

Interdependencies of the Neuronal, Immune and Tumor Microenvironment in Gliomas
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Tumor Components of the TME
2.1. Glioma Stem Cells
2.2. Mutational Landscape of Glioma Cells
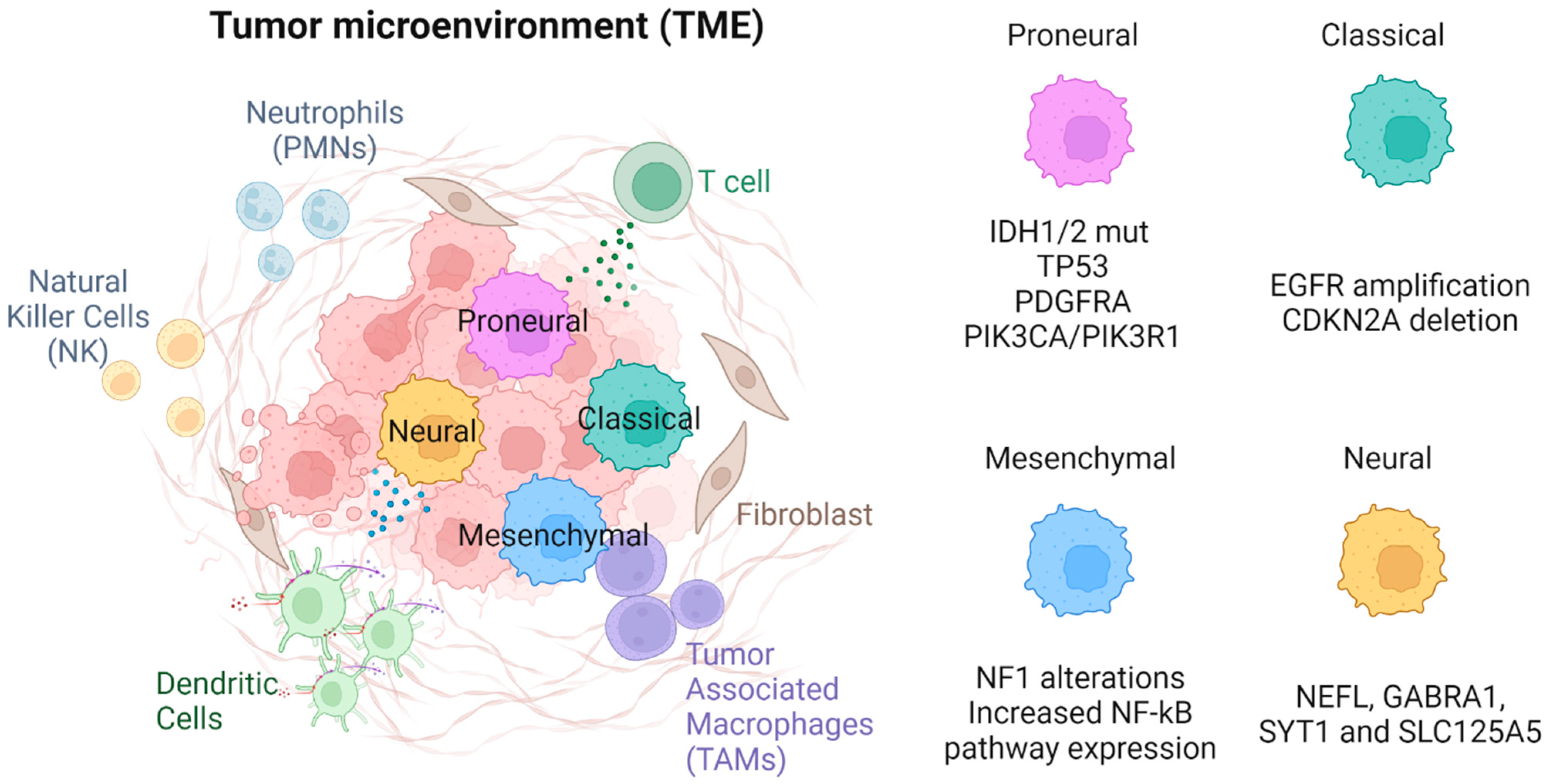
2.3. Tumor Cell Subtypes
2.3.1. Proneural
2.3.2. Classical
2.3.3. Mesenchymal
2.3.4. Neural
3. Immune Components of the TME
3.1. Innate Immune Component
3.1.1. Tumor-Associated Macrophages (TAMs)—Microglia and Bone Marrow Derived Macrophages (BMDM)
3.1.2. Neutrophils (PMNs)
3.1.3. Dendritic Cells
3.1.4. Natural Killer (NK) Cells
3.1.5. Monocyte-Derived Suppressor Cells (MDSCs)
3.2. Adaptive Immune Component
Tumor-Infiltrating lymphocytes (TILs)
4. Non-Tumoral, Non-Immune Components of the TME
4.1. Glial Cells
4.2. Neurons
4.3. The Blood Brain Barrier (BBB) and Vasculature
4.4. The Extra-Cellular Matrix
5. Tumoral Influence on TME
5.1. Tumoral Electrical Signaling
5.2. Effect of GSC on TME
5.3. Influence of Tumor Molecular Phenotype on the Immune Component
5.3.1. Influence of Transcriptional Signatures on Immune Component
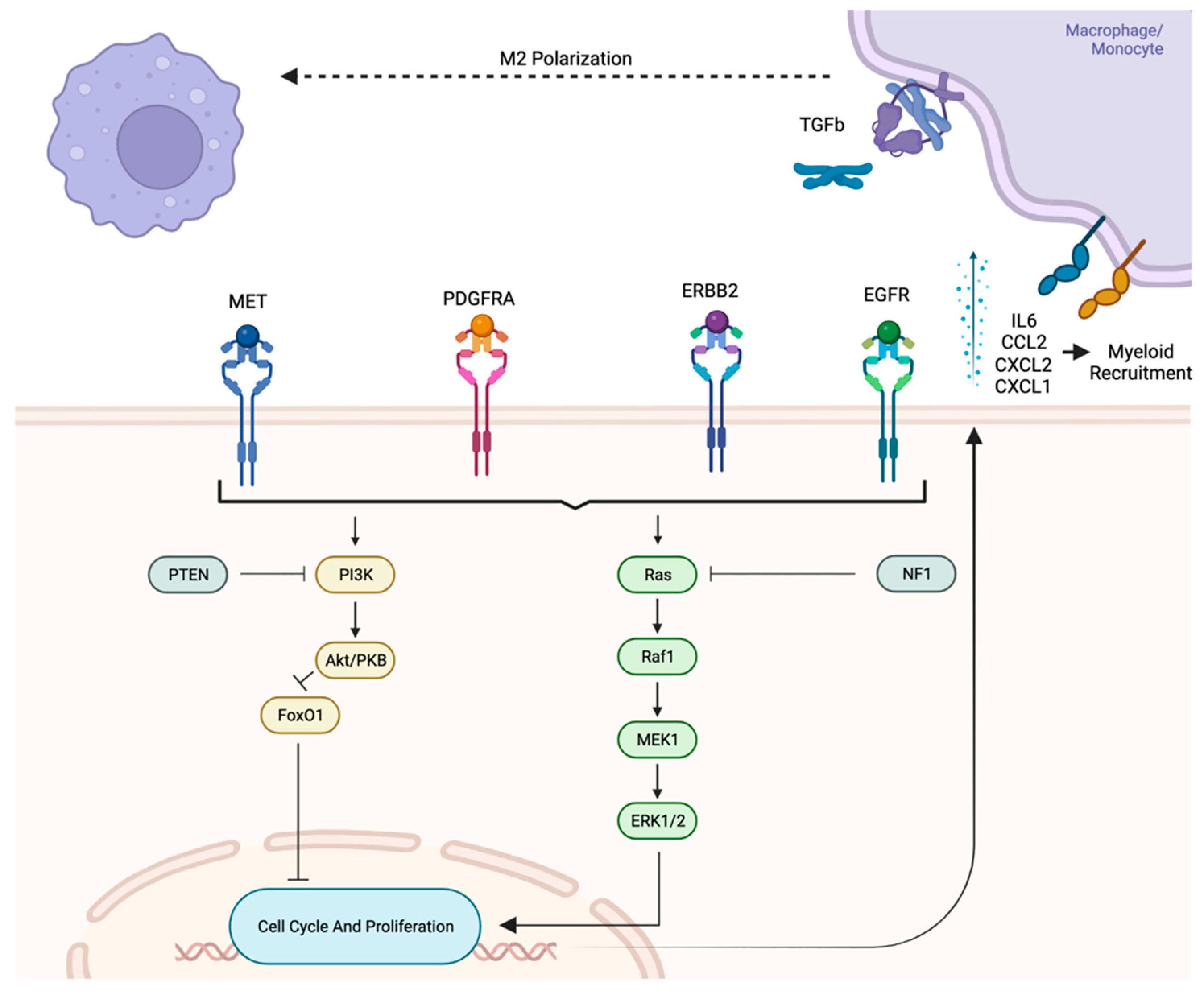
5.3.2. Influence of Oncogenic Pathways on Immune Component
