



Non-Canonical Activin A Signaling Stimulates Context-Dependent and Cellular-Specific Outcomes in CRC to Promote Tumor Cell Migration and Immune Tolerance
 , , , ,
, , , ,  and
and
Abstract
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Immunohistochemistry
2.3. Ts4-Cre; cApcflox; Smad4flox Mouse Model (TcAS)
2.4. Patient Cohort Tissue Microarray
2.5. Digital Spatial Profiling (DSP, NanoString)
2.6. Transwell Migration Assay
2.7. Subcutaneous Tumor Model
2.8. Statistical Analysis
3. Results
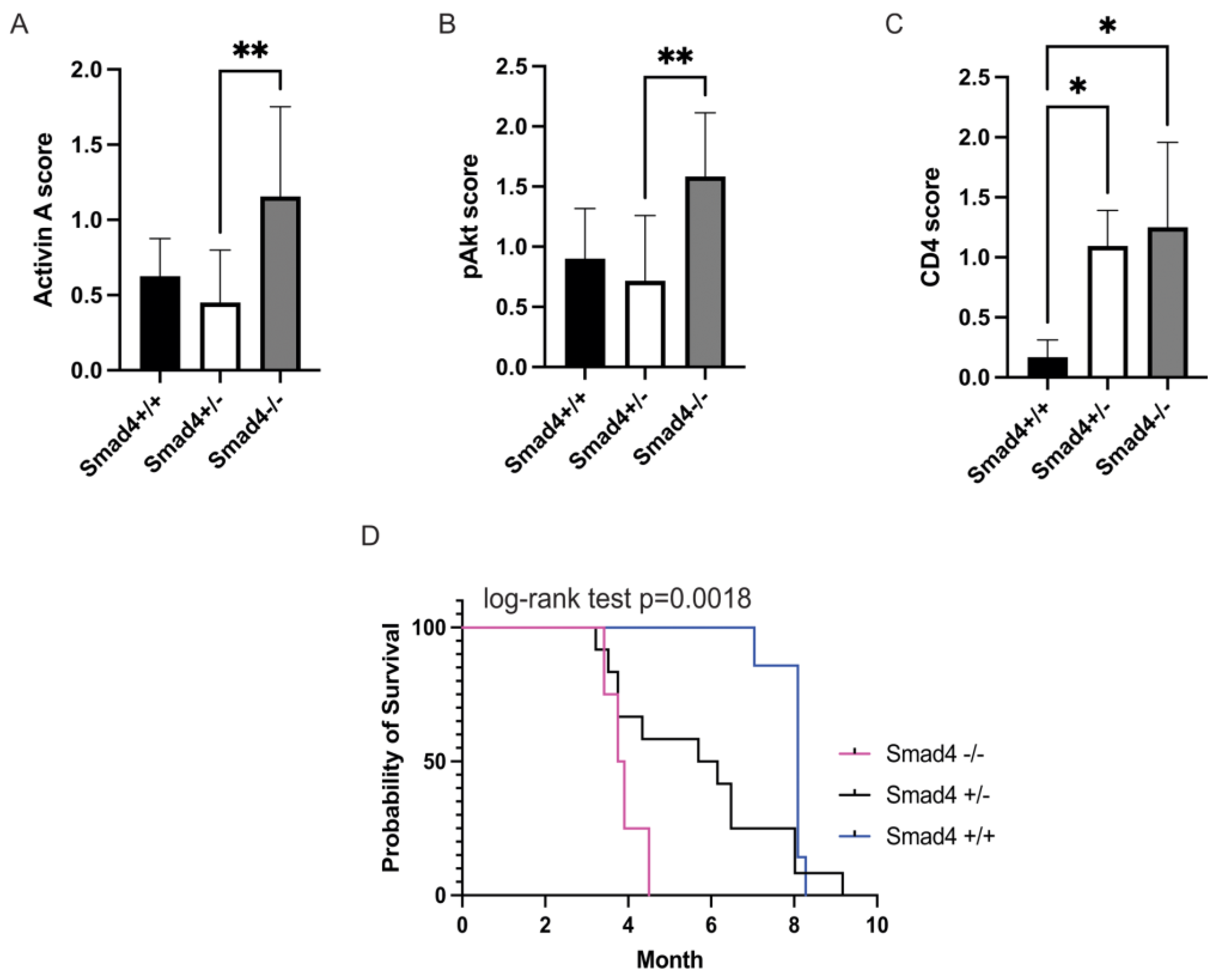
3.1. Loss of SMAD4 in Murine Intestinal Tumors Is Associated with Increased Activin, pAKT, and CD4 and Decreased Survival
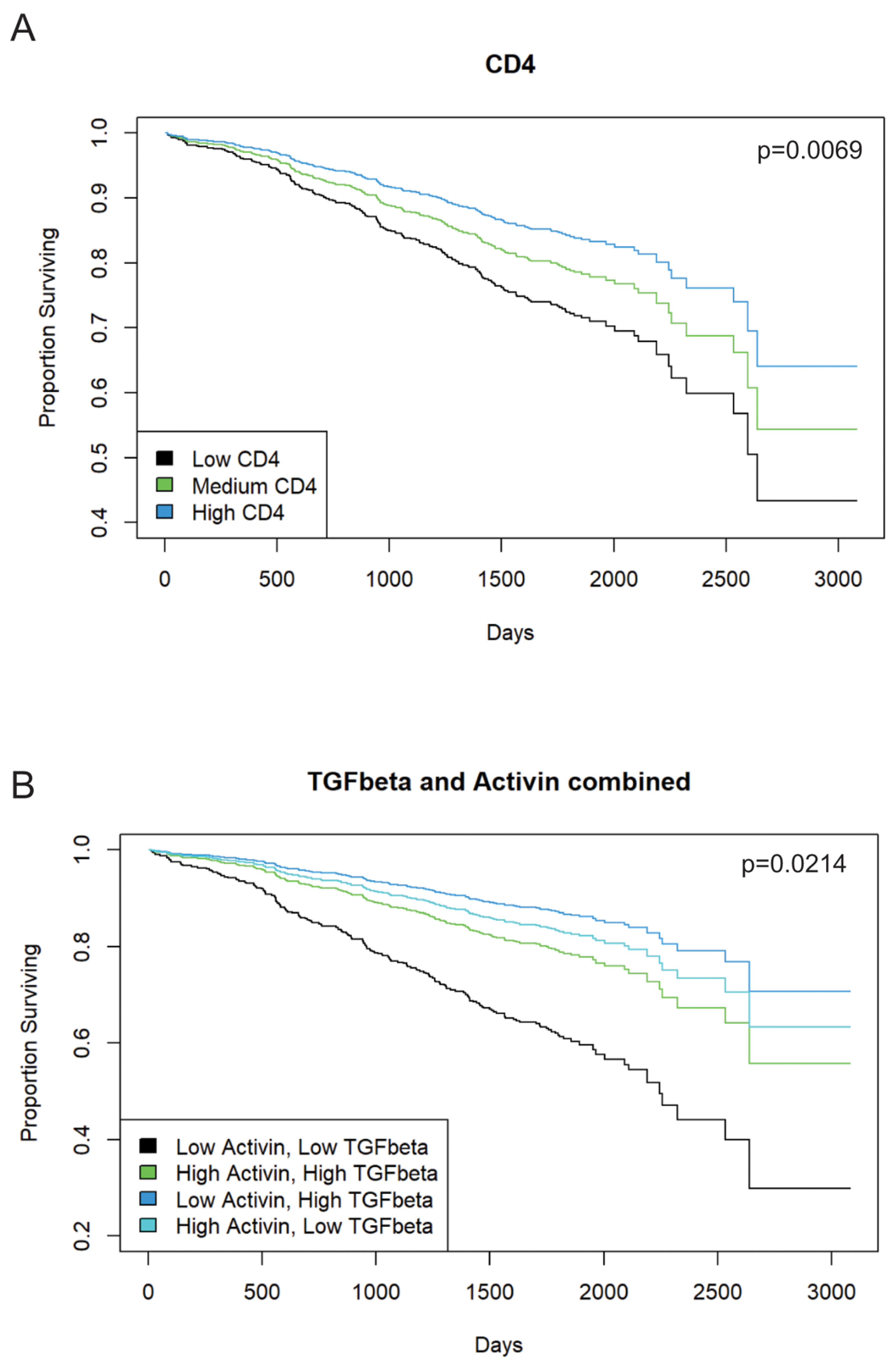
3.2. Increased Activin Expression Is Required for TGF-β-Associated Improvements in Patient Outcome in Stage II or III CRC
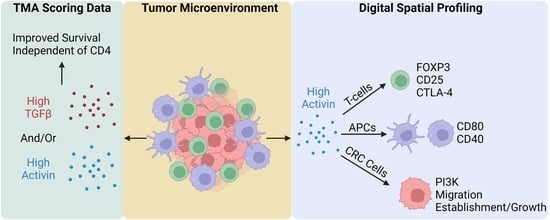
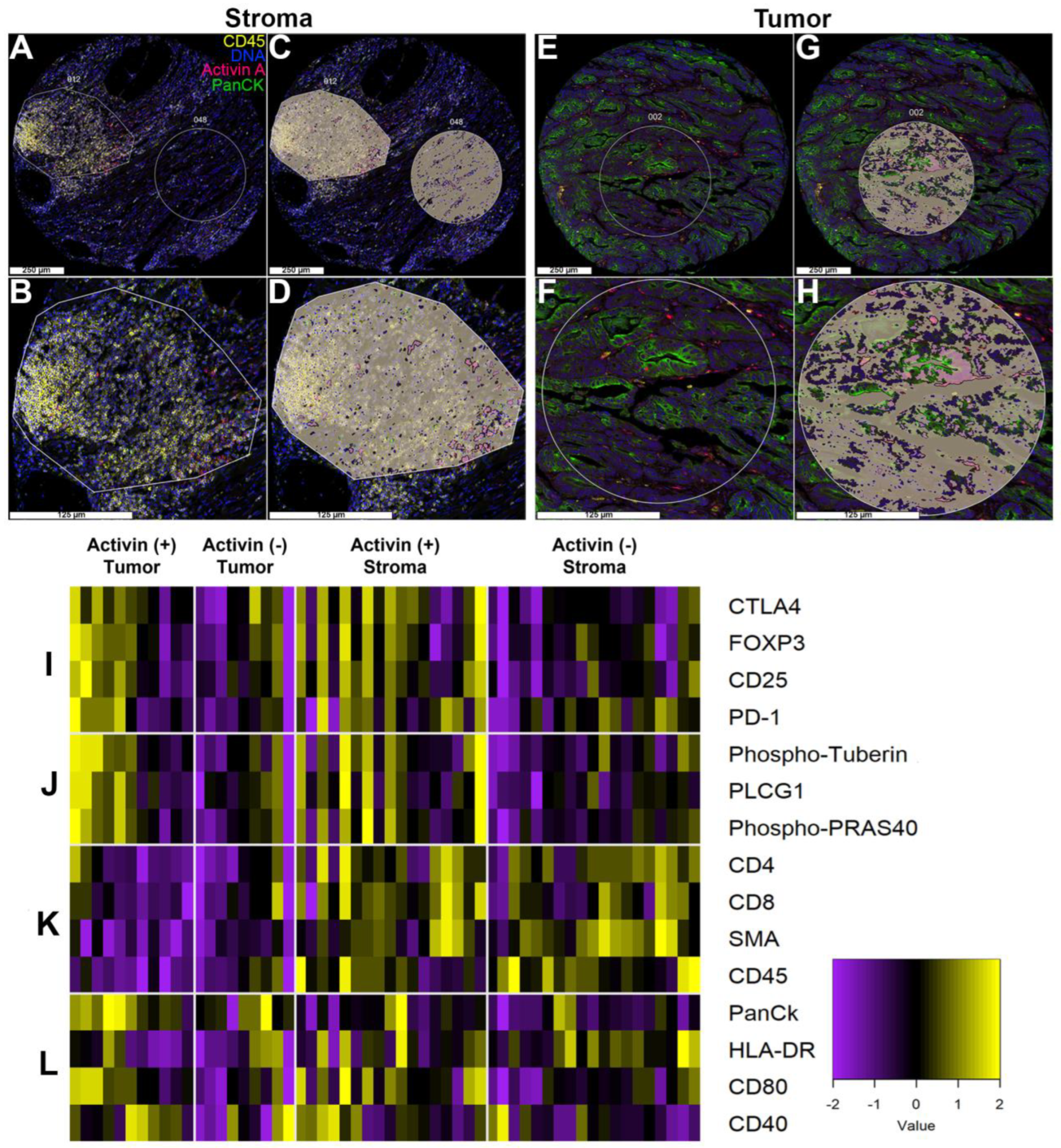
3.3. Activin Co-Localizes with Both Tumor and Immune Cells in the TME to Generate Activin-Dependent Signaling Compartments
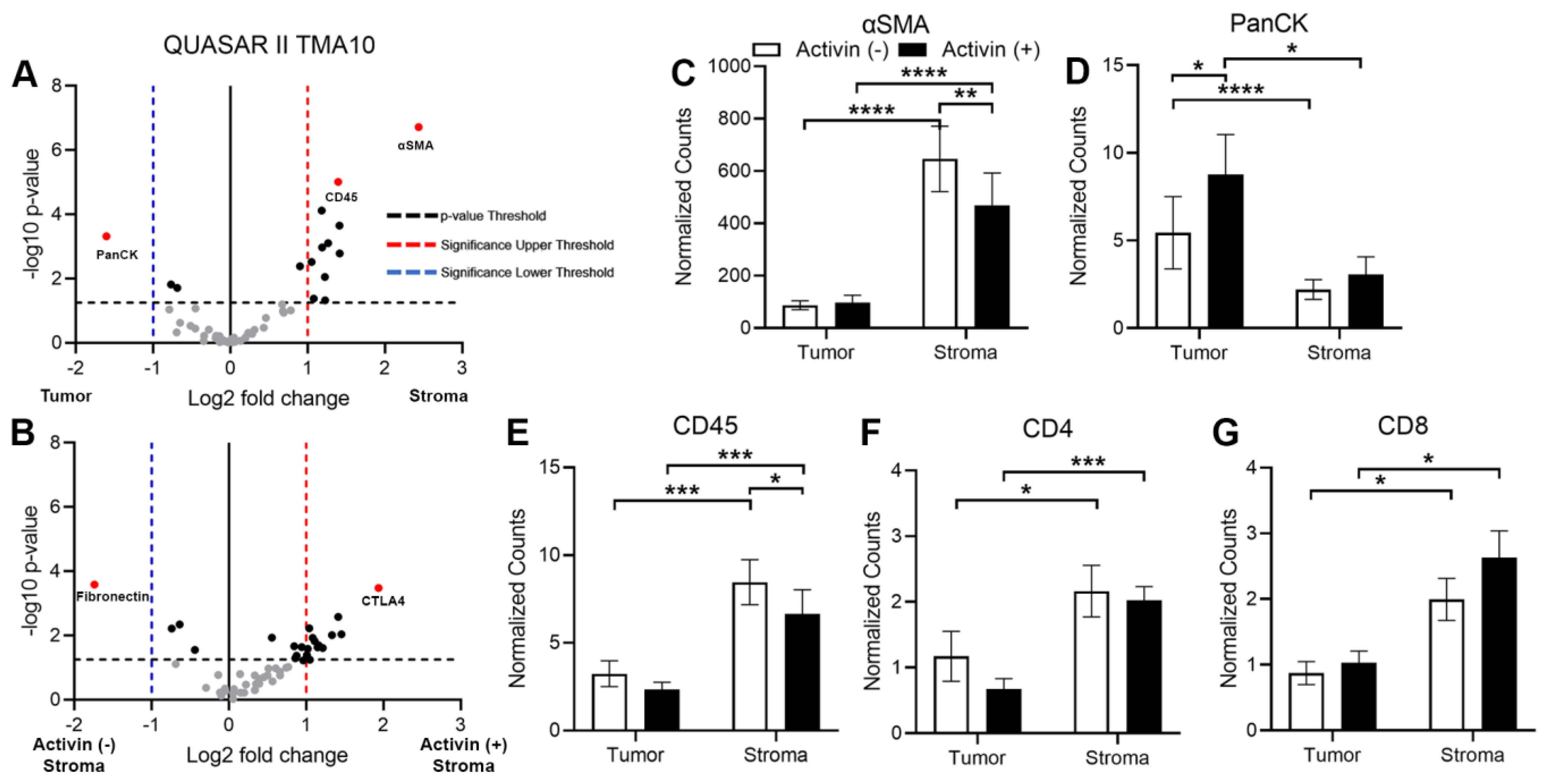
3.4. Markers of Tumoral and Stromal Compartments Confirm Successful Segregation of Regions with an Increase in CD45 Observed in Activin (−) Regions of the Stroma
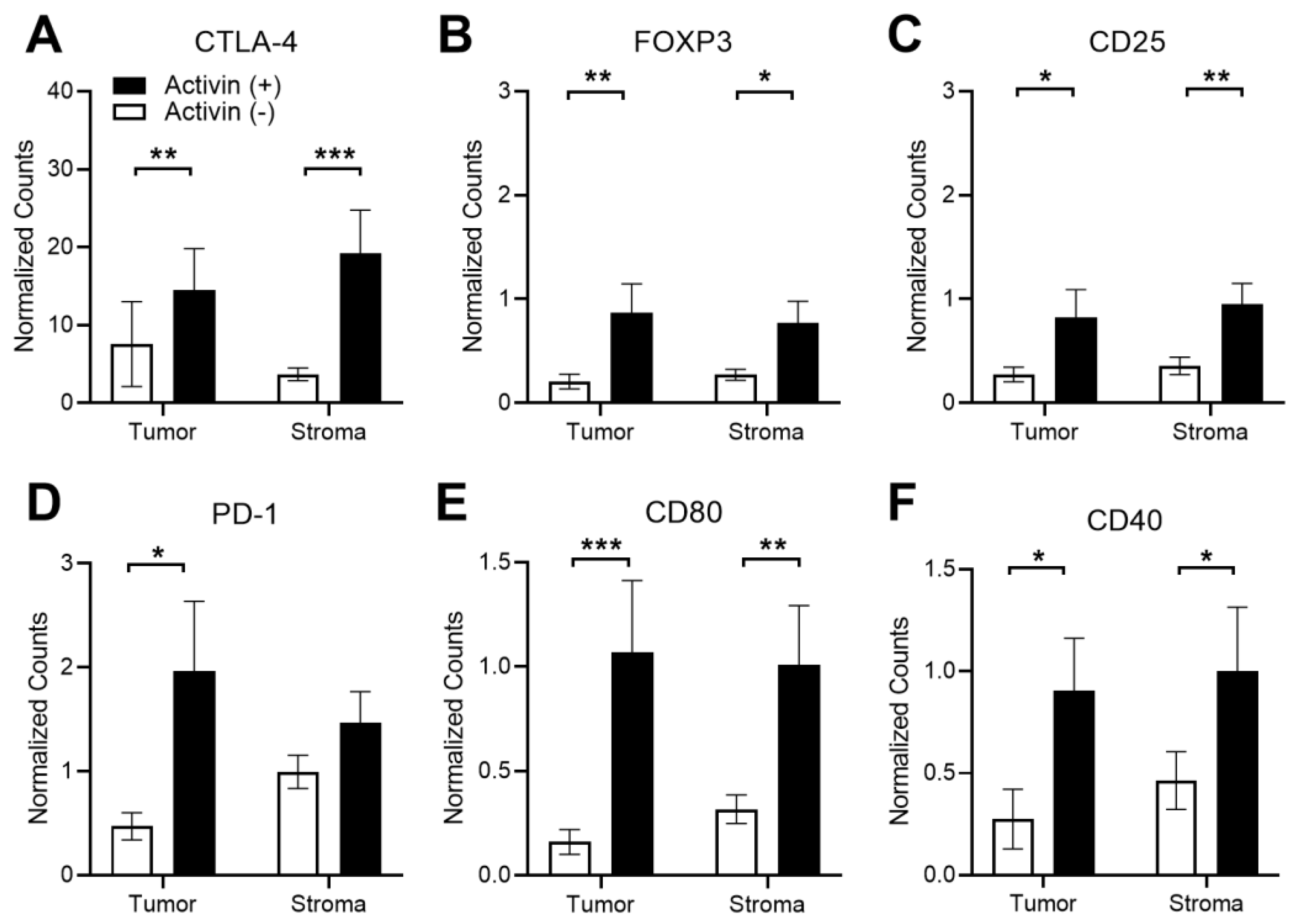
3.5. Activin Stimulates T-Cell Suppression and APC Activation in the Stromal Compartment of the TME in CRC
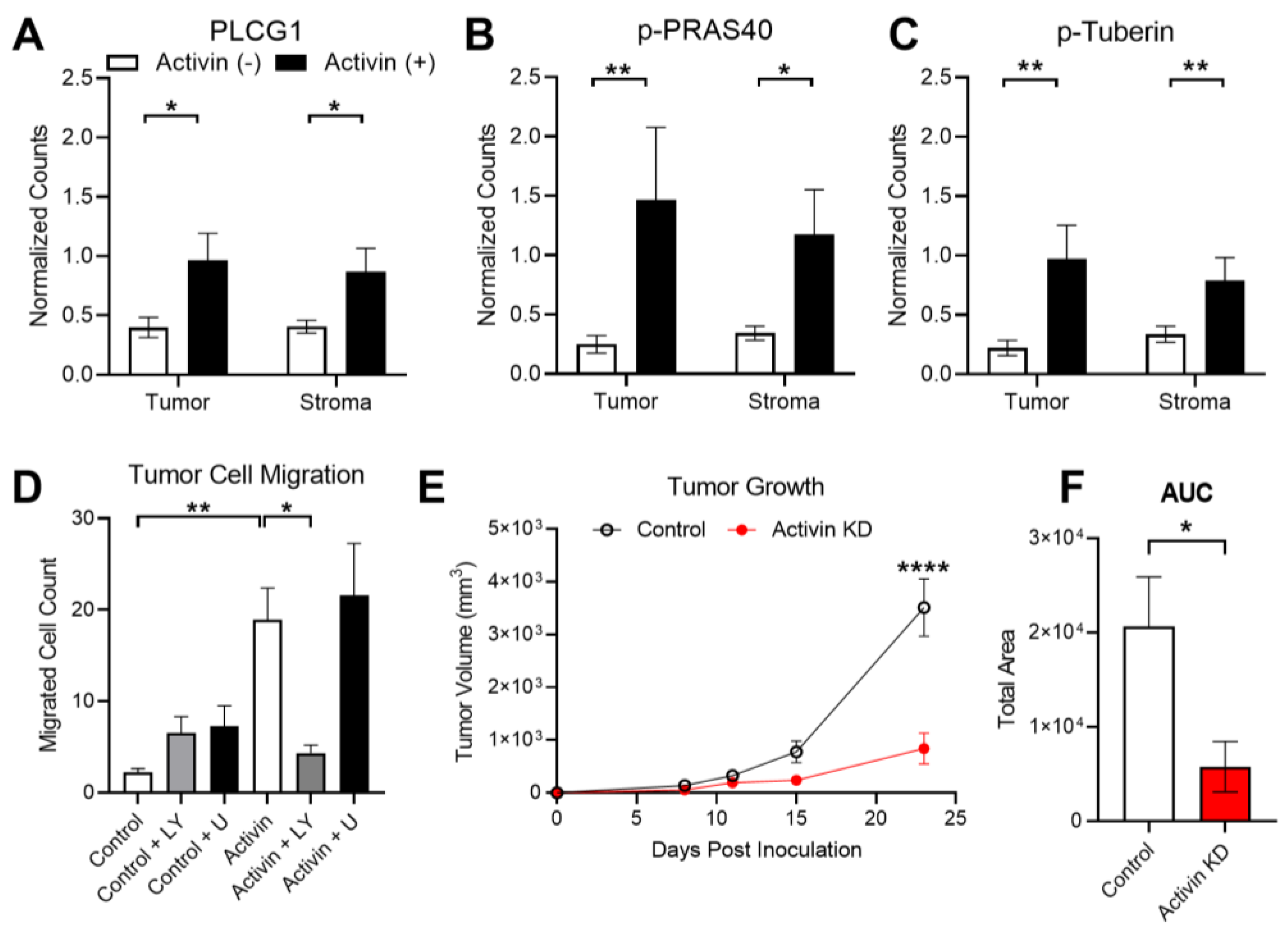
3.6. Activin Co-Localization Is Associated with Increased Activation of PI3K/AKT Signaling to Enhance Tumor Cell Migration and Growth
4. Discussion
4.1. SMAD-Independent Signaling in CRC
4.2. TGF-β, Activin, and CD4 in Human CRC
4.3. Activin Co-Localization with Fibroblasts and Immune Cells
4.4. Activin Signaling in the Tumor Cells
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Loomans-Kropp, H.A.; Umar, A. Increasing Incidence of Colorectal Cancer in Young Adults. J. Cancer Epidemiol. 2019, 2019, 9841295. [Google Scholar] [CrossRef]
