
Combined Blockade of TIGIT and PD-L1 Enhances Anti-Neuroblastoma Efficacy of GD2-Directed Immunotherapy with Dinutuximab Beta

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethic Statement
2.2. Cell Cultivation
2.3. Flow Cytometry
2.4. ADCC
2.5. Evaluation of Antitumor Efficacies of DB-Based Immunotherapies In Vivo
2.6. Assessment of DB Serum Levels in Treated Mice
2.7. Statistics
3. Results
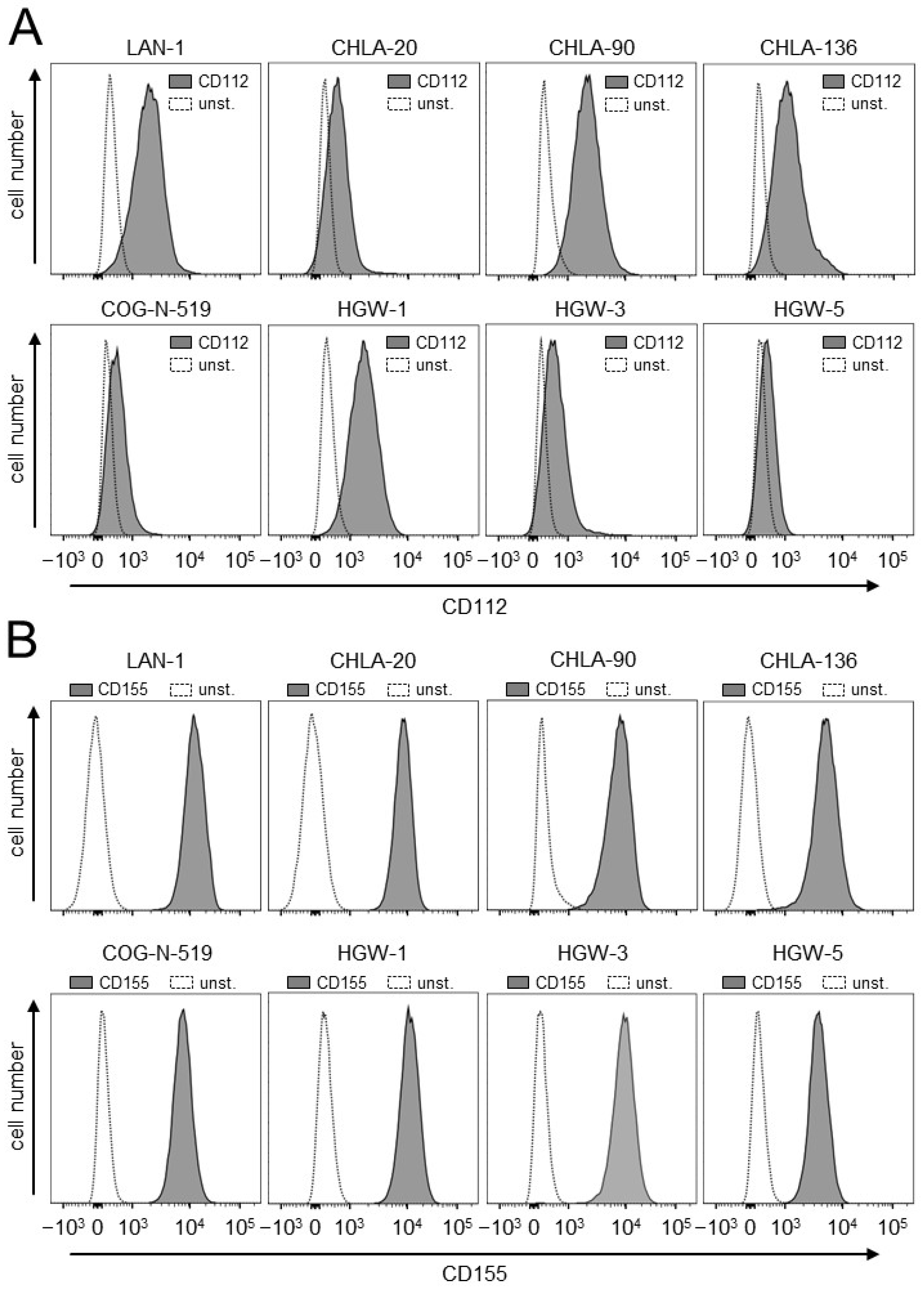
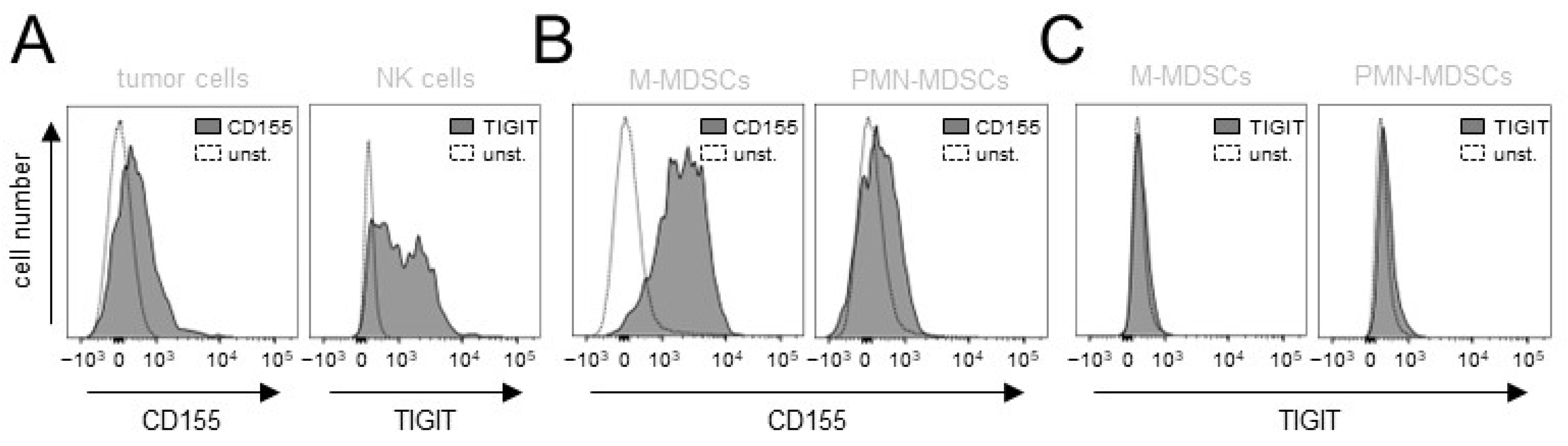
3.1. Evaluation of Expression of the TIGIT Ligands, CD112 and CD155, on NB Cells
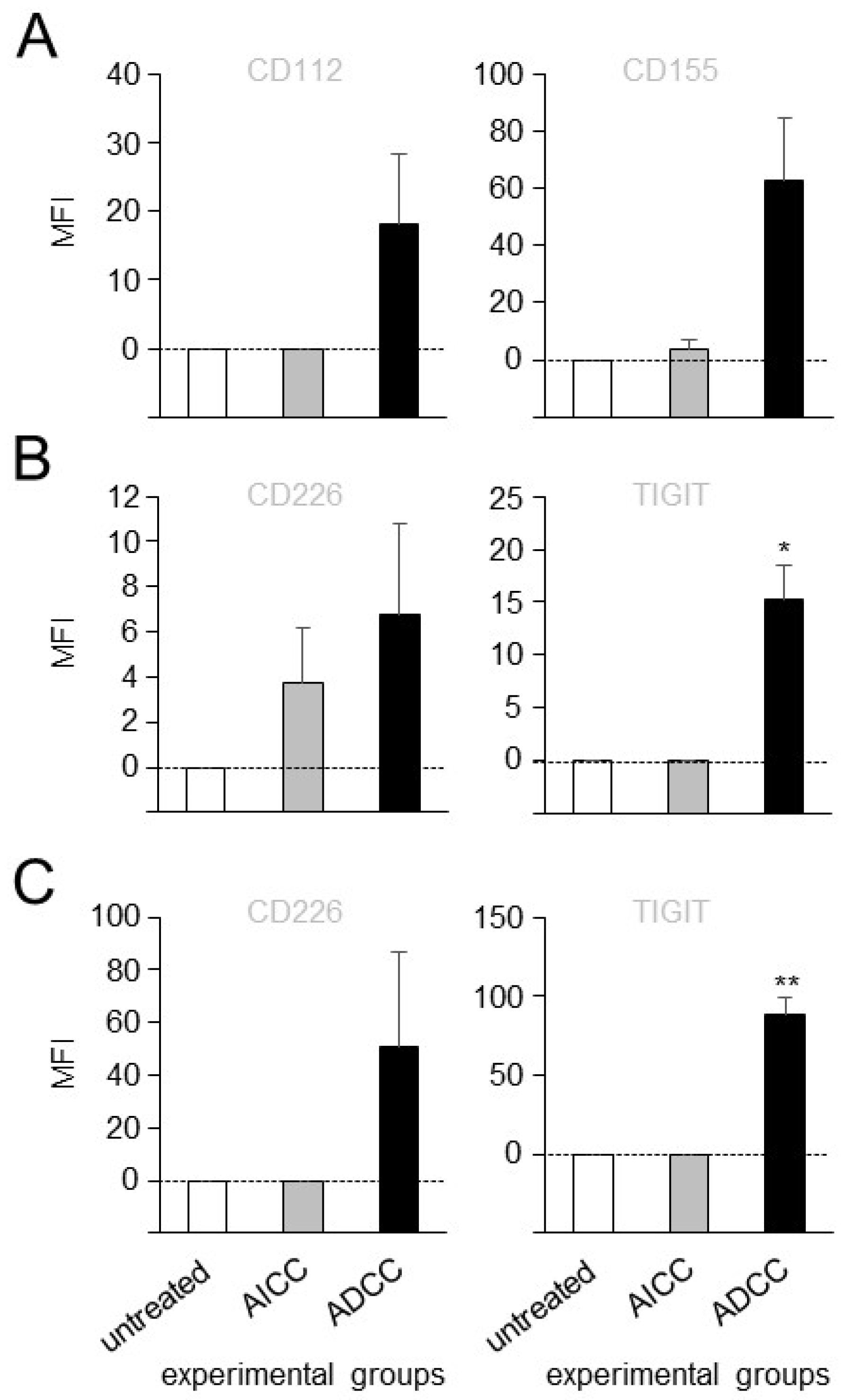
3.2. Effects of ADCC Mediated by DB on Expression of TIGIT and CD226 on Effector Cells as Well as CD112 and CD155 on Tumor Cells
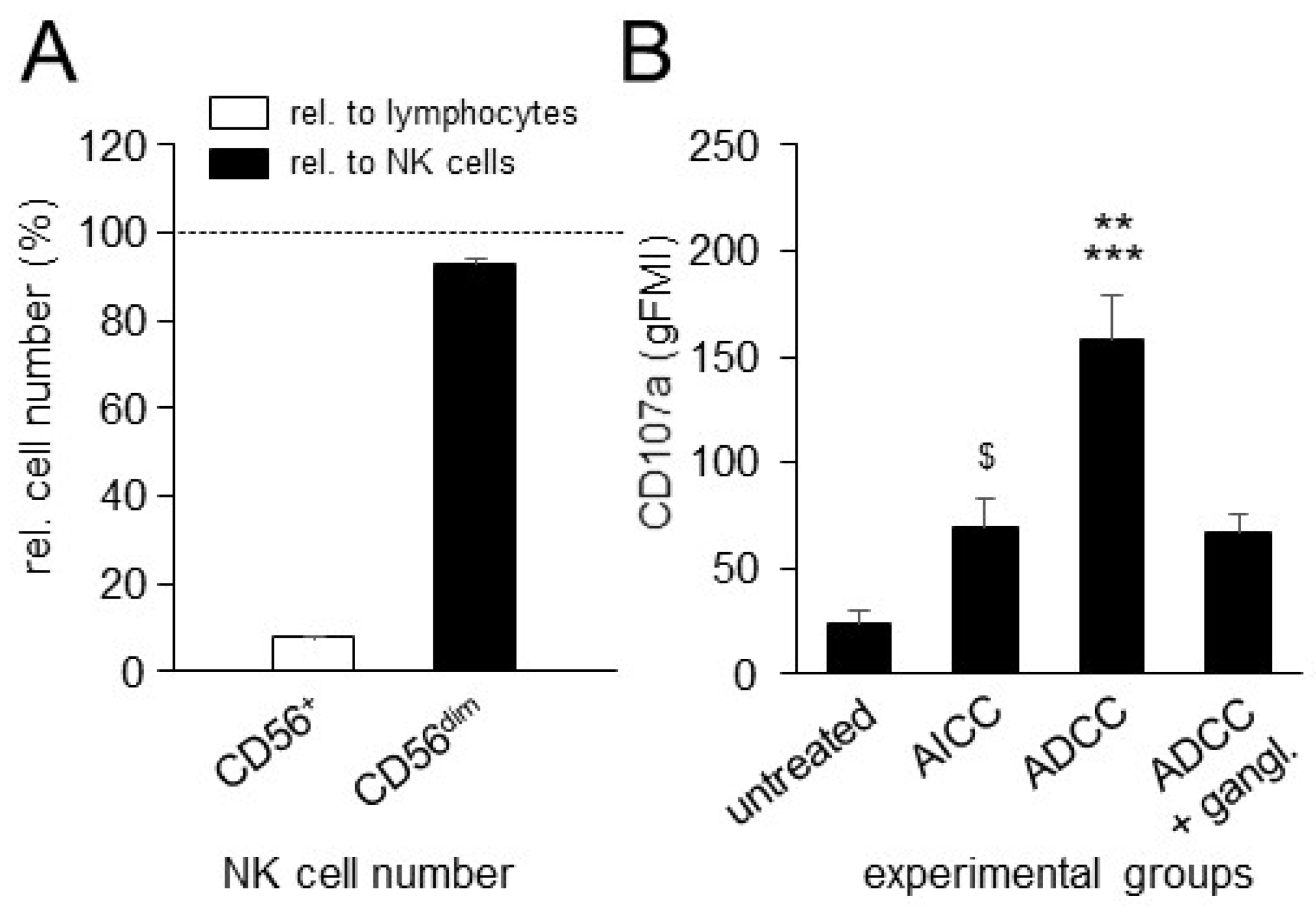
3.3. Effects of ADCC Mediated by DB on NK-Cell Activity
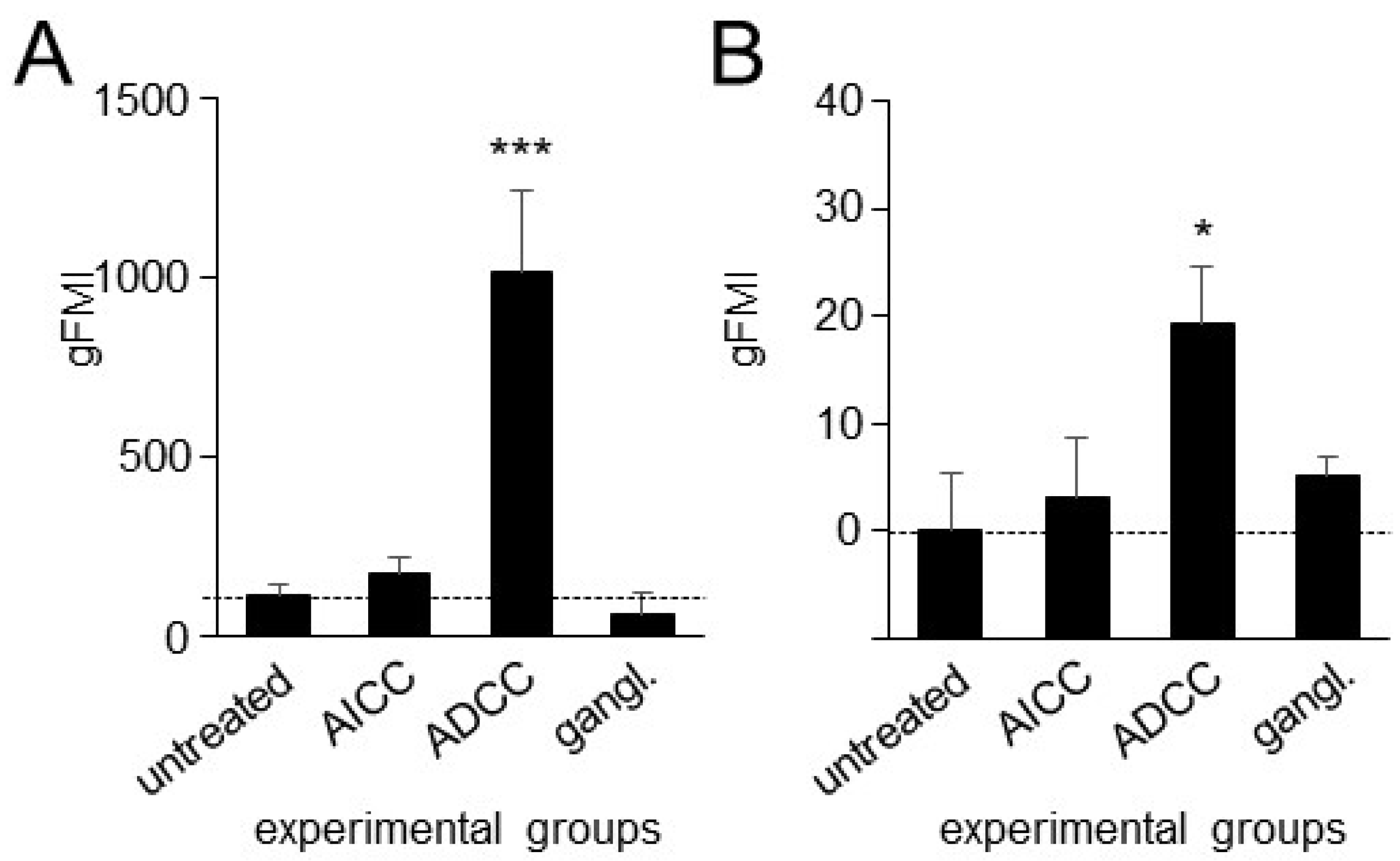
3.4. Effects of ADCC Mediated by DB on Expression of PD-L1 on Tumor Cells and PD-1 on NK Cells
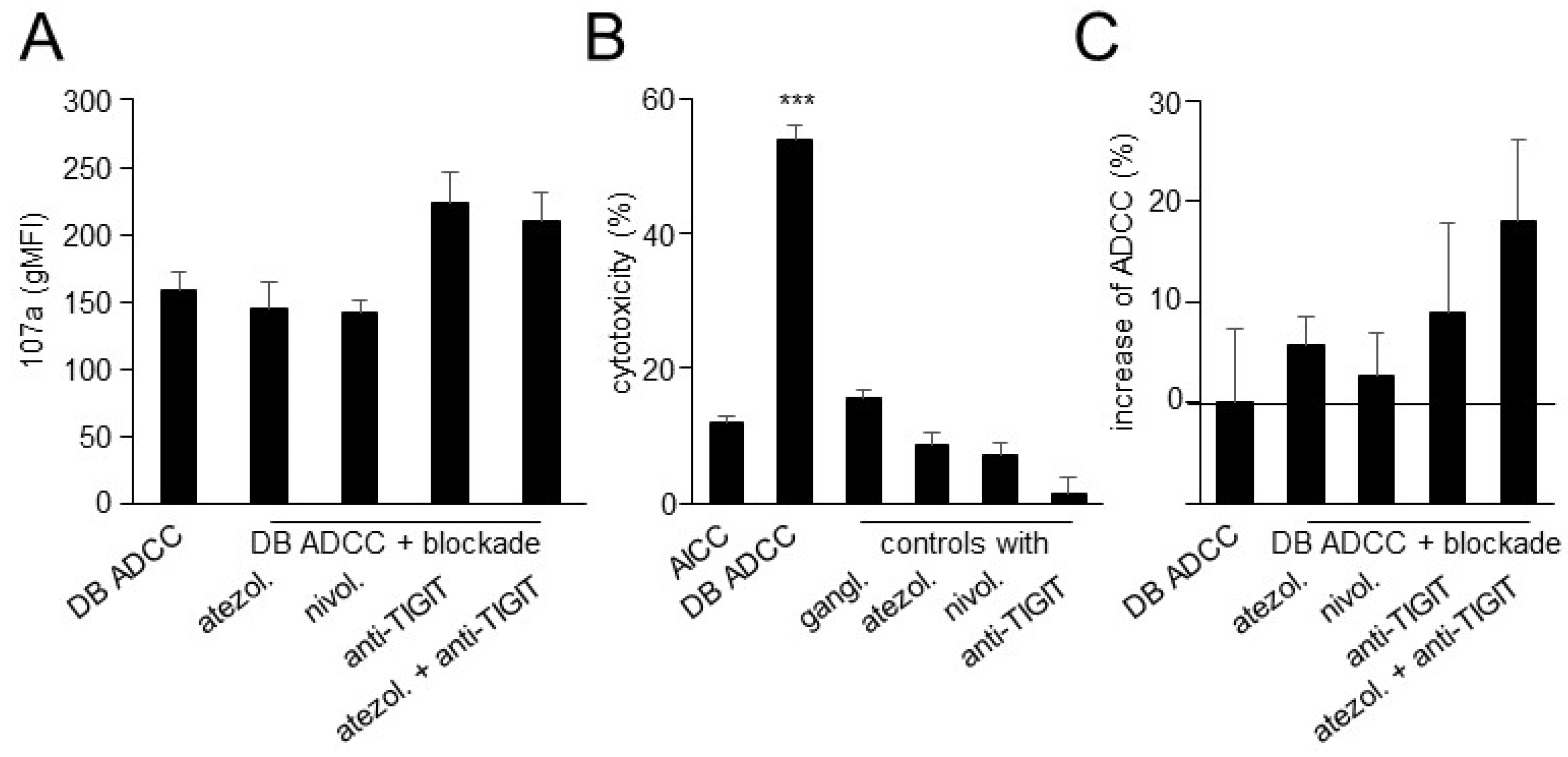
3.5. Effects of TIGIT Blockade, in Combination with Blockade of the PD-1/PD-L1 Pathway, on ADCC Mediated by DB
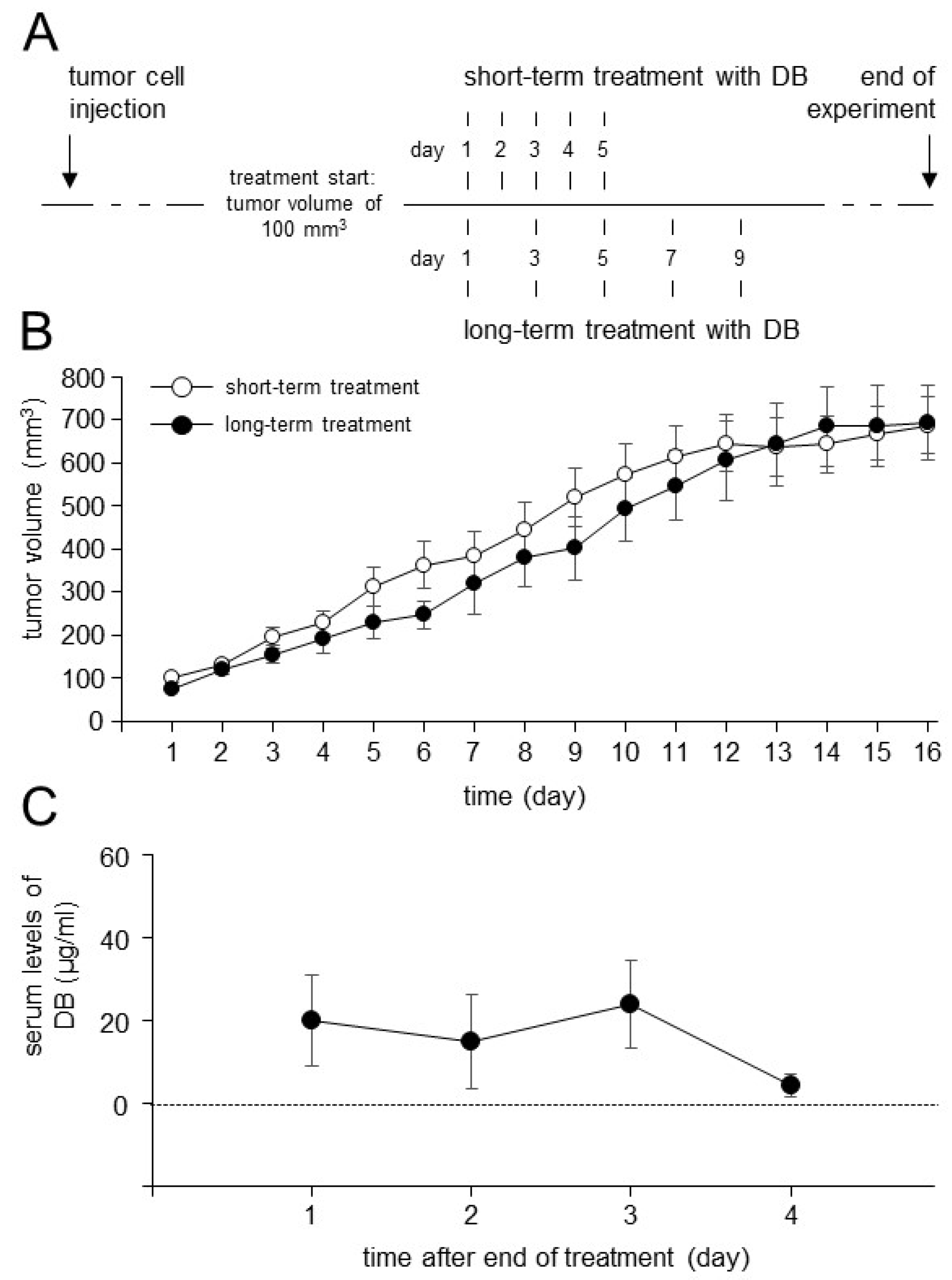
3.6. Establishment of the Dinutuximab Beta Long-Term Treatment Model
