1. Introduction
Immunotherapy is one of the major breakthroughs in oncology, as demonstrated by the spectacular therapeutic efficacy of monoclonal antibodies, CAR T cells and adoptive transfer of tumor-infiltrating T cells [
1,
2,
3,
4,
5,
6].
Several other immunotherapeutic strategies are being developed, among which therapeutic cancer vaccines are emerging as a promising approach [
7]. Recently, the development of efficacious cancer vaccines had been extremely challenging, mostly owing to the difficulties in identifying tumor-specific antigens and effective adjuvants [
8]. Thanks to the discovery that mutations in cancer cells can create immunogenic epitopes (“neo-epitopes”) and that such neo-epitopes induce tumor-specific T cells, vaccines based on cancer neo-epitopes formulated with novel adjuvants have shown high efficacy in preclinical settings and have now reached the clinic with promising results, particularly in combination with other immunotherapies [
9,
10].
Neo-epitope-based cancer vaccines require the up-front identification of the tumor immunogenic mutations and are highly patient specific since most of the mutations vary from patient to patient. A different approach to cancer vaccination is in situ vaccination (ISV), which consists of the intratumoral administration of immunostimulatory molecules (adjuvants). ISV does not envisage the use of specific cancer antigens, the rationale being that antigens are already present at the tumor site and all that is needed is an adjuvant, which can turn the immunosuppressive environment into an immunologically active one. This strategy was conceptualized at the end of the nineteen century by Dr. William Coley. Observing several patients who fully recovered from cancer when an infection occurred at the surgical sites (a frequent outcome at that time), he developed a variety of strategies for treating cancers with live and dead bacteria or with bacterial extracts, later named “Coley’s Toxin”. He treated 896 patients, achieving five-year survival rates of 34% to 73% for inoperable carcinomas and 13% to 79% for inoperable sarcomas [
11]. Although the use of the Coley’s toxin is no longer in the clinical practice, Dr. Coley’s pioneer work paved the way for the Food and Drug Administration (FDA) approval of the tuberculosis BCG vaccine as an ISV treatment of superficial bladder carcinoma and of the TLR7/8 agonist Resiquimod for skin carcinomas. In the January 2018–June 2021 period, 153 clinical trials dealing with the intratumoral injections of a variety of different formulations have been registered on
ClinicalTrials.gov (accessed on 6 June 2021) [
12].
ISV effectiveness relies on the potency of the immunostimulatory components delivered at the tumor site. Different adjuvants are being used, and they include CpG, Hiltonol, TLR4 agonists and agonists of the STING pathway [
12]. As we previously anticipated [
13], bacterial outer membrane vesicles (OMVs) have features particularly attractive for ISV applications. These vesicles, 30 to 300 nm in diameter, have a potent built-in adjuvanticity provided by a number of microbe-associated molecular patterns (MAMPs) naturally present in the outer membrane and in the periplasmic space of Gram-negative bacteria (LPS, lipoproteins, peptidoglycan, etc.). Such components trigger a potent inflammation and a Th1-skewed immune response, which ultimately promote cytotoxic T cell production and recruit phagocytic cells and NK cells. Moreover, the OMVs from several bacteria, including
E. coli, induce immunogenic cancer cell death [
14]. This feature is particularly interesting since the intratumoral OMV administration would favor the dissemination of cancer antigens, making them available for cross-presentation by DCs. Finally, OMVs can be decorated with foreign antigens and OMVs engineered with, or mixed to deliver cancer-specific peptide epitopes elicit anti-tumor immunity [
15,
16,
17,
18]. This offers the opportunity to test whether the inclusion of tumor-specific epitopes to the ISV formulation further potentiates the efficacy of ISV.