- Araghi, M.; Soerjomataram, I.; Bardot, A.; Ferlay, J.; Cabasag, C.J.; Morrison, D.S.; De, P.; Tervonen, H.; Walsh, P.M.; Bucher, O.; et al. Changes in colorectal cancer incidence in seven high-income countries: A population-based study. Lancet Gastroenterol. Hepatol. 2019, 4, 511–518. [Google Scholar] [CrossRef]
- Staudacher, J.J.; Bauer, J.; Jana, A.; Tian, J.; Carroll, T.; Mancinelli, G.; Ozden, O.; Krett, N.; Guzman, G.; Kerr, D.; et al. Activin signaling is an essential component of the TGF-beta induced pro-metastatic phenotype in colorectal cancer. Sci. Rep. 2017, 7, 5569. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, H.; Arao, T.; Matsumoto, K.; De Velasco, M.A.; Tamura, D.; Aomatsu, K.; Kudo, K.; Sakai, K.; Nagai, T.; Fujita, Y.; et al. Activin A inhibits vascular endothelial cell growth and suppresses tumour angiogenesis in gastric cancer. Br. J. Cancer 2011, 105, 1210–1217. [Google Scholar] [CrossRef]
- Zessner-Spitzenberg, J.; Thomas, A.L.; Krett, N.L.; Jung, B. TGFbeta and activin A in the tumor microenvironment in colorectal cancer. Gene Rep. 2019, 17, 100501. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.; Staudacher, J.J.; Beauchamp, D. Transforming Growth Factor beta Superfamily Signaling in Development of Colorectal Cancer. Gastroenterology 2017, 152, 36–52. [Google Scholar] [CrossRef]
- Bauer, J.; Ozden, O.; Akagi, N.; Carroll, T.; Principe, D.R.; Staudacher, J.J.; Spehlmann, M.E.; Eckmann, L.; Grippo, P.J.; Jung, B. Activin and TGFbeta use diverging mitogenic signaling in advanced colon cancer. Mol. Cancer 2015, 14, 182. [Google Scholar] [CrossRef]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef]
- Allen, F.; Maillard, I. Therapeutic Targeting of Notch Signaling: From Cancer to Inflammatory Disorders. Front. Cell Dev. Biol. 2021, 9, 649205. [Google Scholar] [CrossRef]