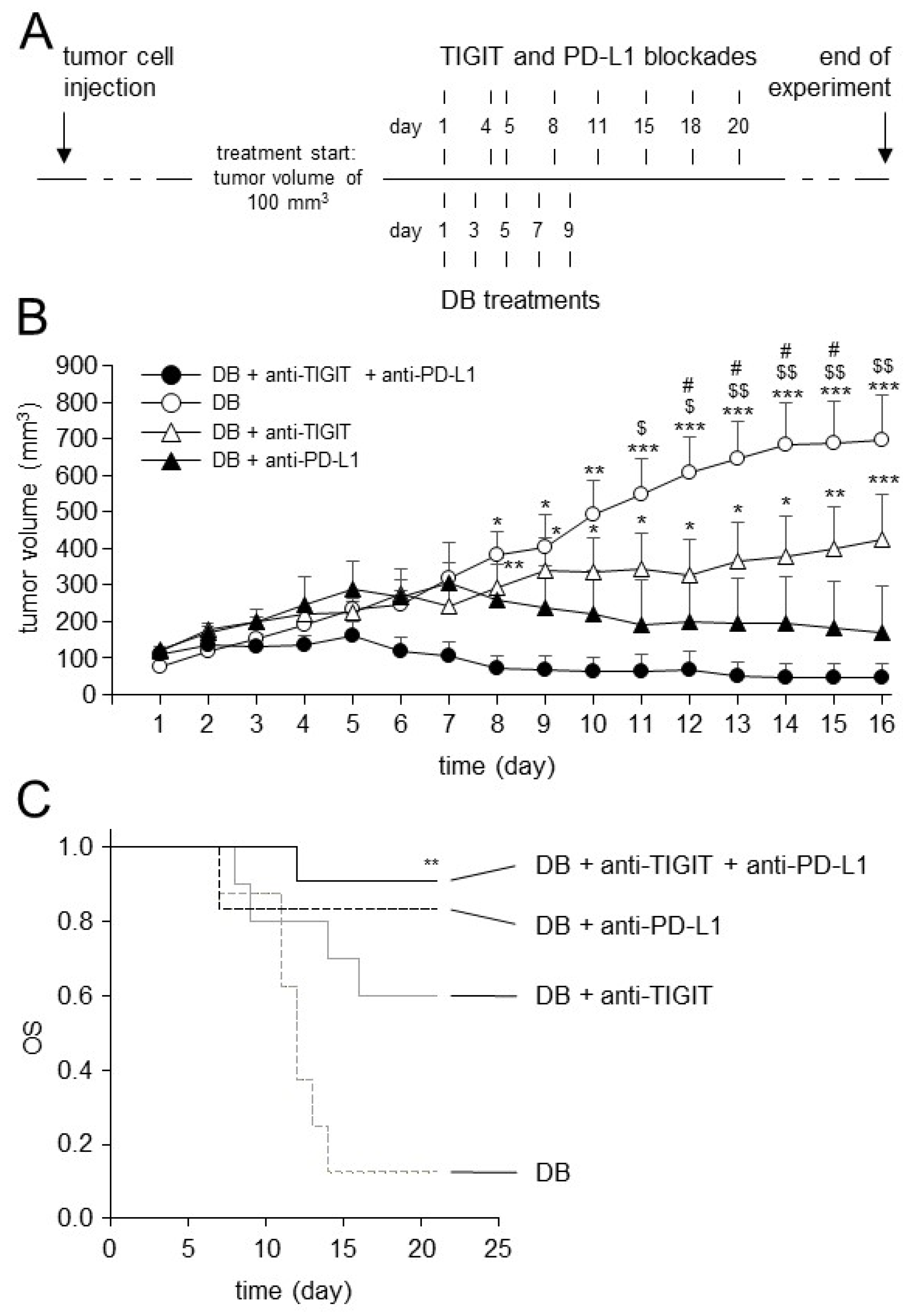
3.7. Effects of TIGIT Blockade in Combination with Blockade of PD-L1 on Antitumor Efficacy of Immunotherapy with DB In Vivo
3.8. Analysis of TIGIT and CD155 Expression in Tumor Tissue
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Paraboschi, I.; Privitera, L.; Kramer-Marek, G.; Anderson, J.; Giuliani, S. Novel Treatments and Technologies Applied to the Cure of Neuroblastoma. Children 2021, 8, 482. [Google Scholar] [CrossRef]
- Ladenstein, R.; Pötschger, U.; Valteau-Couanet, D.; Luksch, R.; Castel, V.; Ash, S.; Laureys, G.; Brock, P.; Michon, J.M.; Owens, C.; et al. Investigation of the Role of Dinutuximab Beta-Based Immunotherapy in the SIOPEN High-Risk Neuroblastoma 1 Trial (HR-NBL1). Cancers 2020, 12, 309. [Google Scholar] [CrossRef] [PubMed]
- Siebert, N.; Leopold, J.; Zumpe, M.; Troschke-Meurer, S.; Biskupski, S.; Zikoridse, A.; Lode, H.N. The Immunocytokine FAP-IL-2v Enhances Anti-Neuroblastoma Efficacy of the Anti-GD(2) Antibody Dinutuximab Beta. Cancers 2022, 14, 4842. [Google Scholar] [CrossRef] [PubMed]
- Ladenstein, R.; Pötschger, U.; Valteau-Couanet, D.; Luksch, R.; Castel, V.; Yaniv, I.; Laureys, G.; Brock, P.; Michon, J.M.; Owens, C.; et al. Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): A multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1617–1629. [Google Scholar] [CrossRef] [PubMed]
