapy with autologous hematopoietic stem-cell rescue for pediatric brain tumor patients: A single institution experience from UCLA. J. Transpl. 2011, 2011, 740673. [Google Scholar] [CrossRef]
- Prados, M.D.; Edwards, M.S.; Chang, S.M.; Russo, C.; Davis, R.; Rabbitt, J.; Page, M.; Lamborn, K.; Wara, W.M. Hyperfractionated craniospinal radiation therapy for primitive neuroectodermal tumors: Results of a Phase II study. Int. J. Radiat. Oncol. Biol. Phys. 1999, 43, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Tate, M.C.; Banerjee, A.; Vandenberg, S.R.; Tihan, T.; Chi, J.H.; Ames, C.P.; Parsa, A.T. Post-radiation reactive changes in a single vertebral body mimicking metastatic pineoblastoma: Case report. J. Neurosurg. Pediatr. 2009, 4, 479–483. [Google Scholar] [CrossRef]
- Tian, Y.; Liu, R.; Qin, J.; Wang, J.; Ma, Z.; Gong, J.; Li, C. Retrospective Analysis of the Clinical Characteristics, Therapeutic Aspects, and Prognostic Factors of 18 Cases of Childhood Pineoblastoma. World Neurosurg. 2018, 116, e162–e168. [Google Scholar] [CrossRef]
- Horiba, A.; Hayashi, M.; Tamura, N.; Chiba, K.; Aihara, Y.; Kawamata, T. Gamma Knife treatment of malignant infantile brain tumors—Case report. J. Radiosurg. SBRT 2018, 5, 249–253. [Google Scholar]
- Selvanathan, S.K.; Hammouche, S.; Smethurst, W.; Salminen, H.J.; Jenkinson, M.D. Outcome and prognostic features in adult pineoblastomas: Analysis of cases from the SEER database. Acta Neurochir. 2012, 154, 863–869. [Google Scholar] [CrossRef]
- Sonabend, A.M.; Bowden, S.; Bruce, J.N. Microsurgical resection of pineal region tumors. J. Neuro-Oncol. 2016, 130, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Hansford, J.R.; Huang, J.; Endersby, R.; Dodgshun, A.J.; Li, B.K.; Hwang, E.; Leary, S.; Gajjar, A.; Von Hoff, K.; Wells, O.; et al. Pediatric pineoblastoma: A pooled outcome study of North American and Australian therapeutic data. Neuro-Oncol. Adv. 2022, 4, vdac056. [Google Scholar] [CrossRef] [PubMed]
- Raleigh, D.R.; Tomlin, B.; Buono, B.D.; Roddy, E.; Sear, K.; Byer, L.; Felton, E.; Banerjee, A.; Torkildson, J.; Samuel, D.; et al. Survival after chemotherapy and stem cell transplant followed by delayed craniospinal irradiation is comparable to upfront craniospinal irradiation in pediatric embryonal brain tumor patients. J. Neuro-Oncol. 2017, 131, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Li, B.K.; Vasiljevic, A.; Dufour, C.; Yao, F.; Ho, B.L.B.; Lu, M.; Hwang, E.I.; Gururangan, S.; Hansford, J.R.; Fouladi, M.; et al. Pineoblastoma segregates into molecular sub-groups with distinct clinico-pathologic features: A Rare Brain Tumor Consortium registry study. Acta Neuropathol. 2020, 139, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.P.Y.; Gudenas, B.; Lin, T.; Orr, B.A.; Klimo, P.; Kumar, R.; Bouffet, E.; Gururangan, S.; Crawford, J.R.; Kellie, S.J.; et al. Risk-adapted therapy and biological heterogeneity in pineoblastoma: Integrated clinico-pathological analysis from the prospective, multi-center SJMB03 and SJYC07 trials. Acta Neuropathol. 2020, 139, 259–271. [Google Scholar] [CrossRef]