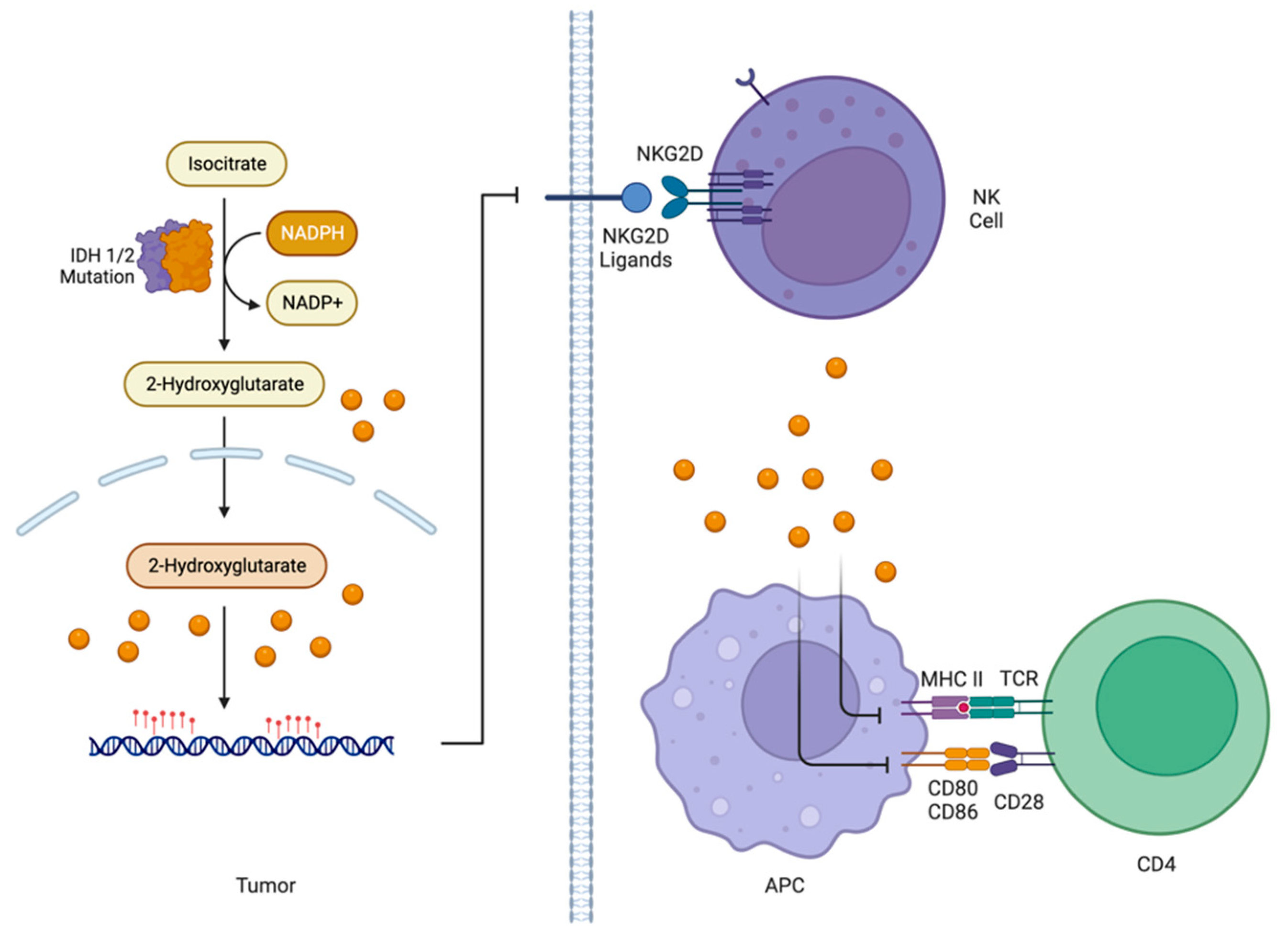
5.3.3. Influence of IDH1/2 Status on Immune Component of TME
5.3.4. Tumoral Influence on Non-Immune TME Components
5.4. Immune Influence on TME
6. Non-Tumoral, Non-Immune Influence on the TME
6.1. Influence of Glial Cells on the TME
6.2. Effect of Neurons on the TME
6.3. Effect of BBB and Vasculature on the TME
6.4. Effect of the ECM on the TME
7. Future Therapeutic Directions
7.1. Interruption of TME Signaling
7.2. Reverse Immunosuppression of TME
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro Oncol. 2021, 23 (Suppl. S3), iii1–iii105. [Google Scholar] [CrossRef] [PubMed]
- Pekmezci, M.; Rice, T.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Hansen, H.; Sicotte, H.; Kollmeyer, T.M.; McCoy, L.S.; Sarkar, G.; et al. Adult infiltrating gliomas with WHO 2016 integrated diagnosis: Additional prognostic roles of ATRX and TERT. Acta Neuropathol. 2017, 133, 1001–1016. [Google Scholar] [CrossRef] [PubMed]
- WHO. Classification of Tumours Editorial Board. Central Nervous System Tumours. 2021. Available online: https://publications.iarc.fr/Book-And-Report-Series/Who-Classification-Of-Tumours/Central-Nervous-System-Tumours-2021 (accessed on 27 January 2023).