- Sierra-Filardi, E.; Puig-Kroger, A.; Blanco, F.J.; Nieto, C.; Bragado, R.; Palomero, M.I.; Bernabeu, C.; Vega, M.A.; Corbi, A.L. Activin A skews macrophage polarization by promoting a proinflammatory phenotype and inhibiting the acquisition of anti-inflammatory macrophage markers. Blood 2011, 117, 5092–5101. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.L.; Castellanos, K.; Mancinelli, G.; Xia, Y.; Bauer, J.; Yazici, C.; Fantuzzi, G.; Hwang, R.F.; Krett, N.L.; Papachristou, G.I.; et al. Activin A Modulates Inflammation in Acute Pancreatitis and Strongly Predicts Severe Disease Independent of Body Mass Index. Clin. Transl. Gastroenterol. 2020, 11, e00152. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cui, X.; Tai, G.; Ge, J.; Li, N.; Chen, F.; Yu, F.; Liu, Z. A critical role of activin A in maturation of mouse peritoneal macrophages in vitro and in vivo. Cell. Mol. Immunol. 2009, 6, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.; Stahl, F.R.; Schrader, J.; Luth, S.; Presser, K.; Carambia, A.; Flavell, R.A.; Werner, S.; Blessing, M.; Herkel, J.; et al. Activin a promotes the TGF-beta-induced conversion of CD4+CD25- T cells into Foxp3+ induced regulatory T cells. J. Immunol. 2009, 182, 4633–4640. [Google Scholar] [CrossRef] [PubMed]
- Morianos, I.; Tsitsopoulou, A.; Potaris, K.; Valakos, D.; Fari, O.; Vatsellas, G.; Bostantzoglou, C.; Photiades, A.; Gaga, M.; Xanthou, G.; et al. Activin-A impedes the establishment of CD4(+) T cell exhaustion and enhances anti-tumor immunity in the lung. J. Exp. Clin. Cancer Res. 2021, 40, 295. [Google Scholar] [CrossRef]
- Andre, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Wang, C.; Fakih, M. Targeting MSS colorectal cancer with immunotherapy: Are we turning the corner? Expert. Opin. Biol. Ther. 2021, 21, 1347–1357. [Google Scholar] [CrossRef]
- Foroutan, M.; Molania, R.; Pfefferle, A.; Behrenbruch, C.; Scheer, S.; Kallies, A.; Speed, T.P.; Cursons, J.; Huntington, N.D. The Ratio of Exhausted to Resident Infiltrating Lymphocytes Is Prognostic for Colorectal Cancer Patient Outcome. Cancer Immunol. Res. 2021, 9, 1125–1140. [Google Scholar] [CrossRef]