- de Kock, L.; Sabbaghian, N.; Druker, H.; Weber, E.; Hamel, N.; Miller, S.; Choong, C.S.; Gottardo, N.G.; Kees, U.R.; Rednam, S.P.; et al. Germ-line and somatic DICER1 mutations in pineoblastoma. Acta Neuropathol. 2014, 128, 583–595. [Google Scholar] [CrossRef]
- Snuderl, M.; Kannan, K.; Pfaff, E.; Wang, S.; Stafford, J.M.; Serrano, J.; Heguy, A.; Ray, K.; Faustin, A.; Aminova, O.; et al. Recurrent homozygous deletion of DROSHA and microduplication of PDE4DIP in pineoblastoma. Nat. Commun. 2018, 9, 2868. [Google Scholar] [CrossRef]
- Lee, J.C.; Mazor, T.; Lao, R.; Wan, E.; Diallo, A.B.; Hill, N.S.; Thangaraj, N.; Wendelsdorf, K.; Samuel, D.; Kline, C.N.; et al. Recurrent KBTBD4 small in-frame insertions and absence of DROSHA deletion or DICER1 mutation differentiate pineal parenchymal tumor of intermediate differentiation (PPTID) from pineoblastoma. Acta Neuropathol. 2019, 137, 851–854. [Google Scholar] [CrossRef]
- Pfaff, E.; Aichmüller, C.; Sill, M.; Stichel, D.; Snuderl, M.; Karajannis, M.A.; Schuhmann, M.U.; Schittenhelm, J.; Hasselblatt, M.; Thomas, C.; et al. Molecular subgrouping of primary pineal parenchymal tumors reveals distinct subtypes correlated with clinical parameters and genetic alterations. Acta Neuropathol. 2020, 139, 243–257. [Google Scholar] [CrossRef]
- Hart, M.N.; Earle, K.M. Primitive neuroectodermal tumors of the brain in children. Cancer 1973, 32, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Cambruzzi, E.; Medeiros, M.S.; da Silva, J.N.A.M.; Nascimento, G.B.C.; Zandoná, N.B.; Kus, W.P. Pineal anlage tumor: A case report and review of the literature. Childs Nerv. Syst. 2022, 38, 1625–1629. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; Ge, M.; Yang, W.; Cai, Y.; Zhang, N. Pineal anlage tumor: A case report and the literature review. Childs Nerv. Syst. 2023, 39, 353–358. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author | Country | Study Design | Num. of PB Patients | Quality † | Risk of Bias † | Female, n | Age (yrs) * | Follow-Up (Month) * | Chemotherapy |
|---|---|---|---|---|---|---|---|---|---|
| Herrick et al., 1979 [46] | USA | Case series | 10 | Low | Serious | 4 (40%) | 9.5 (4.6–39) | 32 (21.4–74.9) | |
| Borit et al., 1980 [6] | UK | Case series | 7 | Low | High | 2 (29%) | 18 (5.5–52) | 18 (3–84) | |
| Jooma et al., 1983 [5] | UK | Case series | 3 | Low | Serious | 2 (67%) | 11 (1.3–23) | 18 (6–24) | |
| Lesnick et al., 1985 [52] | USA | Case series | 2 | Very low | Serious | 2 (100%) | 27.5 (12–43) | 26 | Lomustine |
| Linggood et al., 1992 [53] | USA | Case series | 4 | Low | High | 2 (50%) | 18.5 (3–35) | 23.5 (16–72) | |
| Kellie et al., 1992 [50] | USA | Case series | 1 | Very low | Serious | 1 (100%) | 13 | 28 | Carboplatin and etoposide |
| Ghim et al., 1993 [41] | USA | Case series | 3 | Low | High | Not reported | 5 (3–7) | 24 (5–60) | Cisplatin, etoposide, vincristine |