- Bradshaw, A.; Wickremsekera, A.; Tan, S.T.; Peng, L.; Davis, P.F.; Itinteang, T. Cancer Stem Cell Hierarchy in Glioblastoma Multiforme. Front. Surg. 2016, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. An integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR and NF1. Cancer Cell 2010, 17, 98. [Google Scholar] [CrossRef]
- Medzhitov, R.; Janeway, C.A. Innate immunity: The virtues of a nonclonal system of recognition. Cell 1997, 91, 295–298. [Google Scholar] [CrossRef]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef]
- Desland, F.A.; Hormigo, A. The CNS and the Brain Tumor Microenvironment: Implications for Glioblastoma Immunotherapy. Int. J. Mol. Sci. 2020, 21, 7358. [Google Scholar] [CrossRef]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Long, G.V.; Atkinson, V.; Lo, S.; Sandhu, S.; Guminski, A.D.; Brown, M.P.; Wilmott, J.S.; Edwards, J.; Gonzalez, M.; Scolyer, R.A.; et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: A multicentre randomised phase 2 study. Lancet Oncol. 2018, 19, 672–681. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs. Bevacizumab in Patients With Recurrent Glioblastoma. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Venkataramani, V.; Yang, Y.; Schubert, M.C.; Reyhan, E.; Tetzlaff, S.K.; Wißmann, N.; Botz, M.; Soyka, S.J.; Beretta, C.A.; Pramatarov, R.L.; et al. Glioblastoma hijacks neuronal mechanisms for brain invasion. Cell 2022, 185, 2899–2917. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.P.; Sells, B.E.; Haque, S.J.; Chakravarti, A. Tumor Heterogeneity in Glioblastomas: From Light Microscopy to Molecular Pathology. Cancers 2021, 13, 761. [Google Scholar] [CrossRef]
- Charles, N.; Holland, E.C. The perivascular niche microenvironment in brain tumor progression. Cell Cycle 2010, 9, 3012–3021. [Google Scholar] [CrossRef] [PubMed]
- Pietras, A.; Katz, A.M.; Ekström, E.J.; Wee, B.; Halliday, J.J.; Pitter, K.L.; Werbeck, J.L.; Amankulor, N.M.; Huse, J.T.; Holland, E.C. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell 2014, 14, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Piper, K.; DePledge, L.; Karsy, M.; Cobbs, C. Glioma Stem Cells as Immunotherapeutic Targets: Advancements and Challenges. Front. Oncol. 2021, 11, 615704. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Perez, D.; Chang, L.; Shi, J.; Ma, J.; Kim, S.-H.; Zhao, X.; Malkoc, V.; Wang, X.; Minata, M.; Kwak, K.J.; et al. On-Chip Clonal Analysis of Glioma-Stem-Cell Motility and Therapy Resistance. Nano Lett. 2016, 16, 5326–5332. [Google Scholar] [CrossRef]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.I.S.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.-J.; et al. Clonal Evolution of Glioblastoma under Therapy. Nat. Genet 2016, 48, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Di Cintio, F.; Dal Bo, M.; Baboci, L.; De Mattia, E.; Polano, M.; Toffoli, G. The Molecular and Microenvironmental Landscape of Glioblastomas: Implications for the Novel Treatment Choices. Front. Neurosci. 2020, 14, 603647. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, M.L.; Sherlock, G.; Saldanha, A.J.; Murray, J.I.; Ball, C.A.; Alexander, K.E.; Matese, J.C.; Perou, C.M.; Hurt, M.M.; Brown, P.O.; et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol. Biol. Cell 2002, 13, 1977–2000. [Google Scholar] [CrossRef]
- Noble, M.; Pröschel, C.; Mayer-Pröschel, M. Getting a GR(i)P on oligodendrocyte development. Dev. Biol. 2004, 265, 33–52. [Google Scholar] [CrossRef]
- Glasgow, S.M.; Zhu, W.; Stolt, C.C.; Huang, T.-W.; Chen, F.; LoTurco, J.J.; Neul, J.L.; Wegner, M.; Mohila, C.; Deneen, B. Mutual antagonism between Sox10 and NFIA regulates diversification of glial lineages and glioma subtypes. Nat. Neurosci. 2014, 17, 1322–1329. [Google Scholar] [CrossRef]
- Ligon, K.L.; Huillard, E.; Mehta, S.; Kesari, S.; Liu, H.; Alberta, J.A.; Bachoo, R.M.; Kane, M.; Louis, D.N.; Depinho, R.A.; et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron 2007, 53, 503–517. [Google Scholar] [CrossRef]
- Burnet, F.M. The Clonal Selection Theory of Acquired Immunity; Vanderbilt University Press: Nashville, TN, USA, 1959; p. 232. Available online: https://www.biodiversitylibrary.org/item/34425 (accessed on 17 December 2022).
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The brain tumor microenvironment. Glia 2012, 60, 502–514. [Google Scholar] [CrossRef]
- Engler, J.R.; Robinson, A.E.; Smirnov, I.; Hodgson, J.G.; Berger, M.S.; Gupta, N.; James, C.D.; Molinaro, A.; Phillips, J.J. Increased Microglia/Macrophage Gene Expression in a Subset of Adult and Pediatric Astrocytomas. PLoS ONE 2012, 7, e43339. [Google Scholar] [CrossRef]
- Bowman, R.L.; Joyce, J.A. Therapeutic targeting of tumor-associated macrophages and microglia in glioblastoma. Immunotherapy 2014, 6, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef]
- Müller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef] [PubMed]
- Mysore, V.; Tahir, S.; Furuhashi, K.; Arora, J.; Rosetti, F.; Cullere, X.; Yazbeck, P.; Sekulic, M.; Lemieux, M.E.; Raychaudhuri, S.; et al. Monocytes transition to macrophages within the inflamed vasculature via monocyte CCR2 and endothelial TNFR2. J. Exp. Med. 2022, 219, e20210562. [Google Scholar] [CrossRef]
- Trouplin, V.; Boucherit, N.; Gorvel, L.; Conti, F.; Mottola, G.; Ghigo, E. Bone marrow-derived macrophage production. J. Vis. Exp. 2013, 81, e50966. [Google Scholar] [CrossRef]
- Jung, Y.; Ahn, S.-H.; Park, H.; Park, S.H.; Choi, K.; Choi, C.; Kang, J.L.; Choi, Y.-H. MCP-1 and MIP-3α Secreted from Necrotic Cell-Treated Glioblastoma Cells Promote Migration/Infiltration of Microglia. Cell Physiol. Biochem. 2018, 48, 1332–1346. [Google Scholar] [CrossRef]
- Liu, X.; Liu, Y.; Qi, Y.; Huang, Y.; Hu, F.; Dong, F.; Shu, K.; Lei, T. Signal Pathways Involved in the Interaction Between Tumor-Associated Macrophages/TAMs and Glioblastoma Cells. Front. Oncol. 2022, 12, 822085. [Google Scholar] [CrossRef]
- Chen, Z.; Ross, J.L.; Hambardzumyan, D. Intravital 2-photon imaging reveals distinct morphology and infiltrative properties of glioblastoma-associated macrophages. Proc. Natl. Acad. Sci. USA 2019, 116, 14254–14259. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Cen, J.; Huang, Y.; Shen, H.; Yao, L.; Wang, Y.; Chen, Z. Matrix metalloproteinase-2 and -9 secreted by leukemic cells increase the permeability of blood-brain barrier by disrupting tight junction proteins. PLoS ONE 2011, 6, e20599. [Google Scholar] [CrossRef]