In this work, we first show that the OMVs from
E. coli BL21(DE3)Δ60 strain, a strain deprived of several OMV endogenous proteins [
19], promote a strong anti-tumor activity when intratumorally injected into the tumors of three different mouse models. Tumor inhibition correlates with a rapid infiltration of DCs and NK cells, occurring 24 h from the first OMV injection. We also show that the addition of five CT26 neo-epitopes to OMVs synergizes with the vesicle adjuvanticity, resulting in a potent anti-tumor activity, as judged by a two-tumor mouse model.
Overall, our data support the use of the OMVs in ISV and indicate that ISV efficacy can benefit from the addition of properly selected tumor-specific neo-antigens.
2. Materials and Methods
2.1. Bacterial Strain, Cell Lines and Mouse Strains
E. coli BL21(DE3)Δ60 was produced in our laboratory [
16] and grown at 30 °C under shaking conditions (200 rpm).
The cell lines B16-F10, CT26 and WEHI-164 were from ATCC. All cell lines were cultured in RPMI supplemented with 10% FBS, penicillin/streptomycin/L-glutamine and grown at 37 °C in 5% CO2.
C57BL/6 or BALB/c female 8-week-old mice were purchased from Charles River Laboratories and kept and treated in accordance with the Italian policies on animal research at the animal facilities of Toscana Life Sciences, Siena, Italy and Department of Cellular, Computational and Integrative Biology (CIBIO)—University of Trento, Italy. Mice were caged in groups of 5/8 animals in ventilated cages.
2.2. Synthetic Peptides
The synthetic peptides M03 (DKPLRRNNSYTSYIMAICGMPLDSFRA), M20 (PLLPFYPPDEALEIGLELNSSALPPTE), M26 (VILPQAPSGPSYATYLQPAQAQML TPP), M27 (EHIHRAGGLFVADAIQVGFGRIGKHFW) and M68 (VTSIPSVSNALNWKEFSFIQSTLGYVA) were purchased from GeneScript (Piscataway, NJ, USA) in lyophilic form and solubilized in milliQ water at a final concentration of 5 mg/mL.
2.3. OMV Preparation
OMVs
Δ60 were prepared growing the
E. coli BL21(DE3)Δ60 strain in an EZ control bioreactor (Applikon Biotechnology, Schiedam, The Netherlands) until the end of the exponential phase at 30 °C, pH 6.8 (±0.2), dO
2 > 30%, 280–500 rpm. OMVs were then purified and quantified as previously described [
16]. Briefly, the culture supernatant was separated from biomass by centrifugation at 4000×
g for 20 min. After filtration through a 0.22 μm pore size filter (Millipore, Burlington, MA, USA), OMVs were isolated, concentrated and diafiltrated from the supernatant using Tangential Flow Filtration (TFF) with a Cytiva Äkta Flux system (Marlborough, MA, USA). OMVs were quantified using DC protein assay (Bio-Rad, Hercules, CA, USA). OMV proteins were separated using a 4–12% gradient polyacrylamide gel (Invitrogen, Waltham, MA, USA) and finally stained with Coomassie Blue (Giotto, Sesto Fiorentino, Italy).
2.4. Dynamic Light Scattering Analysis
Size distribution profile of OMVs was determined by Dynamic Light Scattering (DLS) based on laser diffraction method using Zetasizer Nano (Malvern Panalytical, Malvern, UK). The OMV diameter of the batch preparation diluted at a final concentration of 0.5 mg/mL in PBS was determined by measuring the 90° side scatter size at 25 °C. Three measurements (between 15 and 20 experimental runs for each measurement) were averaged to determine the vesicle size.
2.5. Negative Staining Electron Microscopy Analysis
A volume of 5 μL of OMVs diluted at 80 ng/μL in PBS was loaded onto a copper 200-squaremesh grid of carbon/formvar rendered hydrophilic by glow discharge using a Q150R S (Quorum, Laughton, UK). The excess solution was blotted off after 30 s using Whatman filter Paper No.1 (Maidstone, Kent, UK). The grids were negatively stained with NanoW (Nanoprobes, Yaphank, NY, USA) for 30 s, then blotted using Whatman filter Paper No.1 and finally left to air dry. Micrographs were acquired using a G2 Spirit Transmission Electron Microscope (Tecnai, Dawson Creek, NE, USA) equipped with a CCD 2k × 4k camera at a final magnification of 120,000×.