- Troschke-Meurer, S.; Siebert, N.; Marx, M.; Zumpe, M.; Ehlert, K.; Mutschlechner, O.; Loibner, H.; Ladenstein, R.; Lode, H.N. Low CD4+/CD25+/CD127− regulatory T cell- and high INF-γ levels are associated with improved survival of neuroblastoma patients treated with long-term infusion of ch14.18/CHO combined with interleukin-2. Oncoimmunology 2019, 8, 1661194. [Google Scholar] [CrossRef] [PubMed]
- Siebert, N.; Zumpe, M.; Juttner, M.; Troschke-Meurer, S.; Lode, H.N. PD-1 blockade augments anti-neuroblastoma immune response induced by anti-GD2 antibody ch14.18/CHO. Oncoimmunology 2017, 6, e1343775. [Google Scholar] [CrossRef] [PubMed]
- Ehlert, K.; Hansjuergens, I.; Zinke, A.; Otto, S.; Siebert, N.; Henze, G.; Lode, H. Nivolumab and dinutuximab beta in two patients with refractory neuroblastoma. J. Immunother. Cancer 2020, 8, e000540. [Google Scholar] [CrossRef] [PubMed]
- Siebert, N.; Zumpe, M.; von Lojewski, L.; Troschke-Meurer, S.; Marx, M.; Lode, H.N. Reduction of CD11b(+) myeloid suppressive cells augments anti-neuroblastoma immune response induced by the anti-GD(2) antibody ch14.18/CHO. Oncoimmunology 2020, 9, 1836768. [Google Scholar] [CrossRef]