- Ishihara, H.; Kubota, H.; Lindberg, R.L.P.; Leppert, D.; Gloor, S.M.; Errede, M.; Virgintino, D.; Fontana, A.; Yonekawa, Y.; Frei, K. Endothelial cell barrier impairment induced by glioblastomas and transforming growth factor beta2 involves matrix metalloproteinases and tight junction proteins. J. Neuropathol. Exp. Neurol. 2008, 67, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.W.; Ludwig, T.; Tatenhorst, L.; Braune, S.; Oberleithner, H.; Senner, V.; Paulus, W. Glioblastoma cells release factors that disrupt blood-brain barrier features. Acta Neuropathol. 2004, 107, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Pinton, L.; Masetto, E.; Vettore, M.; Solito, S.; Magri, S.; D’Andolfi, M.; Del Bianco, P.; Lollo, G.; Benoit, J.-P.; Okada, H.; et al. The immune suppressive microenvironment of human gliomas depends on the accumulation of bone marrow-derived macrophages in the center of the lesion. J. Immunother. Cancer 2019, 7, 58. [Google Scholar] [CrossRef]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef]
- Li, Y.; Wang, W.; Yang, F.; Xu, Y.; Feng, C.; Zhao, Y. The regulatory roles of neutrophils in adaptive immunity. Cell Commun. Signal. 2019, 17, 147. [Google Scholar] [CrossRef]
- Liang, J.; Piao, Y.; Holmes, L.; Fuller, G.N.; Henry, V.; Tiao, N.; de Groot, J.F. Neutrophils Promote the Malignant Glioma Phenotype through S100A4. Clin. Cancer Res. 2014, 20, 187–198. [Google Scholar] [CrossRef]
- Karimi, E.; Yu, M.W.; Maritan, S.M.; Perus, L.J.M.; Rezanejad, M.; Sorin, M.; Dankner, M.; Fallah, P.; Doré, S.; Zuo, D.; et al. Single-cell spatial immune landscapes of primary and metastatic brain tumours. Nature 2023, 614, 555–563. [Google Scholar] [CrossRef]
- Friebel, E.; Kapolou, K.; Unger, S.; Núñez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642. [Google Scholar] [CrossRef]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.-P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e17. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.; Han, S.J.; Sughrue, M.E.; Tihan, T.; Parsa, A.T. Immune cell infiltrate differences in pilocytic astrocytoma and glioblastoma: Evidence of distinct immunological microenvironments that reflect tumor biology. J. Neurosurg. 2011, 115, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Zhao, Q.; Huang, L.; Zheng, Y.; Li, L.; He, Q.; Zhang, C.; Li, F.; Maimela, N.R.; Sun, Z.; et al. The R132H mutation in IDH1 promotes the recruitment of NK cells through CX3CL1/CX3CR1 chemotaxis and is correlated with a better prognosis in gliomas. Immunol. Cell Biol. 2019, 97, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Gielen, P.R.; Schulte, B.M.; Kers-Rebel, E.D.; Verrijp, K.; Bossman, S.A.J.F.H.; Ter Laan, M.; Wesseling, P.; Adema, G.J. Elevated levels of polymorphonuclear myeloid-derived suppressor cells in patients with glioblastoma highly express S100A8/9 and arginase and suppress T cell function. Neuro Oncol. 2016, 18, 1253–1264. [Google Scholar] [CrossRef]
- Richard, S.A. Explicating the Pivotal Pathogenic, Diagnostic, and Therapeutic Biomarker Potentials of Myeloid-Derived Suppressor Cells in Glioblastoma. Dis. Markers 2020, 2020, 8844313. [Google Scholar] [CrossRef]
- Halle, S.; Halle, O.; Förster, R. Mechanisms and Dynamics of T Cell-Mediated Cytotoxicity In Vivo. Trends Immunol. 2017, 38, 432–443. [Google Scholar] [CrossRef]
- Paladugu, M.; Thakur, A.; Lum, L.G.; Mittal, S.; Parajuli, P. Generation and immunologic functions of Th17 cells in malignant gliomas. Cancer Immunol. Immunother. 2013, 62, 75–86. [Google Scholar] [CrossRef]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef]
- Fecci, P.E.; Sweeney, A.E.; Grossi, P.M.; Nair, S.K.; Learn, C.A.; Mitchell, D.A.; Cui, X.; Cummings, T.J.; Bigner, D.D.; Gilboa, E.; et al. Systemic anti-CD25 monoclonal antibody administration safely enhances immunity in murine glioma without eliminating regulatory T cells. Clin. Cancer Res. 2006, 12 Pt 1, 4294–4305. [Google Scholar] [CrossRef]
- Woroniecka, K.I.; Rhodin, K.E.; Chongsathidkiet, P.; Keith, K.A.; Fecci, P.E. T-cell Dysfunction in Glioblastoma: Applying a New Framework. Clin. Cancer Res. 2018, 24, 3792–3802. [Google Scholar] [CrossRef]
- Han, S.; Ma, E.; Wang, X.; Yu, C.; Dong, T.; Zhan, W.; Wei, X.; Liang, G.; Feng, S. Rescuing defective tumor-infiltrating T-cell proliferation in glioblastoma patients. Oncol. Lett. 2016, 12, 2924–2929. [Google Scholar] [CrossRef]
- Herculano-Houzel, S. The glia/neuron ratio: How it varies uniformly across brain structures and species and what that means for brain physiology and evolution. Glia 2014, 62, 1377–1391. [Google Scholar] [CrossRef]
- Jäkel, S.; Dimou, L. Glial Cells and Their Function in the Adult Brain: A Journey through the History of Their Ablation. Front. Cell Neurosci. 2017, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Wolburg, H.; Noell, S.; Mack, A.; Wolburg-Buchholz, K.; Fallier-Becker, P. Brain endothelial cells and the glio-vascular complex. Cell Tissue Res. 2009, 335, 75–96. [Google Scholar] [CrossRef]
- Khakh, B.S.; Sofroniew, M.V. Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 2015, 18, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Bechmann, I.; Mor, G.; Nilsen, J.; Eliza, M.; Nitsch, R.; Naftolin, F. FasL (CD95L, Apo1L) is expressed in the normal rat and human brain: Evidence for the existence of an immunological brain barrier. Glia 1999, 27, 62–74. [Google Scholar] [CrossRef]
- Quintana, F.J. Astrocytes to the rescue! Glia limitans astrocytic endfeet control CNS inflammation. J. Clin. Investig. 2017, 127, 2897–2899. [Google Scholar] [CrossRef]
- Hickey, W.F. Basic principles of immunological surveillance of the normal central nervous system. Glia 2001, 36, 118–124. [Google Scholar] [CrossRef]
- Miller, R.H. Oligodendrocyte origins. Trends Neurosci. 1996, 19, 92–96. [Google Scholar] [CrossRef]
- Gorczynski, R.M.; Cattral, M.S.; Chen, Z.; Hu, J.; Lei, J.; Min, W.P.; Yu, G.; Ni, J. An immunoadhesin incorporating the molecule OX-2 is a potent immunosuppressant that prolongs allo- and xenograft survival. J. Immunol. 1999, 163, 1654–1660. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Engelhardt, B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 2012, 12, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Deisseroth, K.; Singla, S.; Toda, H.; Monje, M.; Palmer, T.D.; Malenka, R.C. Excitation-neurogenesis coupling in adult neural stem/progenitor cells. Neuron 2004, 42, 535–552. [Google Scholar] [CrossRef]