2.6. Confocal Microscopy
CT26 cells (1.5 × 105/well) were plated on microscope coverslips in 6 multi-wells plate (Corning, New York, NY, USA) and incubated at 37 °C o/n. Subsequently, labelled OMVs with 2 μM BODIPY™ 493/503 (ThermoFisher Scientific, Waltham, MA, USA) were incubated with cells at 37 °C for 24 h and washed twice with 1X sterile PBS. Nuclei were stained blue using DAPI (300 nM). Glass coverslip-plated cells were fixed with 3.7% formaldehyde solution in PBS for 20 min at 4 °C. Then, cells were permeabilized with 0.1% Triton X-100 (ThermoFisher Scientific, Waltham, MA, USA) in PBS, stained with 50 μg/mL of phalloidin-tetramethyl-rhodamine isothiocyanate (TRITC) for 1 h at room temperature (RT) to visualize F-actin and mounted with ProLong™ Gold Antifade Mountant (ThermoFisher Scientific, Waltham, MA, USA). All fluorescence samples were examined at RT using a laser-scanning confocal microscope TCS SP5 (Leica, Mannheim, Germany). Lasers and spectral detection bands were chosen for the optimal imaging of TRITC, and FITC PMT levels were set using control samples. Multicolor images were collected simultaneously in two channels. Images were taken using a 63×, 1.4 NA, oil, HCX Plan APO lens (Leica, Mannheim, Germany). Images were captured using the Leica LAS-AF image acquisition software 1.2. Overlays were generated using LAS-AF software 4.0.
2.7. Lactate Dehydrogenase (LDH) Release Cytotoxicity Assay
CT26 cells were seeded at 1 × 104 cells per well in 100 μL of RPMI1640 without phenol red (Corning, New York, NY, USA), with 10% FBS (heat inactivated) (ThermoFisher Scientific, Waltham, MA, USA), 2 mM L-glutamine (EuroClone, Milan, Italy) and 1X penicillin/streptomycin (EuroClone, Milan, Italy), in a 96-well flat bottom, tissue culture-treated plate (Corning, New York, NY, USA) and then incubated overnight at 37 °C, 5% CO2. The next day, the culture medium was carefully removed so as not to disturb the cells. Treatments and controls were added in a final volume of 200 μL of treatment media (RPMI1640 without phenol red, 2 mM L-glutamine and 1X penicillin/streptomycin and 1.25% FBS). Negative control was treatment media only, positive control was Triton X-100 (ThermoFisher Scientific, Waltham, MA, USA) at 0.1%. OMVs, derived from BL21(DE3)ΔompA and BL21(DE3)Δ60, added in the following concentrations: 1 μg/well; 5 μg/well; 10 μg/well; 50 μg/well. Treated cells were incubated for 24 h at 37 °C 5% CO2. The plate was centrifuged for 10 min at 250× g and 100 μL of cell-free supernatant was transferred to a clear flat bottom 96-well plate. Subsequently, 100 μL of freshly prepared reaction mixture (catalyst and INT dye solution) was added and incubated for 30 min at RT in the dark. Then, the reaction was stopped with 1M HCl. The absorbance of colorimetric reaction was measured at 492 nm (reference wavelength: 620 nm) using an Infinite M200PRO Plate reader (TECAN, Männedorf, Swiss). The percentage cytotoxicity was calculated as follows: cytotoxicity % = (sample value-negative value)/(positive value-negative value) × 100.