- Harjunpää, H.; Guillerey, C. TIGIT as an emerging immune checkpoint. Clin. Exp. Immunol. 2020, 200, 108–119. [Google Scholar] [CrossRef]
- Chauvin, J.M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef]
- Castriconi, R.; Dondero, A.; Corrias, M.V.; Lanino, E.; Pende, D.; Moretta, L.; Bottino, C.; Moretta, A. Natural killer cell-mediated killing of freshly isolated neuroblastoma cells: Critical role of DNAX accessory molecule-1-poliovirus receptor interaction. Cancer Res. 2004, 64, 9180–9184. [Google Scholar] [CrossRef]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Annese, T.; Tamma, R.; Ribatti, D. Update in TIGIT Immune-Checkpoint Role in Cancer. Front. Oncol. 2022, 12, 871085. [Google Scholar] [CrossRef] [PubMed]
- Siebert, N.; Seidel, D.; Eger, C.; Juttner, M.; Lode, H.N. Functional bioassays for immune monitoring of high-risk neuroblastoma patients treated with ch14.18/CHO anti-GD2 antibody. PLoS ONE 2014, 9, e107692. [Google Scholar] [CrossRef]
- Lode, H.N.; Xiang, R.; Varki, N.M.; Dolman, C.S.; Gillies, S.D.; Reisfeld, R.A. Targeted interleukin-2 therapy for spontaneous neuroblastoma metastases to bone marrow. J. Natl. Cancer Inst. 1997, 89, 1586–1594. [Google Scholar] [CrossRef]