- Kougioumtzidou, E.; Shimizu, T.; Hamilton, N.B.; Tohyama, K.; Sprengel, R.; Monyer, H.; Attwell, D.; Richardson, W.D. Signalling through AMPA receptors on oligodendrocyte precursors promotes myelination by enhancing oligodendrocyte survival. Elife 2017, 6, e28080. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Q.; Haydar, T.F.; Bordey, A. Nonsynaptic GABA signaling in postnatal subventricular zone controls proliferation of GFAP-expressing progenitors. Nat. Neurosci. 2005, 8, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- LoTurco, J.J.; Owens, D.F.; Heath, M.J.; Davis, M.B.; Kriegstein, A.R. GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron 1995, 15, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Sadikot, A.F. Glutamate and regulation of proliferation in the developing mammalian telencephalon. Dev. Neurosci. 2004, 26, 218–228. [Google Scholar] [CrossRef] [PubMed]
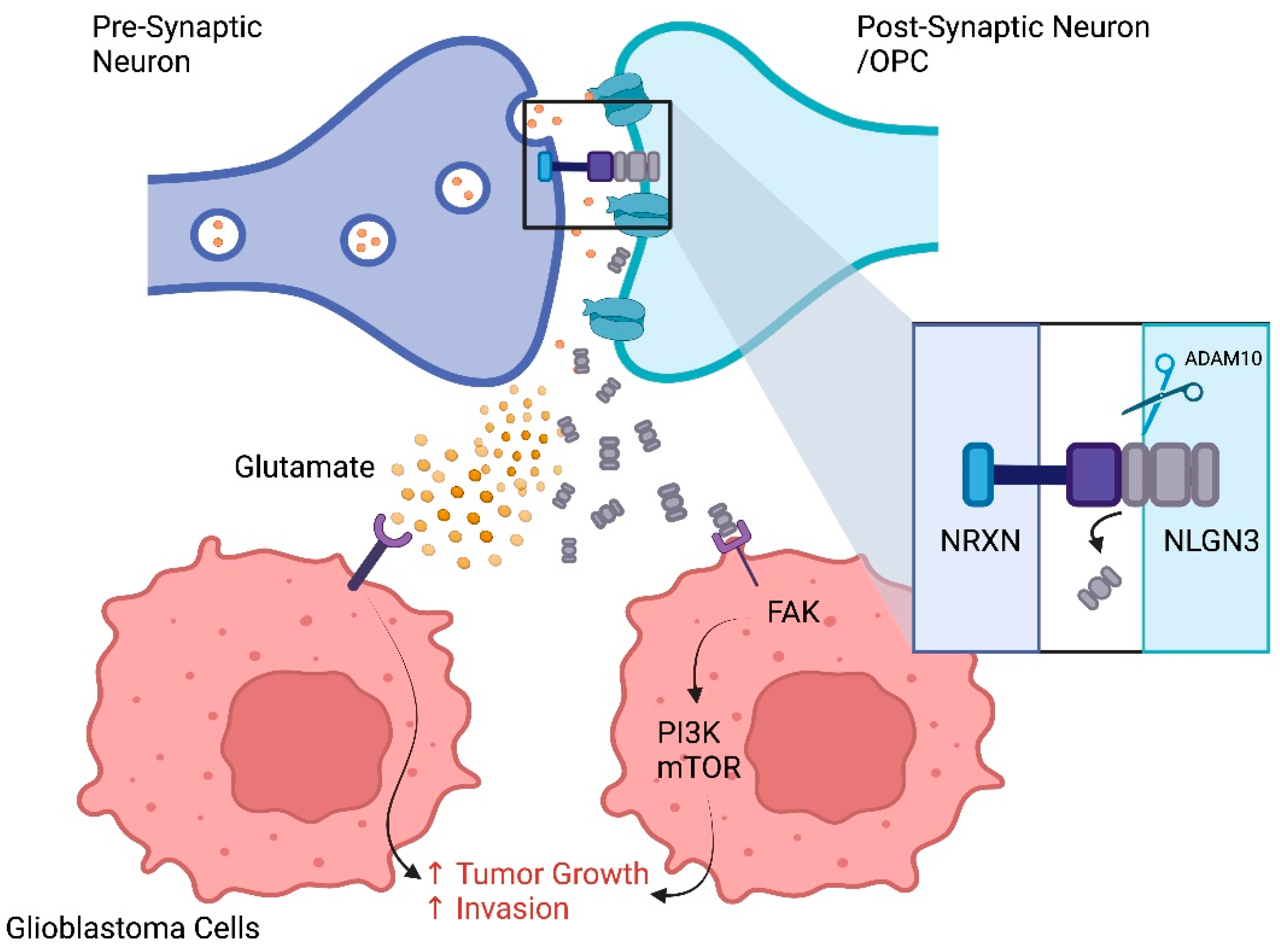
- Südhof, T.C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 2008, 455, 903–911. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Haddad-Tóvolli, R.; Dragano, N.R.V.; Ramalho, A.F.S.; Velloso, L.A. Development and Function of the Blood-Brain Barrier in the Context of Metabolic Control. Front. Neurosci. 2017, 11, 224. [Google Scholar] [CrossRef] [PubMed]
- Reese, T.S.; Karnovsky, M.J. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J. Cell Biol. 1967, 34, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Nico, B.; Crivellato, E.; Artico, M. Development of the blood-brain barrier: A historical point of view. Anat. Rec. B New Anat. 2006, 289, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, E.S.; Tan, A.C.; Anders, C.K.; Pendergast, A.M.; Sipkins, D.A.; Ashley, D.M.; Fecci, P.E.; Khasraw, M. Salting the Soil: Targeting the Microenvironment of Brain Metastases. Mol. Cancer Ther. 2021, 20, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, E.S.; Deshpande, K.; Neman, J.; Winkler, F.; Khasraw, M. The microenvironment of brain metastases from solid tumors. Neurooncol. Adv. 2021, 3 (Suppl. S5), v121–v132. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef]
- Banks, W.A. From blood-brain barrier to blood-brain interface: New opportunities for CNS drug delivery. Nat. Rev. Drug Discov. 2016, 15, 275–292. [Google Scholar] [CrossRef]
- Chow, B.W.; Gu, C. The molecular constituents of the blood-brain barrier. Trends Neurosci. 2015, 38, 598–608. [Google Scholar] [CrossRef]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef]
- Stefanik, D. Vascular Endothelial Growth Factor in Malignant Disease of the Central Nervous System. Madame Curie Bioscience Database; Landes Bioscience. 2013. Available online: https://www.ncbi.nlm.nih.gov/books/NBK6434/ (accessed on 27 January 2023).
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123 Pt 24, 4195–4200. [Google Scholar] [CrossRef] [PubMed]
- Faisal, S.M.; Comba, A.; Varela, M.L.; Argento, A.E.; Brumley, E.; Abel, C.; Castro, M.G.; Lowenstein, P.R. The complex interactions between the cellular and non-cellular components of the brain tumor microenvironmental landscape and their therapeutic implications. Front. Oncol. 2022, 12, 1005069. [Google Scholar] [CrossRef] [PubMed]
- Comba, A.; Faisal, S.M.; Dunn, P.J.; Argento, A.E.; Hollon, T.C.; Al-Holou, W.N.; Varela, M.L.; Zamler, D.B.; Quass, G.L.; Apostolides, P.F.; et al. Spatiotemporal analysis of glioma heterogeneity reveals COL1A1 as an actionable target to disrupt tumor progression. Nat. Commun. 2022, 13, 3606. [Google Scholar] [CrossRef] [PubMed]
- Huijbers, I.J.; Iravani, M.; Popov, S.; Robertson, D.; Al-Sarraj, S.; Jones, C.; Isacke, C.M. A role for fibrillar collagen deposition and the collagen internalization receptor endo180 in glioma invasion. PLoS ONE 2010, 5, e9808. [Google Scholar] [CrossRef] [PubMed]
- DeCordova, S.; Shastri, A.; Tsolaki, A.G.; Yasmin, H.; Klein, L.; Singh, S.K.; Kishore, U. Molecular Heterogeneity and Immunosuppressive Microenvironment in Glioblastoma. Front. Immunol. 2020, 1, 1402. [Google Scholar] [CrossRef] [PubMed]
- Hausmann, D.; Hoffmann, D.C.; Venkataramani, V.; Jung, E.; Horschitz, S.; Tetzlaff, S.K.; Jabali, A.; Hai, L.; Kessler, T.; Azoŕin, D.D.; et al. Autonomous rhythmic activity in glioma networks drives brain tumour growth. Nature 2023, 613, 179–186. [Google Scholar] [CrossRef]
- D’Alessandro, G.; Catalano, M.; Sciaccaluga, M.; Chece, G.; Cipriani, R.; Rosito, M.; Grimaldi, A.; Lauro, C.; Cantore, G.; Santoro, A.; et al. KCa3.1 channels are involved in the infiltrative behavior of glioblastoma in vivo. Cell Death Dis. 2013, 4, e773. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Eyler, C.E.; Foo, W.-C.; LaFiura, K.M.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. Brain cancer stem cells display preferential sensitivity to Akt inhibition. Stem Cells 2008, 26, 3027–3036. [Google Scholar] [CrossRef]
- Wang, X.; Prager, B.C.; Wu, Q.; Kim, L.J.Y.; Gimple, R.C.; Shi, Y.; Yang, K.; Morton, A.R.; Zhou, W.; Zhu, Z.; et al. Reciprocal Signaling between Glioblastoma Stem Cells and Differentiated Tumor Cells Promotes Malignant Progression. Cell Stem Cell 2018, 22, 514–528.e5. [Google Scholar] [CrossRef]
- Wu, A.; Wei, J.; Kong, L.-Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010, 12, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Akgül, S.; Patch, A.-M.; D’Souza, R.C.J.; Mukhopadhyay, P.; Nones, K.; Kempe, S.; Kazakoff, S.H.; Jeffree, R.L.; Stringer, B.W.; Pearson, J.V.; et al. Intratumoural Heterogeneity Underlies Distinct Therapy Responses and Treatment Resistance in Glioblastoma. Cancers 2019, 11, 190. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef]
- Dejaegher, J.; Solie, L.; Hunin, Z.; Sciot, R.; Capper, D.; Siewert, C.; Van Cauter, S.; Wilms, G.; van Loon, J.; Ectors, N.; et al. DNA methylation based glioblastoma subclassification is related to tumoral T-cell infiltration and patient survival. Neuro Oncol. 2021, 23, 240–250. [Google Scholar] [CrossRef]
- Klughammer, J.; Kiesel, B.; Roetzer, T.; Fortelny, N.; Nemc, A.; Nenning, K.-H.; Furtner, J.; Sheffield, N.C.; Datlinger, P.; Peter, N.; et al. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat. Med. 2018, 24, 1611–1624. [Google Scholar] [CrossRef]
- Li, B.; Li, T.; Pignon, J.-C.; Wang, B.; Wang, J.; Shukla, S.A.; Dou, R.; Chen, Q.; Hodi, F.S.; Choueiri, T.K.; et al. Landscape of tumor-infiltrating T cell repertoire of human cancers. Nat. Genet. 2016, 48, 725–732. [Google Scholar] [CrossRef]
- McGillicuddy, L.T.; Fromm, J.A.; Hollstein, P.E.; Kubek, S.; Beroukhim, R.; De Raedt, T.; Johnson, B.W.; Williams, S.M.G.; Nghiemphu, P.; Liau, L.M.; et al. Proteasomal and genetic inactivation of the NF1 tumor suppressor in gliomagenesis. Cancer Cell 2009, 16, 44–54. [Google Scholar] [CrossRef]
- Zhao, W.; Liu, M.; Kirkwood, K.L. p38α Stabilizes Interleukin-6 mRNA via Multiple AU-rich Elements. J. Biol. Chem. 2008, 283, 1778–1785. [Google Scholar] [CrossRef]
- Xiao, Y.Q.; Freire-de-Lima, C.G.; Schiemann, W.P.; Bratton, D.L.; Vandivier, R.W.; Henson, P.M. Transcriptional and Translational Regulation of Transforming Growth Factor-β Production in Response to Apoptotic Cells. J. Immunol. 2008, 181, 3575–3585. [Google Scholar] [CrossRef] [PubMed]