| Chang et al., 1995 [31] | USA | Case series | 9 | Low | High | 3 (33%) | 35 (17–59) | 26 (6–62) | 6-Thioguanine, dibromodulcitol, lomustine, procarbazine, vincristine |
| Duffner et al., 1995 [36] | USA | Case series | 11 | Low | High | 3 (27%) | 0.67 (0.13–3) | 11 (3–34) | Cisplatinum, cyclophosphamide, etoposide, vincristine |
| Jakacki et al., 1995 [9] | USA | Case series | 25 | Low | High | Not reported | 3.1 (1.5–19.6) | Not reported | Lomustine, prednisone, vincristine OR Cisplatin, cytarabine, cyclophosphamide, hydroxurea, lomustine, methylprednisone, procarbazine |
| Ashley et al., 1996 [26] | USA | Case series | 8 | Low | High | 2 (25%) | 21 (3–23) | 70.5 (19–131) | Cyclophosphamide |
| Prados et al., 1999 [58] | USA | Case series | 5 | Low | High | 1 (20%) | 28 (26–30) | 55 (14–64) | |
| Charafe-Jauffret, et al., 2001 [32] | France | Case series | 2 | Low | Serious | 0 | 22 (18–26) | 68.5 (29–108) | |
| Hasegawa et al., 2002 [45] | USA | Case series | 3 | Low | High | 1 (33%) | 43 (15–61) | 17 (7–56) | |
| Broniscer et al., 2004 [29] | USA | Case series | 7 | Low | High | 5 (71%) | 1 (0.9–7.2) | 12 (0.2–101) | Etoposide, thiotepa ± carboplatin |
| Cuccia et al., 2006 [35] | Argentina | Case series | 12 | Low | High | 4 (25%) | 6.6 (0.5–18.1) | 9.6 (6.3–79.3) | Cisplatin, lomustine, vincristine |
| Hinkes et al., 2007 [47] | Germany | Case series | 11 | Low | High | 4 (36%) | 3.6 (0.6–16.9) | 26 (4–130) | Cisplatin, lomustine, vincristine |
| Gilheeney et al., 2008 [42] | USA | Case series | 11 | Low | High | 5 (45%) | 8.3 (0.25–13.6) | 61 (3–135) | |
| Tate et al., 2009 [59] | USA | Case report | 1 | Very low | Serious | 1 (100%) | 18 | 24 | Cisplatin, lomustine, vincristine |
| Maarouf et al., 2010 [54] | Germany | Case series | 7 | Low | High | 1 (17%) | 36 (6–65) | Not reported | |
| Nozza et al., 2010 [56] | Italy | Case report | 1 | Very low | Serious | 0 | 0.75 | 9 | |
| Panosyan et al., 2011 [57] | USA | Case series | 1 | Low | High | 1 (100%) | 10 | Not reported | AHSCR **, temozolomide |
| Ito et al., 2014 [48] | Japan | Case series | 3 | Low | High | 1 (33%) | 36 (30–49) | 61 (45–122) | Cisplatinum, etoposide, ifosfamide |
| Farnia et al., 2014 [8] | USA | Case series | 31 | Low | High | 21 (68%) | 18 (0.25–52) | 38.4 (0.2–332.4) | |
| Raleigh et al., 2014 [10] | USA | Case series | 29 | Low | High | 14 (48%) | 19 (1–57) | 49.2 (1–275) | Multiple different regimens |
| Friedrich et al., 2014 [38] | Germany | Case series | 10 | Low | High | 4 (40%) | 30.5 (22–57) | 32 (21.4–59.5) | Cisplatin, lomustine, vincristine or SKK, methotrexate/leucovorin |
| Ai et al., 2015 [24] | China | Case report | 1 | Very low | Serious | 0 | 46 | 36 | |
| Golbin et al., 2015 [43] | Russia | Case report | 1 | Very low | Serious | 1 (100%) | 23 | 57 | Cisplatin, cyclophosphamide, etoposide |
| Gener et al., 2015 [40] | USA | Case series | 12 | Low | High | 10 (83%) | 44.5 (24–81)) | 55 (0.5–288) | Vincristine |