- Lode, H.N.; Schmidt, M.; Seidel, D.; Huebener, N.; Brackrock, D.; Bleeke, M.; Reker, D.; Brandt, S.; Mueller, H.P.; Helm, C.; et al. Vaccination with anti-idiotype antibody ganglidiomab mediates a GD(2)-specific anti-neuroblastoma immune response. Cancer Immunol. Immunother. 2013, 62, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Mueller, I.; Ehlert, K.; Endres, S.; Pill, L.; Siebert, N.; Kietz, S.; Brock, P.; Garaventa, A.; Valteau-Couanet, D.; Janzek, E.; et al. Tolerability, response and outcome of high-risk neuroblastoma patients treated with long-term infusion of anti-GD(2) antibody ch14.18/CHO. MAbs 2018, 10, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Siebert, N.; Eger, C.; Seidel, D.; Juttner, M.; Zumpe, M.; Wegner, D.; Kietz, S.; Ehlert, K.; Veal, G.J.; Siegmund, W.; et al. Pharmacokinetics and pharmacodynamics of ch14.18/CHO in relapsed/refractory high-risk neuroblastoma patients treated by long-term infusion in combination with IL-2. MAbs 2016, 8, 604–616. [Google Scholar] [CrossRef]
- Siebert, N.; Seidel, D.; Eger, C.; Brackrock, D.; Reker, D.; Schmidt, M.; Lode, H.N. Validated detection of anti-GD2 antibody ch14.18/CHO in serum of neuroblastoma patients using anti-idiotype antibody ganglidiomab. J. Immunol. Methods 2013, 398–399, 51–59. [Google Scholar] [CrossRef]