- Tomaszewski, W.; Sanchez-Perez, L.; Gajewski, T.F.; Sampson, J.H. Brain Tumor Microenvironment and Host State: Implications for Immunotherapy. Clin. Cancer Res. 2019, 25, 4202–4210. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Kim, J.E.; Patel, M.; Ruzevick, J.; Jackson, C.M.; Lim, M. STAT3 Activation in Glioblastoma: Biochemical and Therapeutic Implications. Cancers 2014, 6, 376–395. [Google Scholar] [CrossRef] [PubMed]
- Yeung, Y.T.; McDonald, K.L.; Grewal, T.; Munoz, L. Interleukins in glioblastoma pathophysiology: Implications for therapy. Br. J. Pharm. 2013, 168, 591–606. [Google Scholar] [CrossRef]
- De, I.; Steffen, M.D.; Clark, P.A.; Patros, C.J.; Sokn, E.; Bishop, S.M.; Litscher, S.; Maklakova, V.I.; Kuo, J.S.; Rodriguez, F.J.; et al. CSF1 Overexpression Promotes High-Grade Glioma Formation without Impacting the Polarization Status of Glioma-Associated Microglia and Macrophages. Cancer Res. 2016, 76, 2552–2560. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, M.D.; Dahlrot, R.H.; Boldt, H.B.; Hansen, S.; Kristensen, B.W. Tumour-associated microglia/macrophages predict poor prognosis in high-grade gliomas and correlate with an aggressive tumour subtype. Neuropathol. Appl. Neurobiol. 2018, 44, 185–206. [Google Scholar] [CrossRef]
- Yan, D.; Kowal, J.; Akkari, L.; Schuhmacher, A.J.; Huse, J.T.; West, B.L.; Joyce, J.A. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene 2017, 36, 6049–6058. [Google Scholar] [CrossRef] [PubMed]
- Kohanbash, G.; Carrera, D.A.; Shrivastav, S.; Ahn, B.J.; Jahan, N.; Mazor, T.; Chheda, Z.S.; Downey, K.M.; Watchmaker, P.B.; Beppler, C.; et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J. Clin. Investig. 2017, 127, 1425–1437. [Google Scholar] [CrossRef]
- Bunse, L.; Pusch, S.; Bunse, T.; Sahm, F.; Sanghvi, K.; Friedrich, M.; Alansary, D.; Sonner, J.K.; Green, E.; Deumelandt, K.; et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat. Med. 2018, 24, 1192–1203. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Sankowski, R.; Bunse, L.; Kilian, M.; Green, E.; Ramallo Guevara, C.; Pusch, S.; Poschet, G.; Sanghvi, K.; Hahn, M.; et al. Tryptophan metabolism drives dynamic immunosuppressive myeloid states in IDH-mutant gliomas. Nat. Cancer 2021, 2, 723–740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Rao, A.; Sette, P.; Deibert, C.; Pomerantz, A.; Kim, W.J.; Kohanbash, G.; Chang, Y.; Park, Y.; Engh, J.; et al. IDH mutant gliomas escape natural killer cell immune surveillance by downregulation of NKG2D ligand expression. Neuro Oncol. 2016, 18, 1402–1412. [Google Scholar] [CrossRef] [PubMed]
- Amankulor, N.M.; Kim, Y.; Arora, S.; Kargl, J.; Szulzewsky, F.; Hanke, M.; Margineantu, D.H.; Rao, A.; Bolouri, H.; Delrow, J.; et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017, 31, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef]
- Vidyarthi, A.; Agnihotri, T.; Khan, N.; Singh, S.; Tewari, M.K.; Radotra, B.D.; Chatterjee, D.; Agrewala, J.N. Predominance of M2 macrophages in gliomas leads to the suppression of local and systemic immunity. Cancer Immunol. Immunother. 2019, 68, 1995–2004. [Google Scholar] [CrossRef]
- Richard, Q.; Laurenge, A.; Mallat, M.; Sanson, M.; Castro-Vega, L.J. New insights into the Immune TME of adult-type diffuse gliomas. Curr. Opin. Neurol. 2022, 35, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Mu, L.; Long, Y.; Yang, C.; Jin, L.; Tao, H.; Ge, H.; Chang, Y.E.; Karachi, A.; Kubilis, P.S.; De Leon, G.; et al. The IDH1 Mutation-Induced Oncometabolite, 2-Hydroxyglutarate, May Affect DNA Methylation and Expression of PD-L1 in Gliomas. Front. Mol. Neurosci. 2018, 11, 82. [Google Scholar] [CrossRef]
- Mehani, B.; Asanigari, S.; Chung, H.-J.; Dazelle, K.; Singh, A.; Hannenhalli, S.; Aldape, K. Immune cell gene expression signatures in diffuse glioma are associated with IDH mutation status, patient outcome and malignant cell state, and highlight the importance of specific cell subsets in glioma biology. Acta Neuropathol. Commun. 2022, 10, 19. [Google Scholar] [CrossRef]
- Di Tacchio, M.; Macas, J.; Weissenberger, J.; Sommer, K.; Bähr, O.; Steinbach, J.P.; Senft, C.; Seifert, V.; Glas, M.; Herrlinger, U.; et al. Tumor Vessel Normalization, Immunostimulatory Reprogramming, and Improved Survival in Glioblastoma with Combined Inhibition of PD-1, Angiopoietin-2, and VEGF. Cancer Immunol. Res. 2019, 7, 1910–1927. [Google Scholar] [CrossRef]
- Woodworth, G.F.; Dunn, G.P.; Nance, E.A.; Hanes, J.; Brem, H. Emerging insights into barriers to effective brain tumor therapeutics. Front. Oncol. 2014, 4, 126. [Google Scholar] [CrossRef] [PubMed]
- Brandao, M.; Simon, T.; Critchley, G.; Giamas, G. Astrocytes, the rising stars of the glioblastoma microenvironment. Glia 2019, 67, 779–790. [Google Scholar] [CrossRef]
- Henrik Heiland, D.; Ravi, V.M.; Behringer, S.P.; Frenking, J.H.; Wurm, J.; Joseph, K.; Garrelfs, N.W.C.; Strähle, J.; Heynckes, S.; Grauvogel, J.; et al. Tumor-associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat. Commun. 2019, 10, 2541. [Google Scholar] [CrossRef]
- Errede, M.; Annese, T.; Petrosino, V.; Longo, G.; Girolamo, F.; de Trizio, I.; d’Amati, A.; Uccelli, A.; Kerlero de Rosbo, N.; Virgintino, D. Microglia-derived CCL2 has a prime role in neocortex neuroinflammation. Fluids Barriers CNS 2022, 19, 68. [Google Scholar] [CrossRef] [PubMed]
- Kremlev, S.G.; Roberts, R.L.; Palmer, C. Differential expression of chemokines and chemokine receptors during microglial activation and inhibition. J. Neuroimmunol. 2004, 149, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Verreck, F.A.W.; de Boer, T.; Langenberg, D.M.L.; Hoeve, M.A.; Kramer, M.; Vaisberg, E.; Kastelein, R.; Kolk, A.; de Waal-Malefyt, R.; Ottenhoff, T.H.M. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc. Natl. Acad. Sci. USA 2004, 101, 4560–4565. [Google Scholar] [CrossRef] [PubMed]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef]
- Ravi, V.M.; Neidert, N.; Will, P.; Joseph, K.; Maier, J.P.; Kückelhaus, J.; Vollmer, L.; Goeldner, J.M.; Behringer, S.P.; Scherer, F.; et al. T-cell dysfunction in the glioblastoma microenvironment is mediated by myeloid cells releasing interleukin-10. Nat. Commun. 2022, 13, 925. [Google Scholar] [CrossRef]
- Roberts, A.W.; Lee, B.L.; Deguine, J.; John, S.; Shlomchik, M.J.; Barton, G.M. Tissue-Resident Macrophages Are Locally Programmed for Silent Clearance of Apoptotic Cells. Immunity 2017, 47, 913–927.e6. [Google Scholar] [CrossRef]
