
Withaferin A Increases the Effectiveness of Immune Checkpoint Blocker for the Treatment of Non-Small Cell Lung Cancer



 and
and 
Abstract
Simple Summary
Abstract
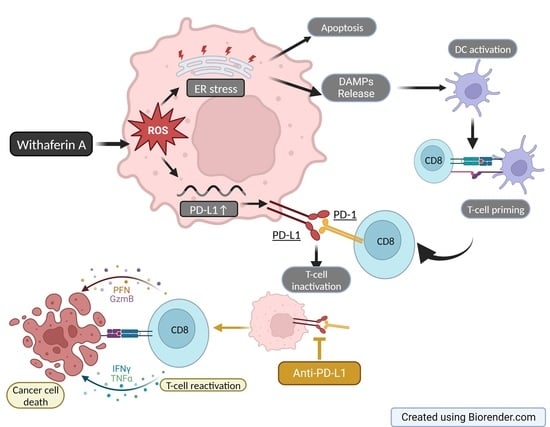
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemotherapeutic Drugs and Antibodies
2.2. Cell Lines
2.3. Cell Titer-Glo® Assay
2.4. Annexin V Assay
2.5. Flow Cytometry
2.6. HMGB-1 Assay
2.7. Ex Vivo Dendritic Cell Activation Assay
2.8. Reverse Transcription and Quantitative PCR
2.9. Protein Extraction and Western Blot
2.10. Ingenuity Pathway Analysis (IPA)
2.11. SiRNA Transfection
2.12. Animal Studies
2.13. Statistical Analysis
3. Results
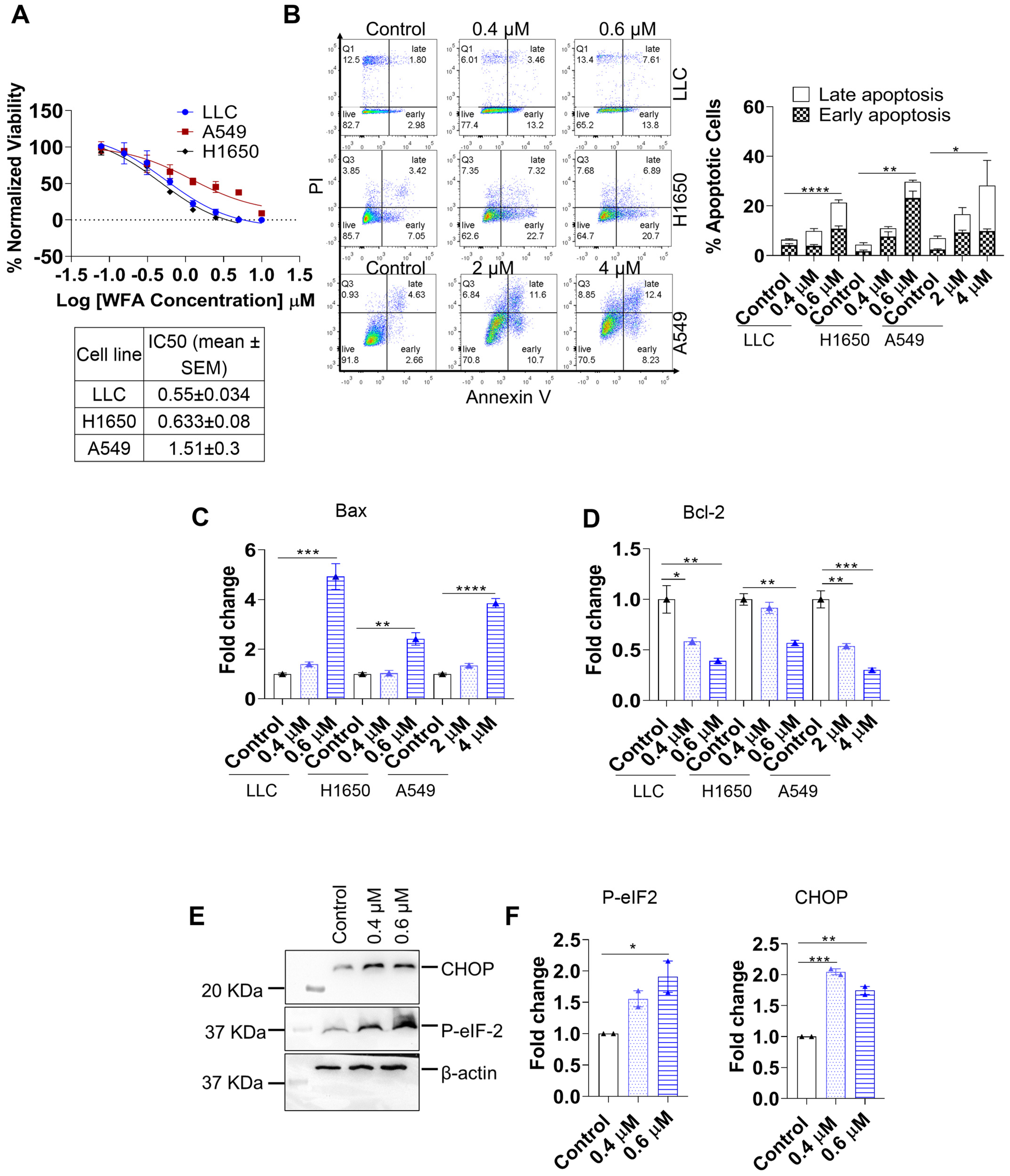