2.8. Mouse Tumor Models
2.8.1. One-Tumor Model
Eight-week-old C57BL/6 and BALB/c female mice were subcutaneously challenged in one flank with 1 × 105 of B16-F10 and with 2.5 × 105 CT26, or 1.5 × 105 WEHI-164 cells, respectively. When tumor size reached a volume of approximately 100 mm3, 10 μg OMVΔ60 in 50 μL PBS, or PBS alone (50 μL), was injected into the tumors, and the treatment was repeated two additional times at two-day intervals. Tumor growth was followed for at least 30 days after the challenge and tumor volumes were determined with a caliper using the formula (A × B2)/2, where A is the largest and B is the smallest diameter of the tumor. The re-challenge experiment was performed by injecting 2.5 × 105 CT26 cells into naïve BALB/c mice and in mice previously cured with OMVsΔ60. Tumor growth was followed as described before. Statistical analysis (unpaired, two-tailed Student’s t-test) and graphs were processed using GraphPad Prism 5.03 software.
2.8.2. Two-Tumor Models
BALB/c female mice were 7 weeks old when the experiments began. A amount of 2.5 × 105 CT26 colon carcinoma cells were injected subcutaneously at sites on both the right and left flank of the mouse. Tumor size was monitored every 2 days on both sides of the animals with a digital caliper and expressed as volume (volume = (width 2 × length)/2). When tumor size reached at ≥50–100 mm3, mice were vaccinated in situ in one of the tumors. Mice were immunized with PBS, OMVsΔ60 (10 μg/injection) or OMVsΔ60 adsorbed with the previously described five synthetic peptides (“pentatope”, 20 μg of each peptide). Mice were vaccinated three times every two days.
2.9. Flow Cytometry Analysis
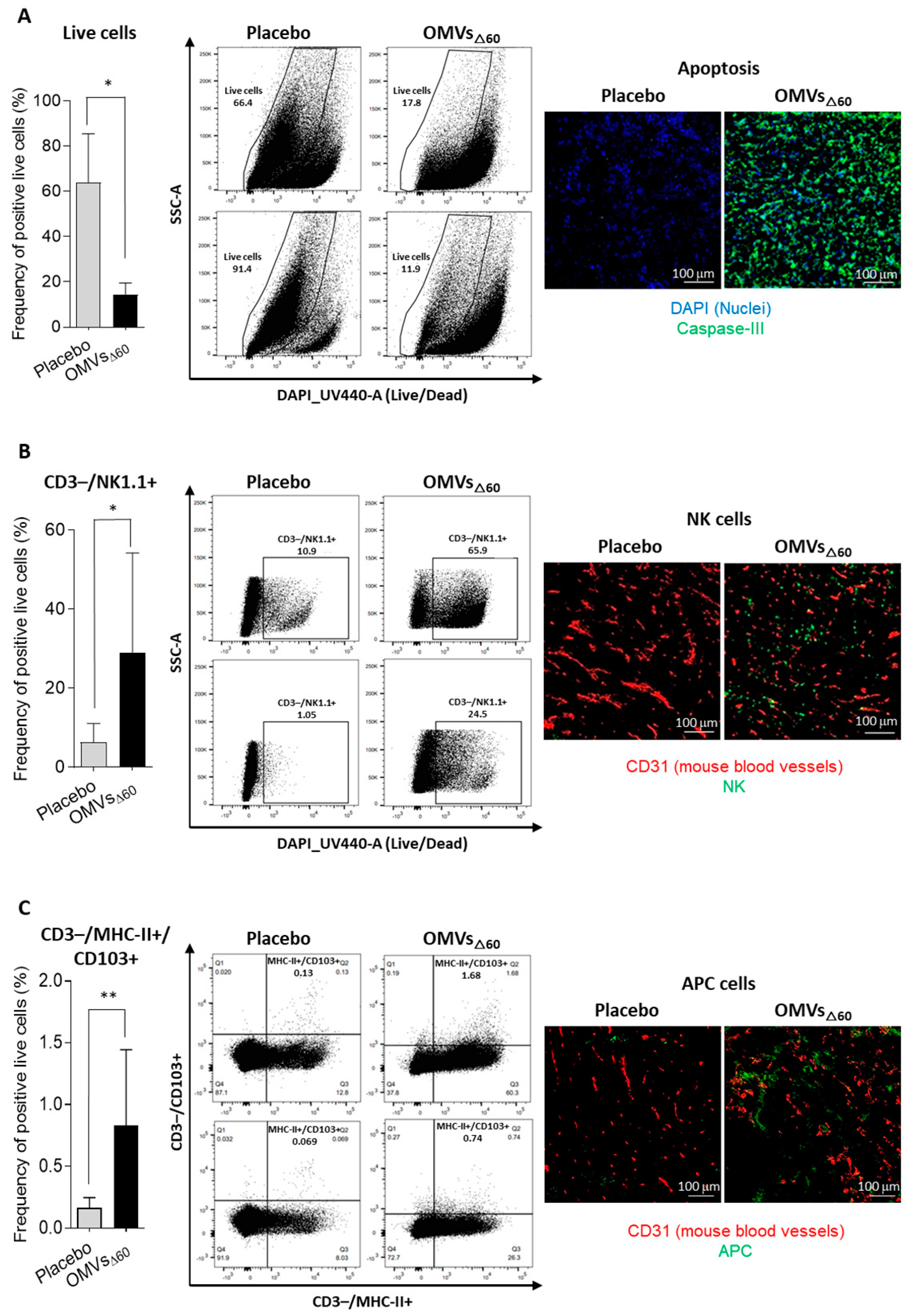
For the analysis of different tumor cell populations, tumor-infiltrating lymphocytes were isolated from subcutaneous CT26 tumors as follows. Two tumors per group were collected and minced into pieces of 1–2 mm in diameter using a sterile scalpel, filtered using a Cell Strainer 70 mm (Miltenyi Biotech, Bergisch Gladbach, Germany) and transferred into 50 mL tubes. Then, the