- da Cruz, L.L.P.; de Souza, P.O.; Dal Pra, M.; Falchetti, M.; de Abreu, A.M.; Azambuja, J.H.; Bertoni, A.P.S.; Paz, A.H.R.; Araujo, A.B.; Visioli, F.; et al. TLR4 expression and functionality are downregulated in glioblastoma cells and in tumor-associated macrophages: A new mechanism of immune evasion? Biochim. Biophys. Acta-Mol. Basis Dis. 2021, 1867, 166155. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yan, Y.; Liu, J.; Huang, X.; Zhang, X.; Kirschning, C.; Xu, H.C.; Lang, P.A.; Dittmer, U.; Zhang, E.; et al. Toll-Like Receptor 7 Activation Enhances CD8+ T Cell Effector Functions by Promoting Cellular Glycolysis. Front. Immunol. 2019, 10, 2191. [Google Scholar] [CrossRef]
- Nouri, Y.; Weinkove, R.; Perret, R. T-cell intrinsic Toll-like receptor signaling: Implications for cancer immunotherapy and CAR T-cells. J. Immunother. Cancer 2021, 9, e003065. [Google Scholar] [CrossRef]
- Quigley, M.; Martinez, J.; Huang, X.; Yang, Y. A critical role for direct TLR2-MyD88 signaling in CD8 T-cell clonal expansion and memory formation following vaccinia viral infection. Blood 2009, 113, 2256–2264. [Google Scholar] [CrossRef] [PubMed]
- Aki, M.; Shimbara, N.; Takashina, M.; Akiyama, K.; Kagawa, S.; Tamura, T.; Tanahashi, N.; Yoshimura, T.; Tanaka, K.; Ichihara, A. Interferon-gamma induces different subunit organizations and functional diversity of proteasomes. J. Biochem. 1994, 115, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Kloetzel, P.-M. Antigen processing by the proteasome. Nat. Rev. Mol. Cell Biol. 2001, 2, 179–188. [Google Scholar] [CrossRef]
- Miyakoshi, J.; Dobler, K.D.; Allalunis-Turner, J.; McKean, J.D.; Petruk, K.; Allen, P.B.; Aronyk, K.N.; Weir, B.; Huyser-Wierenga, D.; Fulton, D. Absence of IFNA and IFNB genes from human malignant glioma cell lines and lack of correlation with cellular sensitivity to interferons. Cancer Res. 1990, 50, 278–283. [Google Scholar]
- Parmigiani, E.; Ivanek, R.; Rolando, C.; Hafen, K.; Turchinovich, G.; Lehmann, F.M.; Gerber, A.; Brkic, S.; Frank, S.; Meyer, S.C.; et al. Interferon-γ resistance and immune evasion in glioma develop via Notch-regulated co-evolution of malignant and immune cells. Dev. Cell 2022, 57, 1847–1865.e9. [Google Scholar] [CrossRef]
- Schartner, J.M.; Hagar, A.R.; Van Handel, M.; Zhang, L.; Nadkarni, N.; Badie, B. Impaired capacity for upregulation of MHC class II in tumor-associated microglia. Glia 2005, 51, 279–285. [Google Scholar] [CrossRef]
- Kumar, V.; Cheng, P.; Condamine, T.; Mony, S.; Languino, L.R.; McCaffrey, J.C.; Hockstein, N.; Guarino, M.; Masters, G.; Penman, E.; et al. CD45 Phosphatase Inhibits STAT3 Transcription Factor Activity in Myeloid Cells and Promotes Tumor-Associated Macrophage Differentiation. Immunity 2016, 44, 303–315. [Google Scholar] [CrossRef]
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J. Immunol. 2009, 182, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Ugolini, A.; Tyurin, V.A.; Tyurina, Y.Y.; Tcyganov, E.N.; Donthireddy, L.; Kagan, V.E.; Gabrilovich, D.I.; Veglia, F. Polymorphonuclear myeloid-derived suppressor cells limit antigen cross-presentation by dendritic cells in cancer. JCI Insight 2020, 5, e138581. [Google Scholar] [CrossRef] [PubMed]
- Raber, P.L.; Thevenot, P.; Sierra, R.; Wyczechowska, D.; Halle, D.; Ramirez, M.E.; Ochoa, A.C.; Fletcher, M.; Velasco, C.; Wilk, A.; et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int. J. Cancer 2014, 134, 2853–2864. [Google Scholar] [CrossRef] [PubMed]
- Hart, K.M.; Byrne, K.T.; Molloy, M.J.; Usherwood, E.M.; Berwin, B. IL-10 immunomodulation of myeloid cells regulates a murine model of ovarian cancer. Front. Immunol. 2011, 2, 29. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef]
- Dubinski, D.; Wölfer, J.; Hasselblatt, M.; Schneider-Hohendorf, T.; Bogdahn, U.; Stummer, W.; Wiendl, H.; Grauer, O.M. CD4+ T effector memory cell dysfunction is associated with the accumulation of granulocytic myeloid-derived suppressor cells in glioblastoma patients. Neuro Oncol. 2016, 18, 807–818. [Google Scholar] [CrossRef]
- Huang, B.; Pan, P.-Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.-H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegué, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef]
- Tanriover, G.; Aytac, G. Mutualistic Effects of the Myeloid-Derived Suppressor Cells and Cancer Stem Cells in the Tumor Microenvironment. Crit Rev. Oncog. 2019, 24, 61–67. [Google Scholar] [CrossRef]
- Basheer, A.S.; Abas, F.; Othman, I.; Naidu, R. Role of Inflammatory Mediators, Macrophages, and Neutrophils in Glioma Maintenance and Progression: Mechanistic Understanding and Potential Therapeutic Applications. Cancers 2021, 13, 4226. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Mittal, S.; McGee, K.; Alfaro-Munoz, K.D.; Majd, N.; Balasubramaniyan, V.; de Groot, J.F. Role of Neutrophils and Myeloid-Derived Suppressor Cells in Glioma Progression and Treatment Resistance. Int. J. Mol. Sci. 2020, 21, 1954. [Google Scholar] [CrossRef] [PubMed]
- Magod, P.; Mastandrea, I.; Rousso-Noori, L.; Agemy, L.; Shapira, G.; Shomron, N.; Friedmann-Morvinski, D. Exploring the longitudinal glioma microenvironment landscape uncovers reprogrammed pro-tumorigenic neutrophils in the bone marrow. Cell Rep. 2021, 36, 109480. [Google Scholar] [CrossRef]
- Chen, Z.; Guo, C.; Chen, J. CTNI-33. CSNO2012001 Study: A Phase Iii Trial on Adjuvant Temozolomide Chemotherapy with or without Interferon-Alpha in Newly Diagnosed High-Grade Gliomas. Neuro Oncol. 2022, 24 (Suppl. 7), vii78. [Google Scholar] [CrossRef]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 2014, 26, 638–652. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Dendritic cells: A link between innate and adaptive immunity. J. Clin. Immunol. 1999, 19, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef]
- Castriconi, R.; Daga, A.; Dondero, A.; Zona, G.; Poliani, P.L.; Melotti, A.; Griffero, F.; Marubbi, D.; Spaziante, R.; Bellora, F.; et al. NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J. Immunol. 2009, 182, 3530–3539. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef]
- Gagliano, N.; Costa, F.; Cossetti, C.; Pettinari, L.; Bassi, R.; Chiriva-Internati, M.; Cobos, E.; Gioia, M.; Pluchino, S. Glioma-astrocyte interaction modifies the astrocyte phenotype in a co-culture experimental model. Oncol. Rep. 2009, 22, 1349–1356. [Google Scholar] [CrossRef]
- Barbero, S.; Bajetto, A.; Bonavia, R.; Porcile, C.; Piccioli, P.; Pirani, P.; Ravetti, J.L.; Zona, G.; Spaziante, R.; Florio, T.; et al. Expression of the Chemokine Receptor CXCR4 and Its Ligand Stromal Cell-Derived Factor 1 in Human Brain Tumors and Their Involvement in Glial Proliferation in Vitro. Ann. N. Y. Acad. Sci. 2002, 973, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Hasan, M.N.; Maniar, S.; Jia, W.; Sun, D. Reactive Astrocytes in Glioblastoma Multiforme. Mol. Neurobiol. 2018, 55, 6927–6938. [Google Scholar] [CrossRef] [PubMed]