3.1. WFA Induces ER-Stress Mediated Apoptosis in NSCLC
3.2. WFA Induces ICD in NSCLC Cell Lines
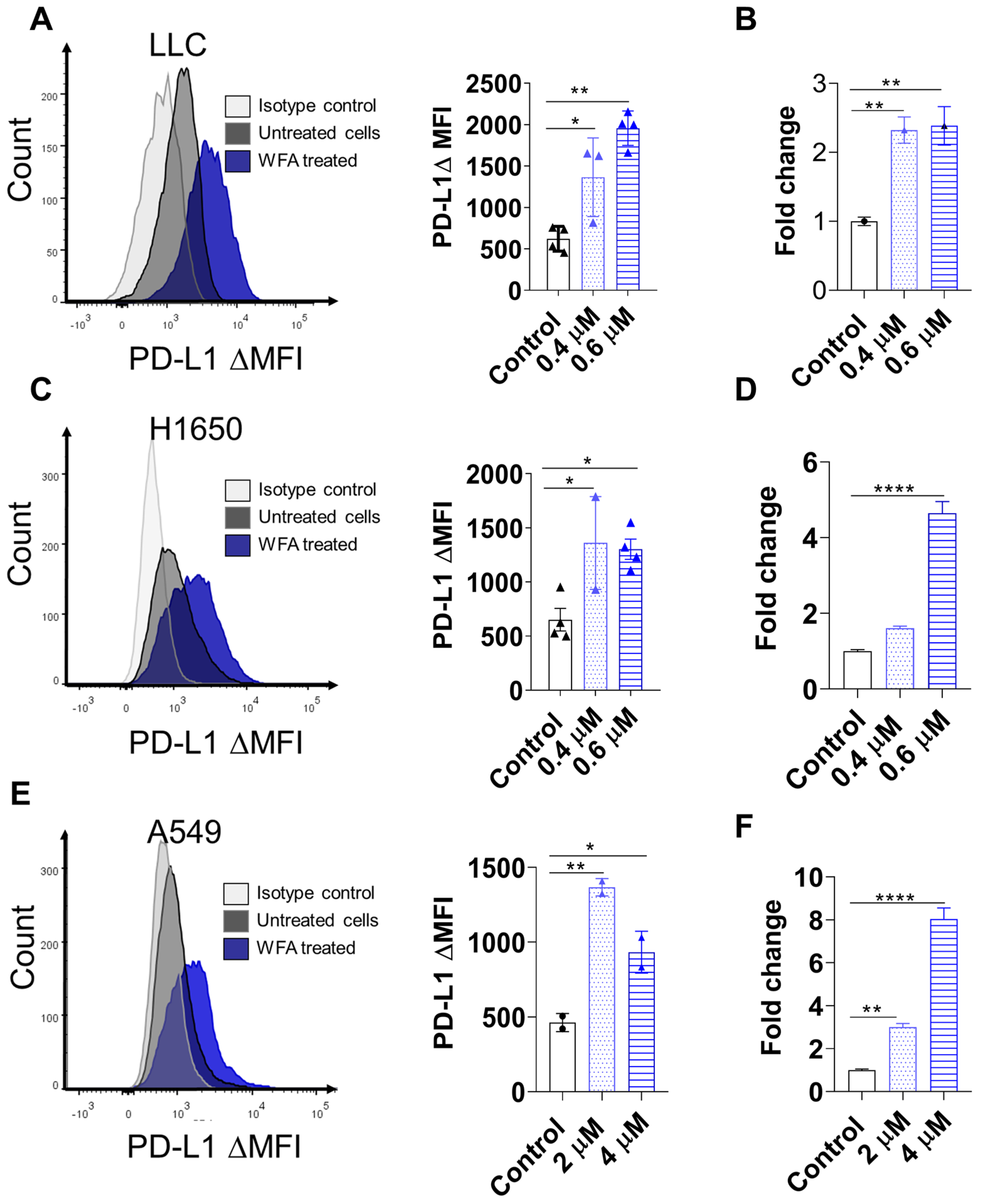
3.3. WFA Increases PD-L1 Expression in NSCLC Cell Lines
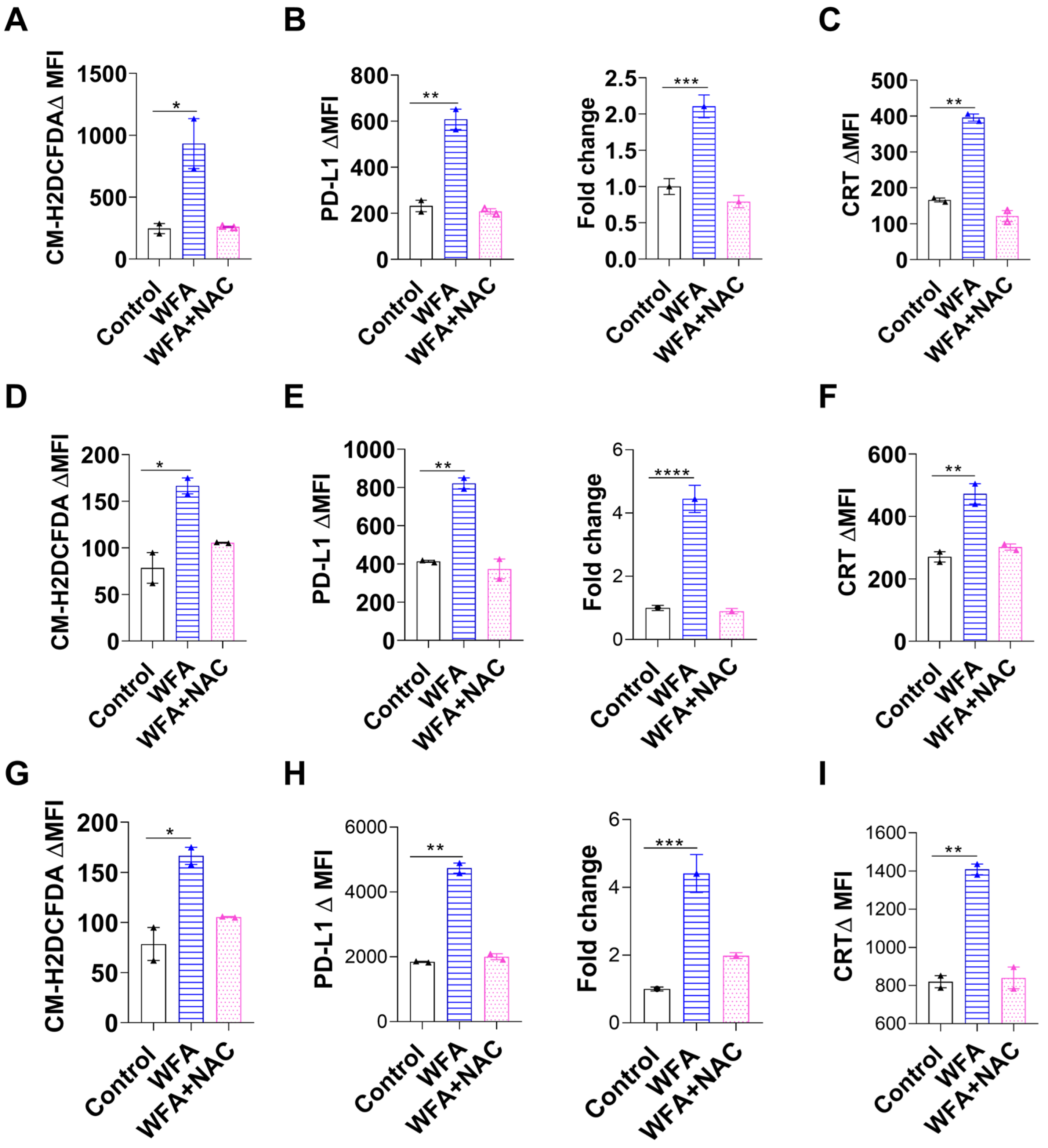
3.4. WFA Mediates PD-L1 Upregulation and ICD by Increasing ROS Production
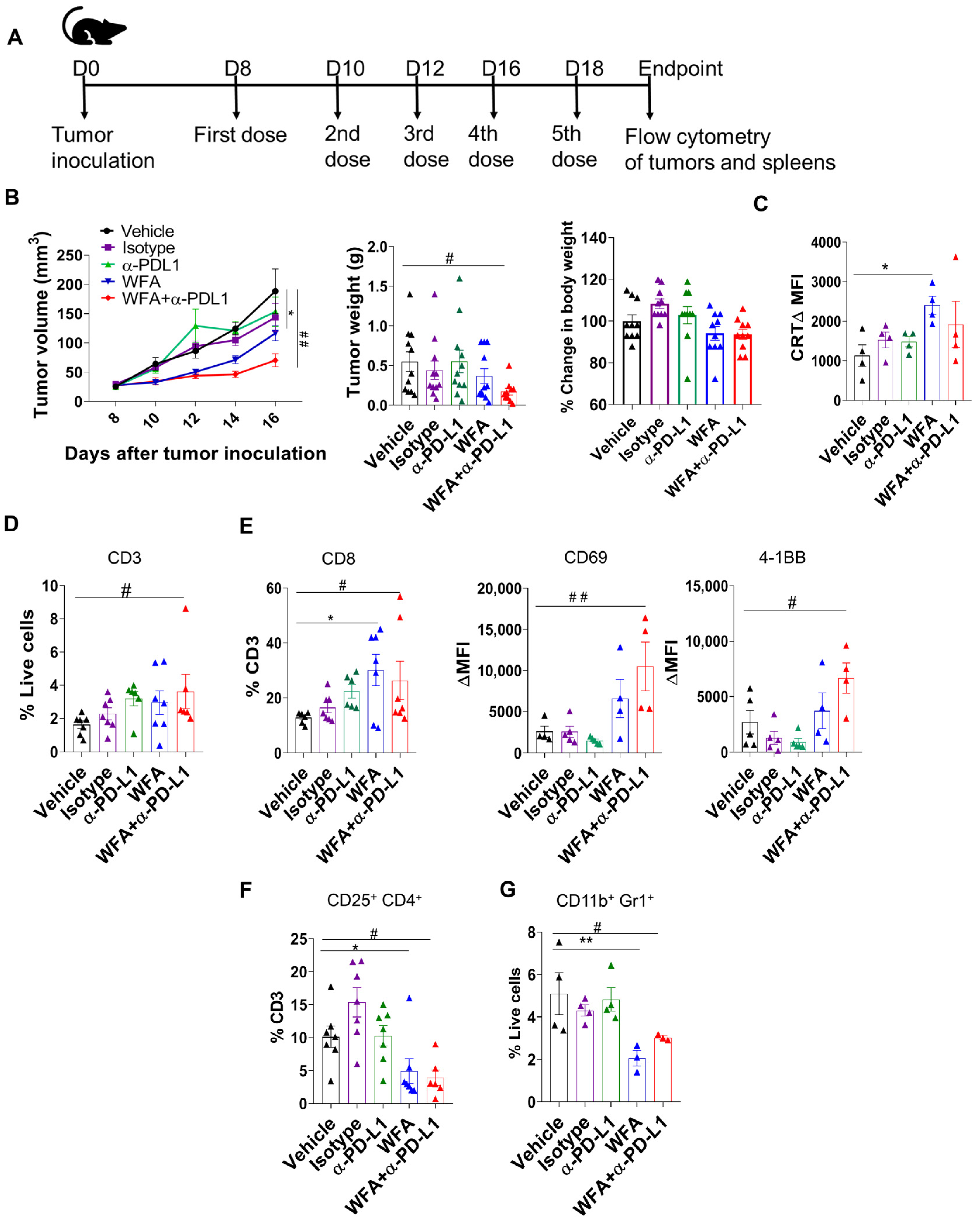