- Principe, D.R.; DeCant, B.; Staudacher, J.; Vitello, D.; Mangan, R.J.; Wayne, E.A.; Mascarinas, E.; Diaz, A.M.; Bauer, J.; McKinney, R.D.; et al. Loss of TGFbeta signaling promotes colon cancer progression and tumor-associated inflammation. Oncotarget 2017, 8, 3826–3839. [Google Scholar] [CrossRef]
- Wiley, M.B.; Bobardt, S.D.; Nordgren, T.M.; Nair, M.G.; DiPatrizio, N.V. Cannabinoid Receptor Subtype-1 Regulates Allergic Airway Eosinophilia During Lung Helminth Infection. Cannabis Cannabinoid Res. 2021, 6, 242–252. [Google Scholar] [CrossRef]
- Gide, T.N.; Silva, I.P.; Quek, C.; Ahmed, T.; Menzies, A.M.; Carlino, M.S.; Saw, R.P.M.; Thompson, J.F.; Batten, M.; Long, G.V.; et al. Close proximity of immune and tumor cells underlies response to anti-PD-1 based therapies in metastatic melanoma patients. Oncoimmunology 2020, 9, 1659093. [Google Scholar] [CrossRef] [PubMed]
- Merritt, C.R.; Ong, G.T.; Church, S.E.; Barker, K.; Danaher, P.; Geiss, G.; Hoang, M.; Jung, J.; Liang, Y.; McKay-Fleisch, J.; et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol. 2020, 38, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Sporn, J.C.; Cabral, J.; Gomez, J.; Jung, B. Effects of activin and TGFbeta on p21 in colon cancer. PLoS ONE 2012, 7, e39381. [Google Scholar] [CrossRef]
- Khazaie, K.; Zadeh, M.; Khan, M.W.; Bere, P.; Gounari, F.; Dennis, K.; Blatner, N.R.; Owen, J.L.; Klaenhammer, T.R.; Mohamadzadeh, M. Abating colon cancer polyposis by Lactobacillus acidophilus deficient in lipoteichoic acid. Proc. Natl. Acad. Sci. USA 2012, 109, 10462–10467. [Google Scholar] [CrossRef] [PubMed]
- Saam, J.R.; Gordon, J.I. Inducible gene knockouts in the small intestinal and colonic epithelium. J. Biol. Chem. 1999, 274, 38071–38082. [Google Scholar] [CrossRef]