| Alsultan et al., 2015 [25] | Saudi Arabia | Case series | 1 | Low | High | 0 | 3 | 13 | Cisplatin, cyclophosphamide, etoposide, vincristine |
| Biswas et al., 2015 [28] | India | Case series | 17 | Low | High | 6 (35%) | 14 (4–47) | 30.3 (1.9–69.3) | Carboplatin, etoposide OR Carboplatin, vincristine, and etoposide |
| Kumar et al., 2018 [51] | India | Case series | 2 | Low | Serious | 0 | 12.5 (12–13) | 31.5 (20–43) | Cisplatin, etoposide |
| Horiba et al., 2018 [61] | Japan | Case report | 1 | Very low | Serious | Not reported | 0.25 | 7 | |
| Kang et al., 2018 [49] | Taiwan | Case series | 11 | Low | High | 6 (55%) | 2.8 (1.5–16.8) | 27 (8–190) | |
| Tian et al., 2018 [60] | China | Case series | 18 | Low | High | 10 (56%) | 2.8 (1.6–13) | Not reported | Doxorubicin, vincristine, ifosfamide |
| Abbassy et al., 2018 [23] | Egypt | Case series | 2 | Low | High | 0 | 3 (2–4) | 22.6 (12–33) | |
| Choque-Velasquez et al., 2019 [33] | Finland | Case series | 3 | Low | High | 3 (100%) | 18 (3–25) | 170 (152–173) | |
| Cuccia et al., 2019 [34] | Italy | Case report | 1 | Very low | Serious | 1 (100%) | 21 | 12 | Cisplatin, lomustine, vincristine |
| Elshahoubi et al., 2019 [37] | Jordan | Case series | 1 | Low | Serious | 1 (100%) | 33 | 17.8 | |
| Gaito et al., 2019 [39] | Italy | Case report | 1 | Very low | Serious | 0 | 46 | 36 | Cisplatin and etoposide |
| Bernstock et al., 2020 [27] | USA | Case series | 1 | Very low | Serious | Not reported | 3.1 | 95 | Cisplatin, cyclophosphamide, etoposide, high-dose methotrexate, vincristine |
| Görgün et al., 2021 [44] | Turkey | Case series | 6 | Low | High | 3 (60%) | 5.8 (2–14) | 66 (12–228) | Cyclophosphamide, etoposide, vincristine OR carboplatin, etoposide, ifosfamide, OR Cyclophsophamide, lomustine, procarbazine, vincristine |
| Nguyen et al., 2021 [55] | USA | Case report | 1 | Very low | Serious | 0 | 22 | 120 | Vincristine |
| Cai et al., 2021 [30] | China | Case series | 1 | Low | High | 0 | 5 | 30 |
| Variable | Value/Pts. with Data | Pre-2012 | 2012 Onward | p-Value |
|---|---|---|---|---|
| Total Patients | 298 | 144 | 154 | |
| Female | 127/270 (47.0%) | 46/117 (39.3%) | 81/153 (52.9%) | 0.0276 |
| Follow up, median (range) | 27.1 (0.2–288.0) mo | |||
| Age, median (range) | 11.7 (0.13–81.0) yrs | 8.55 (0.13–65.0) | 16.0 (0.25–81.0) | 0.0012 |
| Pediatric | 186/298 (62.4%) | |||
| Metastatic at treatment | 65/226 (28.8%) | 40/126 (31.7%) | 25/99 (25.3%) | 0.3033 |
| Extent of resection | <0.0001 | |||
| None | 11/256 (4.3%) | 6/121 (5.0%) | 5/135 (3.7%) | |
| Biopsy | 52/256 (20.3%) | 28/121 (23.1%) | 24/135 (17.8%) | |
| Subtotal | 127/256 (49.2%) | 72/121 (59.5%) | 55/135 (40.7%) | |
| Gross total | 66/256 (25.8%) | 15/121 (12.4%) | 51/135 (37.8%) | |
| Adjuvant RT | 274/298 (91.9%) | 107/132 (81.1%) | 116/142 (81.7%) | 0.3373 |
| EBRT | 213/274 (77.7%) | 105/132 (79.5%) | 108/142 (75.8%) | |
| SRS | 3/274 (1.1%) | 1/132 (0.01%) | 2/142 (1.4%) | |