3.5. WFA Sensitizes LLC Syngeneic Mouse Tumors to α-PD-L1 In Vivo
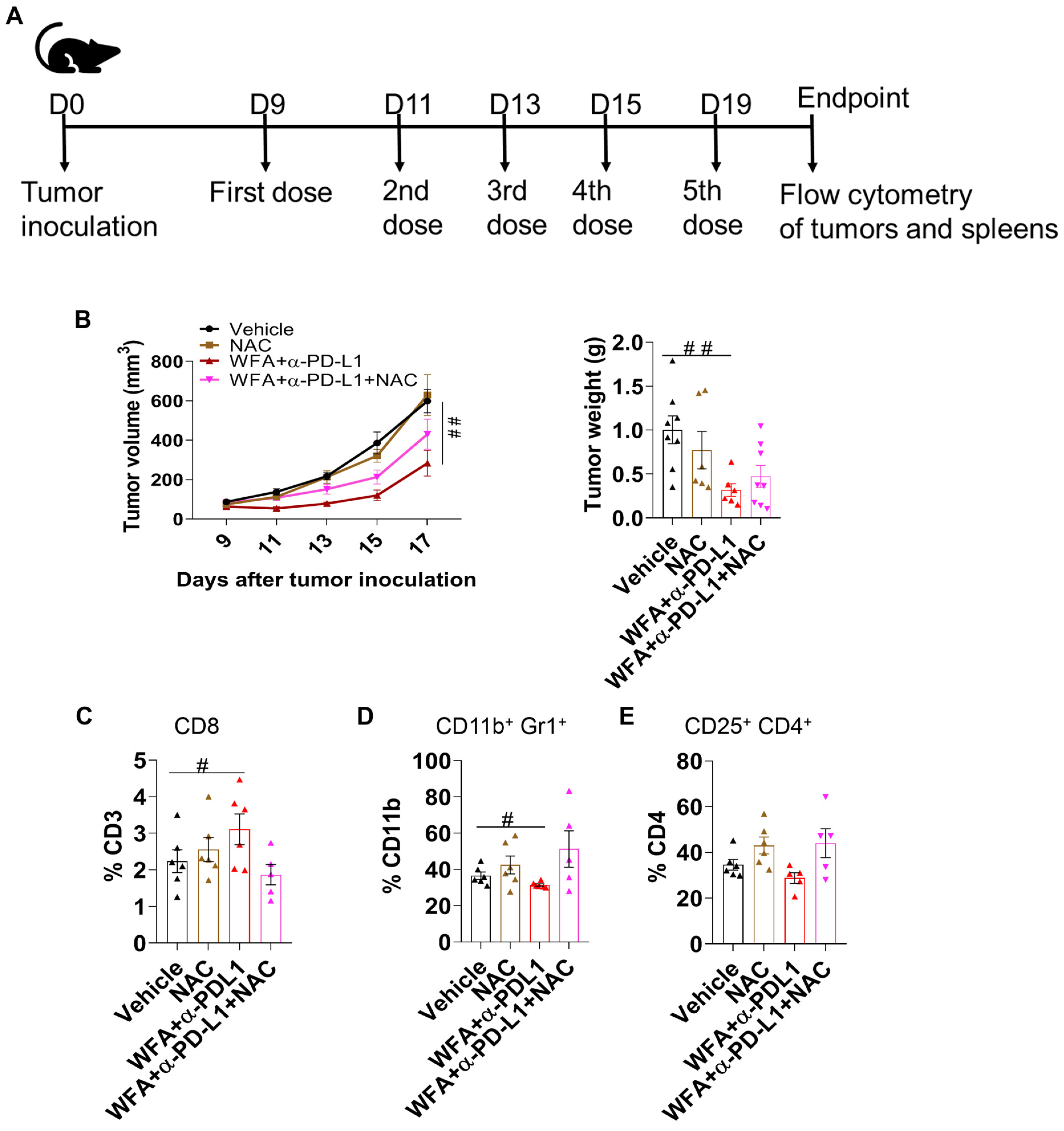
3.6. ROS Plays a Role in Inducing the Effectiveness of WFA+α-PD-L1 Combination
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. U.S. Cancer Statistics Lung Cancer Stat Bite; US Department of Health and Human Services: Washington, DC, USA, 2022.