- Kerr, R.S.; Love, S.; Segelov, E.; Johnstone, E.; Falcon, B.; Hewett, P.; Weaver, A.; Church, D.; Scudder, C.; Pearson, S.; et al. Adjuvant capecitabine plus bevacizumab versus capecitabine alone in patients with colorectal cancer (QUASAR 2): An open-label, randomised phase 3 trial. Lancet Oncol. 2016, 17, 1543–1557. [Google Scholar] [CrossRef]
- Kleppe, A.; Skrede, O.J.; De Raedt, S.; Hveem, T.S.; Askautrud, H.A.; Jacobsen, J.E.; Church, D.N.; Nesbakken, A.; Shepherd, N.A.; Novelli, M.; et al. A clinical decision support system optimising adjuvant chemotherapy for colorectal cancers by integrating deep learning and pathological staging markers: A development and validation study. Lancet Oncol. 2022, 23, 1221–1232. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Farooqi, M.S.; Habeebu, S.S.; Gonzalez, E.; Flatt, T.G.; Wilson, A.L.; Barr, F.G. NanoString Digital Molecular Profiling of Protein and microRNA in Rhabdomyosarcoma. Cancers 2022, 14, 522. [Google Scholar] [CrossRef]
- Wiley, M.B.; DiPatrizio, N.V. Diet-Induced Gut Barrier Dysfunction Is Exacerbated in Mice Lacking Cannabinoid 1 Receptors in the Intestinal Epithelium. Int. J. Mol. Sci. 2022, 23, 549. [Google Scholar] [CrossRef]
- Omilian, A.R.; Sheng, H.; Hong, C.C.; Bandera, E.V.; Khoury, T.; Ambrosone, C.B.; Yao, S. Multiplexed digital spatial profiling of invasive breast tumors from Black and White women. Mol. Oncol. 2022, 16, 54–68. [Google Scholar] [CrossRef]
- Jensen, M.M.; Jorgensen, J.T.; Binderup, T.; Kjaer, A. Tumor volume in subcutaneous mouse xenografts measured by microCT is more accurate and reproducible than determined by 18F-FDG-microPET or external caliper. BMC Med. Imaging 2008, 8, 16. [Google Scholar] [CrossRef]
- Jana, A.; Das, A.; Krett, N.L.; Guzman, G.; Thomas, A.; Mancinelli, G.; Bauer, J.; Ushio-Fukai, M.; Fukai, T.; Jung, B. Nuclear translocation of Atox1 potentiates activin A-induced cell migration and colony formation in colon cancer. PLoS ONE 2020, 15, e0227916. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Emon, M.A.B.; Staudacher, J.J.; Thomas, A.L.; Zessner-Spitzenberg, J.; Mancinelli, G.; Krett, N.; Saif, M.T.; Jung, B. Increased stiffness of the tumor microenvironment in colon cancer stimulates cancer associated fibroblast-mediated prometastatic activin A signaling. Sci. Rep. 2020, 10, 50. [Google Scholar] [CrossRef] [PubMed]