- Chiang, E.Y.; Mellman, I. TIGIT-CD226-PVR axis: Advancing immune checkpoint blockade for cancer immunotherapy. J. Immunother. Cancer 2022, 10, e004711. [Google Scholar] [CrossRef]
- Ceylan, K.; Jahns, L.J.; Lode, B.N.; Ehlert, K.; Kietz, S.; Troschke-Meurer, S.; Siebert, N.; Lode, H.N. Inflammatory response and treatment tolerance of long-term infusion of the anti-GD2 antibody ch14.18/CHO in combination with interleukin-2 in patients with high-risk neuroblastoma. Pediatr. Blood Cancer 2018, 65, e26967. [Google Scholar] [CrossRef] [PubMed]
- Keyel, M.E.; Reynolds, C.P. Spotlight on dinutuximab in the treatment of high-risk neuroblastoma: Development and place in therapy. Biologics 2019, 13, 1–12. [Google Scholar] [CrossRef]
- Dutta, S.; Ganguly, A.; Chatterjee, K.; Spada, S.; Mukherjee, S. Targets of Immune Escape Mechanisms in Cancer: Basis for Development and Evolution of Cancer Immune Checkpoint Inhibitors. Biology 2023, 12, 218. [Google Scholar] [CrossRef]
- Vetsika, E.K.; Koukos, A.; Kotsakis, A. Myeloid-Derived Suppressor Cells: Major Figures that Shape the Immunosuppressive and Angiogenic Network in Cancer. Cells 2019, 8, 1647. [Google Scholar] [CrossRef]