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef]
- Sánchez-Paulete, A.R.; Teijeira, A.; Cueto, F.J.; Garasa, S.; Pérez-Gracia, J.L.; Sánchez-Arráez, A.; Sancho, D.; Melero, I. Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann. Oncol. 2017, 28, xii44–xii55. [Google Scholar] [CrossRef]
- Chambers, C.A.; Allison, J.P. Costimulatory regulation of T cell function. Curr. Opin. Cell Biol. 1999, 11, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Principe, N.; Kidman, J.; Goh, S.; Tilsed, C.M.; Fisher, S.A.; Fear, V.S.; Forbes, C.A.; Zemek, R.M.; Chopra, A.; Watson, M.; et al. Tumor Infiltrating Effector Memory Antigen-Specific CD8(+) T Cells Predict Response to Immune Checkpoint Therapy. Front. Immunol. 2020, 11, 584423. [Google Scholar] [CrossRef]
- Tang, S.; Qin, C.; Hu, H.; Liu, T.; He, Y.; Guo, H.; Yan, H.; Zhang, J.; Tang, S.; Zhou, H. Immune Checkpoint Inhibitors in Non-Small Cell Lung Cancer: Progress, Challenges, and Prospects. Cells 2022, 11, 320. [Google Scholar] [CrossRef] [PubMed]
- Wellenstein, M.D.; de Visser, K.E. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Chen, N.; Li, L.; Du, N.; Bai, L.; Lv, Z.; Tian, H.; Cui, J. Mechanisms of Cancer Resistance to Immunotherapy. Front. Oncol. 2020, 10, 1290. [Google Scholar] [CrossRef]
- Bai, R.; Lv, Z.; Xu, D.; Cui, J. Predictive biomarkers for cancer immunotherapy with immune checkpoint inhibitors. Biomark. Res. 2020, 8, 34. [Google Scholar] [CrossRef]
- Kowanetz, M.; Zou, W.; Gettinger Scott, N.; Koeppen, H.; Kockx, M.; Schmid, P.; Kadel Edward, E.; Wistuba, I.; Chaft, J.; Rizvi Naiyer, A.; et al. Differential regulation of PD-L1 expression by immune and tumor cells in NSCLC and the response to treatment with atezolizumab (anti–PD-L1). Proc. Natl. Acad. Sci. USA 2018, 115, E10119–E10126. [Google Scholar] [CrossRef]
- Wang, L.; Hu, Y.; Wang, S.; Shen, J.; Wang, X. Biomarkers of immunotherapy in non-small cell lung cancer (Review). Oncol. Lett. 2020, 20, 139. [Google Scholar] [CrossRef]
- Sahin, I.H.; Akce, M.; Alese, O.; Shaib, W.; Lesinski, G.B.; El-Rayes, B.; Wu, C. Immune checkpoint inhibitors for the treatment of MSI-H/MMR-D colorectal cancer and a perspective on resistance mechanisms. Br. J. Cancer 2019, 121, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Spaas, M.; Lievens, Y. Is the Combination of Immunotherapy and Radiotherapy in Non-small Cell Lung Cancer a Feasible and Effective Approach? Front. Med. 2019, 6, 244. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Wang, X.; Guo, X.; Jiang, K.; Ye, T.; Chen, J.; Fang, J.; Gu, L.; Wang, S.; Zhang, G.; et al. STAT3 Contributes To Oncolytic Newcastle Disease Virus-Induced Immunogenic Cell Death in Melanoma Cells. Front. Oncol. 2019, 9, 436. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.T.; Sun, Z.J. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics 2021, 11, 5365–5386. [Google Scholar] [CrossRef]
- Luo, M.; Wang, F.; Zhang, H.; To, K.K.W.; Wu, S.; Chen, Z.; Liang, S.; Fu, L. Mitomycin C enhanced the efficacy of PD-L1 blockade in non-small cell lung cancer. Signal Transduct. Target. Ther. 2020, 5, 141. [Google Scholar] [CrossRef]
- Fournel, L.; Wu, Z.; Stadler, N.; Damotte, D.; Lococo, F.; Boulle, G.; Ségal-Bendirdjian, E.; Bobbio, A.; Icard, P.; Trédaniel, J.; et al. Cisplatin increases PD-L1 expression and optimizes immune check-point blockade in non-small cell lung cancer. Cancer Lett. 2019, 464, 5–14. [Google Scholar] [CrossRef]
- Florescu, M.; Cinteza, M.; Vinereanu, D. Chemotherapy-induced Cardiotoxicity. Maedica 2013, 8, 59–67. [Google Scholar] [PubMed]