- Galeano Nino, J.L.; Wu, H.; LaCourse, K.D.; Kempchinsky, A.G.; Baryiames, A.; Barber, B.; Futran, N.; Houlton, J.; Sather, C.; Sicinska, E.; et al. Effect of the intratumoral microbiota on spatial and cellular heterogeneity in cancer. Nature 2022, 611, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Bashir, M.; Damineni, S.; Mukherjee, G.; Kondaiah, P. Activin-A signaling promotes epithelial-mesenchymal transition, invasion, and metastatic growth of breast cancer. NPJ Breast Cancer 2015, 1, 15007. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.Y.; Luan, Y.; Tang, S.; Abazarikia, A.; Dong, R.; Caffrey, T.C.; Hollingsworth, M.A.; Oupicky, D.; Kim, S.Y. Uncovering Tumor-Promoting Roles of Activin A in Pancreatic Ductal Adenocarcinoma. Adv. Sci. 2023, e2207010. [Google Scholar] [CrossRef] [PubMed]
- Stastna, M.; Janeckova, L.; Hrckulak, D.; Kriz, V.; Korinek, V. Human Colorectal Cancer from the Perspective of Mouse Models. Genes 2019, 10, 788. [Google Scholar] [CrossRef]
- Bauer, J.; Staudacher, J.J.; Krett, N.L.; Jung, B. Commentary: Activin and TGFbeta use diverging mitogenic signaling in advanced colon cancer. J. Rare Dis. Res. Treat. 2016, 1, 43–45. [Google Scholar] [CrossRef]
- Staudacher, J.J.; Arnold, A.; Kuhl, A.A.; Potzsch, M.; Daum, S.; Winterfeld, M.; Berg, E.; Hummel, M.; Rau, B.; Stein, U.; et al. Prognostic impact of activin subunit inhibin beta A in gastric and esophageal adenocarcinomas. BMC Cancer 2022, 22, 953. [Google Scholar] [CrossRef]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.; Chen, Y.; Suo, L.; Chen, H.; Zhu, L.; Wan, G.; Han, X. Activin a promotes myofibroblast differentiation of endometrial mesenchymal stem cells via STAT3-dependent Smad/CTGF pathway. Cell Commun. Signal. 2019, 17, 45. [Google Scholar] [CrossRef]