tumor tissue was enzymatically digested using the Tumor Dissociation kit (Miltenyi Biotech, Bergisch Gladbach, Germany) according to the manufacturer’s protocol, and the gentleMACS™ Dissociators (Miltenyi Biotech, Bergisch Gladbach, Germany) were used for the mechanical dissociation steps. After dissociation, the sample was passed through to a 30 mm filter (Miltenyi Biotech, Bergisch Gladbach, Germany) to remove larger particles from the single-cell suspension. At the end of the dissociation protocol, 1–2 × 106 cells from tumors were incubated with Fixable Viability Stain UV440 (BD Bioscience, San Jose, CA, USA) for 15 min at RT in a 96-well plate. Then, cells were washed twice in PBS and incubated with 25 µL of an anti-mouse CD16/CD32-Fc/Block (BD Bioscience, San Jose, CA, USA) 20 min on ice in the dark. Later, 25 µL of the following mixture of fluorescent-labeled antibodies was added to the samples: CD3-APC (Biolegend, San Diego, CA, USA), CD4-BV510 (Biolegend, San Diego, CA, USA) and CD8a-PECF594 (BD Bioscience, San Jose, CA, USA) CD44-APC-Vio770 (Miltenyi Biotech, Bergisch Gladbach, Germany), CD25-PE-Vio770 (Miltenyi Biotech, Bergisch Gladbach, Germany), aNK1.1-PerCP-Vio770 (Miltenyi Biotech, Bergisch Gladbach, Germany), αMHC II (I-Ek)-VioBright (Miltenyi Biotech, Bergisch Gladbach, Germany), CD103-VioBright 515 (Miltenyi Biotech, Bergisch Gladbach, Germany) and CD62L-VioBlue (Miltenyi Biotech, Bergisch Gladbach, Germany). Cells were stained at RT in the dark for 20 min. After two washes with PBS, cells were fixed using the Fixation/Permeabilization Solution of FoxP3 Staining Buffer Set (Miltenyi Biotech, Bergisch Gladbach, Germany) for 20 min at RT, and cells were then washed twice and re-suspended in 50 µL of Anti-FoxP3-PE (Miltenyi Biotech, Bergisch Gladbach, Germany) diluted in permeabilization buffer and stained 20 min at RT. Cells were finally washed twice in permeabilization buffer and then in PBS. Samples were analyzed using a Symphony A3 (BD Bioscience, San Jose, CA, USA), and the raw data were elaborated using FlowJo software V10 6.1.