- Li, C.; Bhatti, S.A.; Ying, J. Immune Checkpoint Inhibitors-Associated Cardiotoxicity. Cancers 2022, 14, 1145. [Google Scholar] [CrossRef]
- Dutta, R.; Khalil, R.; Green, R.; Mohapatra, S.S.; Mohapatra, S. Withania Somnifera (Ashwagandha) and Withaferin A: Potential in Integrative Oncology. Int. J. Mol. Sci. 2019, 20, 5310. [Google Scholar] [CrossRef]
- Kakar, S.S.; Parte, S.; Carter, K.; Joshua, I.G.; Worth, C.; Rameshwar, P.; Ratajczak, M.Z. Withaferin A (WFA) inhibits tumor growth and metastasis by targeting ovarian cancer stem cells. Oncotarget 2017, 8, 74494–74505. [Google Scholar] [CrossRef]
- Kyakulaga, A.H.; Aqil, F.; Munagala, R.; Gupta, R.C. Withaferin A inhibits Epithelial to Mesenchymal Transition in Non-Small Cell Lung Cancer Cells. Sci. Rep. 2018, 8, 15737. [Google Scholar] [CrossRef] [PubMed]
- Ting, L.-L.; Chou, A.S.-B.; Hsieh, C.-H.; Hsiung, S.-C.; Pang, S.-T.; Liao, S.-K. Withaferin A targeting both cancer stem cells and metastatic cancer stem cells in the UP-LN1 carcinoma cell model. J. Cancer Metastasis Treat. 2016, 2, 29–40. [Google Scholar] [CrossRef]
- Shiragannavar, V.D.; Gowda, N.G.S.; Kumar, D.P.; Mirshahi, F.; Santhekadur, P.K. Withaferin A Acts as a Novel Regulator of Liver X Receptor-α in HCC. Front. Oncol. 2020, 10, 628506. [Google Scholar] [CrossRef] [PubMed]
- Heyninck, K.; Lahtela-Kakkonen, M.; Van der Veken, P.; Haegeman, G.; Vanden Berghe, W. Withaferin A inhibits NF-kappaB activation by targeting cysteine 179 in IKKβ. Biochem. Pharm. 2014, 91, 501–509. [Google Scholar] [CrossRef]
- Lee, I.C.; Choi, B.Y. Withaferin-A—A Natural Anticancer Agent with Pleitropic Mechanisms of Action. Int. J. Mol. Sci. 2016, 17, 290. [Google Scholar] [CrossRef]
- Kunimasa, K.; Nagano, T.; Shimono, Y.; Dokuni, R.; Kiriu, T.; Tokunaga, S.; Tamura, D.; Yamamoto, M.; Tachihara, M.; Kobayashi, K.; et al. Glucose metabolism-targeted therapy and withaferin A are effective for epidermal growth factor receptor tyrosine kinase inhibitor-induced drug-tolerant persisters. Cancer Sci. 2017, 108, 1368–1377. [Google Scholar] [CrossRef]
- Ghosh, K.; De, S.; Das, S.; Mukherjee, S.; Sengupta Bandyopadhyay, S. Withaferin A Induces ROS-Mediated Paraptosis in Human Breast Cancer Cell-Lines MCF-7 and MDA-MB-231. PLoS ONE 2016, 11, e0168488. [Google Scholar] [CrossRef]
- Sehrawat, A.; Samanta, S.K.; Hahm, E.R.; St Croix, C.; Watkins, S.; Singh, S.V. Withaferin A-mediated apoptosis in breast cancer cells is associated with alterations in mitochondrial dynamics. Mitochondrion 2019, 47, 282–293. [Google Scholar] [CrossRef]
- Guo, R.; Gan, L.; Lau, W.B.; Yan, Z.; Xie, D.; Gao, E.; Christopher, T.A.; Lopez, B.L.; Ma, X.; Wang, Y. Withaferin A Prevents Myocardial Ischemia/Reperfusion Injury by Upregulating AMP-Activated Protein Kinase-Dependent B-Cell Lymphoma2 Signaling. Circ. J. 2019, 83, 1726–1736. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, L.; Liang, T.; Tian, X.D.; Liu, Y.; Zhang, T. Withaferin A induces mitochondrial-dependent apoptosis in non-small cell lung cancer cells via generation of reactive oxygen species. J. BUON 2017, 22, 244–250. [Google Scholar]
- Malik, F.; Singh, J.; Khajuria, A.; Suri, K.A.; Satti, N.K.; Singh, S.; Kaul, M.K.; Kumar, A.; Bhatia, A.; Qazi, G.N. A standardized root extract of Withania somnifera and its major constituent withanolide-A elicit humoral and cell-mediated immune responses by up regulation of Th1-dominant polarization in BALB/c mice. Life Sci. 2007, 80, 1525–1538. [Google Scholar] [CrossRef] [PubMed]
- Tharakan, A.; Shukla, H.; Benny, I.R.; Tharakan, M.; George, L.; Koshy, S. Immunomodulatory Effect of Withania somnifera (Ashwagandha) Extract-A Randomized, Double-Blind, Placebo Controlled Trial with an Open Label Extension on Healthy Participants. J. Clin. Med. 2021, 10, 3644. [Google Scholar] [CrossRef]