- Ge, J.; Wang, Y.; Feng, Y.; Liu, H.; Cui, X.; Chen, F.; Tai, G.; Liu, Z. Direct effects of activin A on the activation of mouse macrophage RAW264.7 cells. Cell. Mol. Immunol. 2009, 6, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Esensten, J.H.; Helou, Y.A.; Chopra, G.; Weiss, A.; Bluestone, J.A. CD28 Costimulation: From Mechanism to Therapy. Immunity 2016, 44, 973–988. [Google Scholar] [CrossRef]
- Halliday, N.; Williams, C.; Kennedy, A.; Waters, E.; Pesenacker, A.M.; Soskic, B.; Hinze, C.; Hou, T.Z.; Rowshanravan, B.; Janman, D.; et al. CD86 Is a Selective CD28 Ligand Supporting FoxP3+ Regulatory T Cell Homeostasis in the Presence of High Levels of CTLA-4. Front. Immunol. 2020, 11, 600000. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.; Waters, E.; Rowshanravan, B.; Hinze, C.; Williams, C.; Janman, D.; Fox, T.A.; Booth, C.; Pesenacker, A.M.; Halliday, N.; et al. Differences in CD80 and CD86 transendocytosis reveal CD86 as a key target for CTLA-4 immune regulation. Nat. Immunol. 2022, 23, 1365–1378. [Google Scholar] [CrossRef] [PubMed]
- Chaput, N.; Louafi, S.; Bardier, A.; Charlotte, F.; Vaillant, J.C.; Menegaux, F.; Rosenzwajg, M.; Lemoine, F.; Klatzmann, D.; Taieb, J. Identification of CD8+CD25+Foxp3+ suppressive T cells in colorectal cancer tissue. Gut 2009, 58, 520–529. [Google Scholar] [CrossRef]
- Jung, B.; Doctolero, R.T.; Tajima, A.; Nguyen, A.K.; Keku, T.; Sandler, R.S.; Carethers, J.M. Loss of activin receptor type 2 protein expression in microsatellite unstable colon cancers. Gastroenterology 2004, 126, 654–659. [Google Scholar] [CrossRef]
- Jung, B.; Gomez, J.; Chau, E.; Cabral, J.; Lee, J.K.; Anselm, A.; Slowik, P.; Ream-Robinson, D.; Messer, K.; Sporn, J.; et al. Activin signaling in microsatellite stable colon cancers is disrupted by a combination of genetic and epigenetic mechanisms. PLoS ONE 2009, 4, e8308. [Google Scholar] [CrossRef]