2.10. Immunofluorescence Assay
CT26 tumors were excised 24 h after a single intratumoral injection of 10 μg OMVΔ60 in 50 μL PBS, or PBS alone (50 μL). Tumors were embedded in Tissue-Tek® O.C.T. (Leica, Mannheim, Germany) and rapidly frozen in dry ice. Cryosections of 8 μm in size were obtained from frozen tumors at Cryostat (Leica, Mannheim, Germany). CT26 tumors were stained with primary antibodies for rabbit anti-mouse Caspase 3 (Invitrogen, Waltham, MA, USA), rat anti-mouse NKp46 (Biolegend, San Diego, CA, USA), rat anti-mouse Dendritic Cell marker 33D1 (Biolegend, San Diego, CA, USA), and goat anti-mouse CD31 (R&D System, Minneapolis, MN, USA). Three secondary antibodies were used: donkey anti-rat IgG Alexa Fluor 488 (Invitrogen, Waltham, MA, USA), donkey anti-goat IgG Alexa Fluor 594 (Invitrogen, Waltham, MA, USA) and goat anti-rabbit IgG Alexa Fluor 488. DAPI (ThermoFisher Scientific, Waltham, MA, USA) was used to detect the nuclei. Stained sections were mounted with Dako fluorescence mounting medium (Agilent, Santa Clara, CA, USA) and examined with a Eclipse Ti2 microscope (Nikon, Minato, Tokyo, Japan). Images were analyzed with Fiji for Mac OS X software.
4. Discussion
OMVs are emerging as a promising platform for vaccine development. Because of their ability to elicit a potent Th1-skewed immune response, OMVs are being used not only for prophylactic vaccines against infectious diseases but also for designing therapeutic cancer vaccines, the efficacy of which depends on the elicitation of tumor-specific cytotoxic CD8+ T cells and CD4+ Th1 cells. For instance, we previously showed that the s.c. immunization of immunocompetent mice with
E. coli OMVs engineered with tumor-specific B and T cell epitopes inhibited tumor development [
15,
16]. Moreover, Kim et al. reported that the i.v. delivery of vesicles purified from both Gram-negative and Gram-positive bacteria resulted in an anti-tumor activity which correlated with the infiltration of T cells in the experimental tumors [
21]. In addition, Cheng et al. chemically associated tumor T cell epitopes to
E. coli OMVs and showed that such OMVs protected mice against the challenge with tumors expressing such epitopes [
22]. Finally, OMVs expressing specific tumor neo-epitopes either released by intestinal commensal bacteria or administered by oral gavage were shown to protect mice from tumor challenge [
18,
23].
In this work, we further expand the applicability of OMVs to cancer therapy by demonstrating that their intratumoral injection has potent anti-tumor activity. Three different tumor mouse models were used, and in all of them, a relevant tumor-inhibitory effect was observed. Such an effect was mediated by the potent inflammation induced by OMV injection, which promoted a prompt infiltration of NK cells and DCs in the tumor microenvironment. The rapid migration of MHC-II+/CD103+ DCs appears particularly interesting since these cells are expected to internalize the tumor antigens released at the tumor site by dying cells and to promote the production of effector T cells in the draining lymph nodes. The accumulation of NK cells can partially explain the relevant tumor cell death observed in vaccinated mice. However, such killing is also mediated by OMVs, which have been shown to induce immunogenic cell death (pyroptosis), and our in vitro data confirm this observation.