- Mao, L.; Xiao, Y.; Yang, Q.C.; Yang, S.C.; Yang, L.L.; Sun, Z.J. TIGIT/CD155 blockade enhances anti-PD-L1 therapy in head and neck squamous cell carcinoma by targeting myeloid-derived suppressor cells. Oral Oncol. 2021, 121, 105472. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Lu, P.H.; Liu, L.; Fang, Z.M.; Duan, W.; Liu, Z.L.; Wang, C.Y.; Zhou, P.; Yu, X.F.; He, W.T. TIGIT negatively regulates inflammation by altering macrophage phenotype. Immunobiology 2016, 221, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Brauneck, F.; Fischer, B.; Witt, M.; Muschhammer, J.; Oelrich, J.; da Costa Avelar, P.H.; Tsoka, S.; Bullinger, L.; Seubert, E.; Smit, D.J.; et al. TIGIT blockade repolarizes AML-associated TIGIT(+) M2 macrophages to an M1 phenotype and increases CD47-mediated phagocytosis. J. Immunother. Cancer 2022, 10, e004794. [Google Scholar] [CrossRef] [PubMed]
- Tseng, D.; Volkmer, J.P.; Willingham, S.B.; Contreras-Trujillo, H.; Fathman, J.W.; Fernhoff, N.B.; Seita, J.; Inlay, M.A.; Weiskopf, K.; Miyanishi, M.; et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc. Natl. Acad. Sci. USA 2013, 110, 11103–11108. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siebert, N.; Zumpe, M.; Schwencke, C.H.; Biskupski, S.; Troschke-Meurer, S.; Leopold, J.; Zikoridse, A.; Lode, H.N. Combined Blockade of TIGIT and PD-L1 Enhances Anti-Neuroblastoma Efficacy of GD2-Directed Immunotherapy with Dinutuximab Beta. Cancers 2023, 15, 3317. https://doi.org/10.3390/cancers15133317
Siebert N, Zumpe M, Schwencke CH, Biskupski S, Troschke-Meurer S, Leopold J, Zikoridse A, Lode HN. Combined Blockade of TIGIT and PD-L1 Enhances Anti-Neuroblastoma Efficacy of GD2-Directed Immunotherapy with Dinutuximab Beta. Cancers. 2023; 15(13):3317. https://doi.org/10.3390/cancers15133317
Chicago/Turabian StyleSiebert, Nikolai, Maxi Zumpe, Christian Heinrich Schwencke, Simon Biskupski, Sascha Troschke-Meurer, Justus Leopold, Alexander Zikoridse, and Holger N. Lode. 2023. "Combined Blockade of TIGIT and PD-L1 Enhances Anti-Neuroblastoma Efficacy of GD2-Directed Immunotherapy with Dinutuximab Beta" Cancers 15, no. 13: 3317. https://doi.org/10.3390/cancers15133317
APA StyleSiebert, N., Zumpe, M., Schwencke, C. H., Biskupski, S., Troschke-Meurer, S., Leopold, J., Zikoridse, A., & Lode, H. N. (2023). Combined Blockade of TIGIT and PD-L1 Enhances Anti-Neuroblastoma Efficacy of GD2-Directed Immunotherapy with Dinutuximab Beta. Cancers, 15(13), 3317. https://doi.org/10.3390/cancers15133317