- Sinha, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cell function is reduced by Withaferin A, a potent and abundant component of Withania somnifera root extract. Cancer Immunol. Immunother. 2013, 62, 1663–1673. [Google Scholar] [CrossRef] [PubMed]
- Hossain, D.M.S.; Javaid, S.; Cai, M.; Zhang, C.; Sawant, A.; Hinton, M.; Sathe, M.; Grein, J.; Blumenschein, W.; Pinheiro, E.M.; et al. Dinaciclib induces immunogenic cell death and enhances anti-PD1-mediated tumor suppression. J. Clin. Investig. 2018, 128, 644–654. [Google Scholar] [CrossRef]
- Kepp, O.; Menger, L.; Vacchelli, E.; Locher, C.; Adjemian, S.; Yamazaki, T.; Martins, I.; Sukkurwala, A.Q.; Michaud, M.; Senovilla, L.; et al. Crosstalk between ER stress and immunogenic cell death. Cytokine Growth Factor Rev. 2013, 24, 311–318. [Google Scholar] [CrossRef]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020, 11, 1013. [Google Scholar] [CrossRef]
- Nath, S.; Obaid, G.; Hasan, T. The Course of Immune Stimulation by Photodynamic Therapy: Bridging Fundamentals of Photochemically Induced Immunogenic Cell Death to the Enrichment of T-Cell Repertoire. Photochem. Photobiol. 2019, 95, 1288–1305. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Hett, E.; Kroemer, G.; Marincola, F.M. Immunogenic cell death in cancer: Concept and therapeutic implications. J. Transl. Med. 2023, 21, 162. [Google Scholar] [CrossRef]
- Xia, S.; Miao, Y.; Liu, S. Withaferin A induces apoptosis by ROS-dependent mitochondrial dysfunction in human colorectal cancer cells. Biochem. Biophys. Res. Commun. 2018, 503, 2363–2369. [Google Scholar] [CrossRef]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Di Blasio, S.; Wortel, I.M.; van Bladel, D.A.; de Vries, L.E.; Duiveman-de Boer, T.; Worah, K.; de Haas, N.; Buschow, S.I.; de Vries, I.J.; Figdor, C.G.; et al. Human CD1c+ DCs are critical cellular mediators of immune responses induced by immunogenic cell death. Oncoimmunology 2016, 5, e1192739. [Google Scholar] [CrossRef] [PubMed]
- Batumalaie, K.; Amin, M.A.; Murugan, D.D.; Sattar, M.Z.A.; Abdullah, N.A. Withaferin A protects against palmitic acid-induced endothelial insulin resistance and dysfunction through suppression of oxidative stress and inflammation. Sci. Rep. 2016, 6, 27236. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Xiang, W.; Jiang, Y.; Tian, N.; Wei, Z.; Wen, X.; Wang, W.; Liao, W.; Xia, X.; Li, Q.; et al. Withaferin A alleviates traumatic brain injury induced secondary brain injury via suppressing apoptosis in endothelia cells and modulating activation in the microglia. Eur. J. Pharmacol. 2020, 874, 172988. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C. Regulation of PD-L1 expression on cancer cells with ROS-modulating drugs. Life Sci. 2020, 246, 117403. [Google Scholar] [CrossRef]
- Choi, E.J.; Jeon, C.H.; Lee, I.-K. Ferric Ammonium Citrate Upregulates PD-L1 Expression through Generation of Reactive Oxygen Species. J. Immunol. Res. 2022, 2022, 6284124. [Google Scholar] [CrossRef]
- Roux, C.; Jafari, S.M.; Shinde, R.; Duncan, G.; Cescon, D.W.; Silvester, J.; Chu, M.F.; Hodgson, K.; Berger, T.; Wakeham, A.; et al. Reactive oxygen species modulate macrophage immunosuppressive phenotype through the up-regulation of PD-L1. Proc. Natl. Acad. Sci. USA 2019, 116, 4326–4335. [Google Scholar] [CrossRef]