As previously pointed out, ISV typically involves the intratumoral administration of immunostimulatory molecules (adjuvants) assuming that the relevant tumor antigens are readily available in the tumor microenvironment. This makes the therapy particularly attractive since, differently from personalized vaccines, it does not have to be adapted to patients. However, one interesting result emerging from our study is that OMV-based ISV is more effective if the injected formulation includes tumor neo-epitopes. Since s.c. immunization with OMVs decorated with neo-epitopes elicits epitope-specific T cells [
16], the tumor microenvironment should be populated with a high concentration of selected effector T cells, which are expected to amplify the effect of the OMV-mediated inflammation at the tumor site.
It remains to be tested whether the tumor antigens have to be co-injected into the tumor or if they can be delivered separately. For instance, it would be interesting to combine personalized cancer vaccines with OMV-based ISV. Our expectation is that the two strategies synergize, enhancing the infiltration of T cells elicited by personalized vaccines into the tumor microenvironment.
In addition to combining OMV-based ISV with cancer vaccines, other combinations should be tested, including in situ vaccination and checkpoint inhibitors (anti-CTLA4 and anti-PD1/PDL1 mAbs). This combination is particularly attractive since if effective, lower doses of mAbs could be used, thus reducing toxicity and costs.
5. Conclusions
ISV offers several advantages over systemic immunotherapy strategies. First, it is difficult to imagine any better way to induce a potent inflammation within the tumor microenvironment. Such inflammation, taking place where cancer antigens have the maximal concentration, should guarantee the highest possible polyclonal production of anti-tumor T cells, which can reach metastatic sites via systemic circulation (“Abscopal” effect). Second, ISV should mitigate the typical (often severe) adverse reactions of systemically delivered formulations by virtue of the fact that the active substances are administered at a lower dosage exactly where needed (avoiding off-target toxicity).
The effectiveness of ISV relies on the optimal selection of immunostimulatory components. Several PRR agonists are under evaluation in clinical studies. OMVs are naturally decorated with a number of MAMPs, which confer them a potent Th1-skewed adjuvanticity. The local release of IFNγ, IL12 and TNFα is the type of response needed to promote cytotoxic T cell production and to activate phagocytic cells and NK cells. Moreover, E. coli OMVs induce immunogenic death of cancer cells, thus facilitating the up-take of tumor antigens by APCs and the production of effector T cells. Finally, the production process of OMVs involves a simple bacterial fermentation, and OMVs are purified by tangential flow filtration of the supernatant with a yield of 50–100 mg/Lt. Assuming that a full therapy would involve a few injections of 5–20 μg of OMVs, 1 Lt fermentation would be sufficient to treat several hundreds of patients.
The most indicated cancers for ISV are obviously those that are easily accessible, such as melanoma, head and neck cancers, breast cancer and lymphomas. They represent approximately one-third of all cancer cases in the USA [
24]. However, with the current clinical technologies, all tumors can be treated with ISV. Indeed, most oncological units are able reach any tumor lesion with a 1 mm needle to establish a cancer diagnosis, and the same modalities can be used for in situ vaccination.
One of the foreseeable opportunities with high therapy potential is performing ISV on a primary tumor before surgery [
24]. This could be performed as neoadjuvant therapy with no interference with the current standard of care, a key prerequisite to ease regulatory approval. Pre-surgery ISV would offer at least two major advantages. First, the shrinkage of the tumor mass would make the surgical intervention less invasive. Second, it would facilitate the proper activation of the immune responses before tumor removal. Immunologically speaking, it makes little sense to try to force the immune system to recognize and kill cancer cells, and eventually prevent tumor rebound and metastasis formation, if the major source of tumor antigens has been removed.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}