- Yerly, L.; Pich-Bavastro, C.; Di Domizio, J.; Wyss, T.; Tissot-Renaud, S.; Cangkrama, M.; Gilliet, M.; Werner, S.; Kuonen, F. Integrated multi-omics reveals cellular and molecular interactions governing the invasive niche of basal cell carcinoma. Nat. Commun. 2022, 13, 4897. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiley, M.B.; Bauer, J.; Mehrotra, K.; Zessner-Spitzenberg, J.; Kolics, Z.; Cheng, W.; Castellanos, K.; Nash, M.G.; Gui, X.; Kone, L.; et al. Non-Canonical Activin A Signaling Stimulates Context-Dependent and Cellular-Specific Outcomes in CRC to Promote Tumor Cell Migration and Immune Tolerance. Cancers 2023, 15, 3003. https://doi.org/10.3390/cancers15113003
Wiley MB, Bauer J, Mehrotra K, Zessner-Spitzenberg J, Kolics Z, Cheng W, Castellanos K, Nash MG, Gui X, Kone L, et al. Non-Canonical Activin A Signaling Stimulates Context-Dependent and Cellular-Specific Outcomes in CRC to Promote Tumor Cell Migration and Immune Tolerance. Cancers. 2023; 15(11):3003. https://doi.org/10.3390/cancers15113003
Chicago/Turabian StyleWiley, Mark B., Jessica Bauer, Kunaal Mehrotra, Jasmin Zessner-Spitzenberg, Zoe Kolics, Wenxuan Cheng, Karla Castellanos, Michael G. Nash, Xianyong Gui, Lyonell Kone, and et al. 2023. "Non-Canonical Activin A Signaling Stimulates Context-Dependent and Cellular-Specific Outcomes in CRC to Promote Tumor Cell Migration and Immune Tolerance" Cancers 15, no. 11: 3003. https://doi.org/10.3390/cancers15113003
APA StyleWiley, M. B., Bauer, J., Mehrotra, K., Zessner-Spitzenberg, J., Kolics, Z., Cheng, W., Castellanos, K., Nash, M. G., Gui, X., Kone, L., Maker, A. V., Qiao, G., Reddi, D., Church, D. N., Kerr, R. S., Kerr, D. J., Grippo, P. J., & Jung, B. (2023). Non-Canonical Activin A Signaling Stimulates Context-Dependent and Cellular-Specific Outcomes in CRC to Promote Tumor Cell Migration and Immune Tolerance. Cancers, 15(11), 3003. https://doi.org/10.3390/cancers15113003