| Brachytherapy | 3/274 (1.1%) | 3/132 (2.3%) | 0 | |
| Not defined | 55/274 (20.1%) | 35/132 (26.5%) | 22/142 (16.7%) | |
| Adjuvant RT dose, Gy | 80.1 (38.0–181.4) | 88.0 (38.0–181.4) | 56.5 (40.0–158.0) | 0.0019 |
| Chemotherapy | 201/240 (83.8%) | 22/125 (17.6%) | 17/115 (14.8%) | 0.6021 |
| Variable | 1-Year OS | 2-Year OS | 5-Year OS |
|---|---|---|---|
| Overall | 78.5% | 64.2% | 43.1% |
| EOR | |||
| Biopsy/no resection | 69.1% | 43.4% | 30.0% |
| GTR | 88.7% | 85.4% | 72.6% |
| STR | 80.4% | 70.1% | 38.2% |
| Adjuvant therapy | |||
| Chemo | 75.0% | 46.9% | 0% |
| RT | 100% | 86.7% | 58.2% |
| CSI | 78.3% | 65.3% | 45.7% |
| IFRT | 88.3% | 71.5% | 46.1% |
| RT and chemo | 85.3% | 81.5% | 64.9% |
| Metastases | |||
| No | 79.0% | 67.7% | 50.2% |
| Yes | 72.9% | 52.1% | 23.8% |
| Age | |||
| ≥18 years old | 87.6% | 76.9% | 52.1% |
| <18 years old | 71.1% | 53.1% | 35.0% |
| ≥3 years old | 87.6% | 76.9% | 52.1% |
| <3 years old | 34.6% | 22.7% | 3.8% |
| Publication year | |||
| Pre-2012 | 70.8% | 51.6% | 56.1% |
| 2012 Onward | 87.8% | 77.2% | 32.8% |
| Variable | HR | HR 95% CI | p-Value |
|---|---|---|---|
| STR/Biopsy/None * | 2.0 | 1.1–3.9 | 0.0265 |
| Female sex | 1.4 | 0.9–2.2 | 0.1496 |
| Age per year older | 0.99 | 0.97–0.99 | 0.0460 |
| Surgery + Chemo ** | 1.3 | 0.7–2.2 | 0.4691 |
| Publication year pre-2012 | 1.7 | 1.1–2.7 | 0.0286 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nandoliya, K.R.; Sadagopan, N.S.; Thirunavu, V.; Houskamp, E.J.; Karras, C.L.; Chaliparambil, R.K.; Sriram, N.; Jamshidi, P.; Raleigh, D.R.; Lukas, R.V.; et al. Post-Surgical Prognosis of Patients with Pineoblastoma: A Systematic Review and Individual Patient Data Analysis with Trends over Time. Cancers 2023, 15, 3374. https://doi.org/10.3390/cancers15133374
Nandoliya KR, Sadagopan NS, Thirunavu V, Houskamp EJ, Karras CL, Chaliparambil RK, Sriram N, Jamshidi P, Raleigh DR, Lukas RV, et al. Post-Surgical Prognosis of Patients with Pineoblastoma: A Systematic Review and Individual Patient Data Analysis with Trends over Time. Cancers. 2023; 15(13):3374. https://doi.org/10.3390/cancers15133374
Chicago/Turabian StyleNandoliya, Khizar R., Nishanth S. Sadagopan, Vineeth Thirunavu, Ethan J. Houskamp, Constantine L. Karras, Rahul K. Chaliparambil, Nikhil Sriram, Pouya Jamshidi, David R. Raleigh, Rimas V. Lukas, and et al. 2023. "Post-Surgical Prognosis of Patients with Pineoblastoma: A Systematic Review and Individual Patient Data Analysis with Trends over Time" Cancers 15, no. 13: 3374. https://doi.org/10.3390/cancers15133374
APA StyleNandoliya, K. R., Sadagopan, N. S., Thirunavu, V., Houskamp, E. J., Karras, C. L., Chaliparambil, R. K., Sriram, N., Jamshidi, P., Raleigh, D. R., Lukas, R. V., & Magill, S. T. (2023). Post-Surgical Prognosis of Patients with Pineoblastoma: A Systematic Review and Individual Patient Data Analysis with Trends over Time. Cancers, 15(13), 3374. https://doi.org/10.3390/cancers15133374