- Xue, J.; Li, L.; Li, N.; Li, F.; Qin, X.; Li, T.; Liu, M. Metformin suppresses cancer cell growth in endometrial carcinoma by inhibiting PD-L1. Eur. J. Pharmacol. 2019, 859, 172541. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, W.; Wang, C.; Zheng, C.; Xu, X.; Ni, X.; Hu, S.; Cai, B.; Sun, L.; Shi, K.; et al. DPP4 Inhibitor Attenuates Severe Acute Pancreatitis-Associated Intestinal Inflammation via Nrf2 Signaling. Oxidative Med. Cell. Longev. 2019, 2019, 6181754. [Google Scholar] [CrossRef]
- Zhu, S.; Liu, S.; Wang, L.; Ding, W.; Sha, J.; Qian, H.; Lu, Y. Brusatol Protects HepG2 Cells against Oxygen-Glucose Deprivation-Induced Injury via Inhibiting Mitochondrial Reactive Oxygen Species-Induced Oxidative Stress. Pharmacology 2019, 105, 416–423. [Google Scholar] [CrossRef]
- Kotsafti, A.; MScarpa; Castagliuolo, I.; Scarpa, M. Reactive Oxygen Species and Antitumor Immunity-From Surveillance to Evasion. Cancers 2020, 12, 1748. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Kuang, Z.; Zhang, D.; Gao, Y.; Ying, M.; Wang, T. Reactive oxygen species in immune cells: A new antitumor target. Biomed. Pharmacother. 2021, 133, 110978. [Google Scholar] [CrossRef] [PubMed]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayır, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef]
- Ohl, K.; Tenbrock, K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front. Immunol. 2018, 9, 2499. [Google Scholar] [CrossRef]
- Shi, C.; Liu, T.; Guo, Z.; Zhuang, R.; Zhang, X.; Chen, X. Reprogramming Tumor-Associated Macrophages by Nanoparticle-Based Reactive Oxygen Species Photogeneration. Nano Lett. 2018, 18, 7330–7342. [Google Scholar] [CrossRef]
- Zhu, B.; Tang, L.; Chen, S.; Yin, C.; Peng, S.; Li, X.; Liu, T.; Liu, W.; Han, C.; Stawski, L.; et al. Targeting the upstream transcriptional activator of PD-L1 as an alternative strategy in melanoma therapy. Oncogene 2018, 37, 4941–4954. [Google Scholar] [CrossRef]
- Luo, F.; Luo, M.; Rong, Q.-X.; Zhang, H.; Chen, Z.; Wang, F.; Zhao, H.-Y.; Fu, L.-W. Niclosamide, an antihelmintic drug, enhances efficacy of PD-1/PD-L1 immune checkpoint blockade in non-small cell lung cancer. J. Immunother. Cancer 2019, 7, 245. [Google Scholar] [CrossRef]
- Veronica Olivo, P.; Damiënne, M.; Alexander, M.A.v.d.W.; Natasja, G.L.; Rianne, B.; Relinde, I.Y.L.; Dario, N.; Jan, T.; Ala, Y.; Ludwig, J.D.; et al. Releasing the brakes of tumor immunity with anti-PD-L1 and pushing its accelerator with L19–IL2 cures poorly immunogenic tumors when combined with radiotherapy. J. Immunother. Cancer 2021, 9, e001764. [Google Scholar] [CrossRef]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalil, R.; Green, R.J.; Sivakumar, K.; Varandani, P.; Bharadwaj, S.; Mohapatra, S.S.; Mohapatra, S. Withaferin A Increases the Effectiveness of Immune Checkpoint Blocker for the Treatment of Non-Small Cell Lung Cancer. Cancers 2023, 15, 3089. https://doi.org/10.3390/cancers15123089
Khalil R, Green RJ, Sivakumar K, Varandani P, Bharadwaj S, Mohapatra SS, Mohapatra S. Withaferin A Increases the Effectiveness of Immune Checkpoint Blocker for the Treatment of Non-Small Cell Lung Cancer. Cancers. 2023; 15(12):3089. https://doi.org/10.3390/cancers15123089
Chicago/Turabian StyleKhalil, Roukiah, Ryan J. Green, Kavya Sivakumar, Payal Varandani, Srinivas Bharadwaj, Shyam S. Mohapatra, and Subhra Mohapatra. 2023. "Withaferin A Increases the Effectiveness of Immune Checkpoint Blocker for the Treatment of Non-Small Cell Lung Cancer" Cancers 15, no. 12: 3089. https://doi.org/10.3390/cancers15123089
APA StyleKhalil, R., Green, R. J., Sivakumar, K., Varandani, P., Bharadwaj, S., Mohapatra, S. S., & Mohapatra, S. (2023). Withaferin A Increases the Effectiveness of Immune Checkpoint Blocker for the Treatment of Non-Small Cell Lung Cancer. Cancers, 15(12), 3089. https://doi.org/10.3390/cancers15123089