- Le, D.M.; Besson, A.; Fogg, D.K.; Choi, K.-S.; Waisman, D.M.; Goodyer, C.G.; Rewcastle, B.; Yong, V.W. Exploitation of Astrocytes by Glioma Cells to Facilitate Invasiveness: A Mechanism Involving Matrix Metalloproteinase-2 and the Urokinase-Type Plasminogen Activator–Plasmin Cascade. J. Neurosci. 2003, 23, 4034–4043. [Google Scholar] [CrossRef]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Kim, J.-S.; Park, E.S.; Lee, J.-S.; Lin, Q.; Langley, R.R.; Maya, M.; He, J.; Kim, S.-W.; Weihua, Z.; et al. Astrocytes upregulate survival genes in tumor cells and induce protection from chemotherapy. Neoplasia 2011, 13, 286–298. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, S.; Yao, J.; Lowery, F.J.; Zhang, Q.; Huang, W.-C.; Li, P.; Li, M.; Wang, X.; Zhang, C.; et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 2015, 527, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Barthel, F.P.; Johnson, K.C.; Varn, F.S.; Moskalik, A.D.; Tanner, G.; Kocakavuk, E.; Anderson, K.J.; Abiola, O.; Aldape, K.; Alfaro, K.D.; et al. Longitudinal Molecular Trajectories of Diffuse Glioma in Adults. Nature 2019, 576, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Sage, J.C.; Miller, M.R.; Verhaak, R.G.W.; Hippenmeyer, S.; Vogel, H.; Foreman, O.; Bronson, R.T.; Nishiyama, A.; Luo, L.; et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 2011, 146, 209–221. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Tam, L.T.; Woo, P.J.; Lennon, J.; Nagaraja, S.; Gillespie, S.M.; Ni, J.; Duveau, D.Y.; Morris, P.J.; Zhao, J.J.; et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 2017, 549, 533–537. [Google Scholar] [CrossRef]
- Ishiuchi, S.; Yoshida, Y.; Sugawara, K.; Aihara, M.; Ohtani, T.; Watanabe, T.; Saito, N.; Tsuzuki, K.; Okado, H.; Miwa, A.; et al. Ca2+-permeable AMPA receptors regulate growth of human glioblastoma via Akt activation. J. Neurosci. 2007, 27, 7987–8001. [Google Scholar] [CrossRef] [PubMed]
- Sontheimer, H. A role for glutamate in growth and invasion of primary brain tumors. J. Neurochem. 2008, 105, 287–295. [Google Scholar] [CrossRef]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef]
- Gibson, E.M.; Purger, D.; Mount, C.W.; Goldstein, A.K.; Lin, G.L.; Wood, L.S.; Inema, I.; Miller, S.E.; Bieri, G.; Zuchero, J.B.; et al. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 2014, 344, 1252304. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Casazza, A.; Laoui, D.; Wenes, M.; Rizzolio, S.; Bassani, N.; Mambretti, M.; Deschoemaeker, S.; Van Ginderachter, J.A.; Tamagnone, L.; Mazzone, M. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 2013, 24, 695–709. [Google Scholar] [CrossRef]
- Charles, N.; Ozawa, T.; Squatrito, M.; Bleau, A.-M.; Brennan, C.W.; Hambardzumyan, D.; Holland, E.C. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell 2010, 6, 141–152. [Google Scholar] [CrossRef]
- Mazanet, M.M.; Hughes, C.C.W. B7-H1 is expressed by human endothelial cells and suppresses T cell cytokine synthesis. J. Immunol. 2002, 169, 3581–3588. [Google Scholar] [CrossRef] [PubMed]
- Pittet, C.L.; Newcombe, J.; Prat, A.; Arbour, N. Human brain endothelial cells endeavor to immunoregulate CD8 T cells via PD-1 ligand expression in multiple sclerosis. J. Neuroinflammation 2011, 8, 155. [Google Scholar] [CrossRef]
- Bellail, A.C.; Hunter, S.B.; Brat, D.J.; Tan, C.; Van Meir, E.G. Microregional extracellular matrix heterogeneity in brain modulates glioma cell invasion. Int. J. Biochem. Cell Biol. 2004, 36, 1046–1069. [Google Scholar] [CrossRef] [PubMed]
- Tamai, S.; Ichinose, T.; Tsutsui, T.; Tanaka, S.; Garaeva, F.; Sabit, H.; Nakada, M. Tumor Microenvironment in Glioma Invasion. Brain Sci 2022, 12, 505. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.F.; Carter, T.J.; Ottaviani, D.; Mulholland, P. Harnessing the immune system in glioblastoma. Br. J. Cancer 2018, 119, 1171–1181. [Google Scholar] [CrossRef]
- Lorusso, G.; Rüegg, C.; Kuonen, F. Targeting the Extra-Cellular Matrix-Tumor Cell Crosstalk for Anti-Cancer Therapy: Emerging Alternatives to Integrin Inhibitors. Front. Oncol. 2020, 10, 1231. [Google Scholar] [CrossRef] [PubMed]
- Laklai, H.; Miroshnikova, Y.A.; Pickup, M.W.; Collisson, E.A.; Kim, G.E.; Barrett, A.S.; Hill, R.C.; Lakins, J.N.; Schlaepfer, D.D.; Mouw, J.K.; et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat. Med. 2016, 22, 497–505. [Google Scholar] [CrossRef] [PubMed]
- The Fibronectin Expression Determines the Distinct Progressions of Malignant Gliomas via Transforming Growth Factor-Beta Pathway—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/33917452/ (accessed on 30 March 2023).
- Yu, Q.; Xue, Y.; Liu, J.; Xi, Z.; Li, Z.; Liu, Y. Fibronectin Promotes the Malignancy of Glioma Stem-Like Cells Via Modulation of Cell Adhesion, Differentiation, Proliferation and Chemoresistance—PMC. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5908975/ (accessed on 29 March 2023).
- Weathington, N.M.; van Houwelingen, A.H.; Noerager, B.D.; Jackson, P.L.; Kraneveld, A.D.; Galin, F.S.; Folkerts, G.; Nijkamp, F.P.; Blalock, J.E. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat. Med 2006, 12, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Houghton, A.M.; Quintero, P.A.; Perkins, D.L.; Kobayashi, D.K.; Kelley, D.G.; Marconcini, L.A.; Mecham, R.P.; Senior, R.M.; Shapiro, S.D. Elastin fragments drive disease progression in a murine model of emphysema. J. Clin. Investig. 2006, 116, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Rygiel, T.P.; Stolte, E.H.; de Ruiter, T.; van de Weijer, M.L.; Meyaard, L. Tumor-expressed collagens can modulate immune cell function through the inhibitory collagen receptor LAIR-1. Mol. Immunol 2011, 49, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Jürgensen, H.J.; van Putten, S.; Nørregaard, K.S.; Bugge, T.H.; Engelholm, L.H.; Behrendt, N.; Madsen, D.H. Cellular uptake of collagens and implications for immune cell regulation in disease. Cell Mol. Life Sci. 2020, 77, 3161–3176. [Google Scholar] [CrossRef]
- Mohiuddin, E.; Wakimoto, H. Extracellular matrix in glioblastoma: Opportunities for emerging therapeutic approaches. Am. J. Cancer Res. 2021, 11, 3742–3754. [Google Scholar]
- Lee, A.; Arasaratnam, M.; Chan, D.L.H.; Khasraw, M.; Howell, V.M.; Wheeler, H. Anti-epidermal growth factor receptor therapy for glioblastoma in adults. Cochrane Database Syst. Rev. 2020. [Google Scholar] [CrossRef]
- Wen, P.Y.; Touat, M.; Alexander, B.M.; Mellinghoff, I.K.; Ramkissoon, S.; McCluskey, C.S.; Pelton, K.; Haidar, S.; Basu, S.S.; Gaffey, S.C.; et al. Buparlisib in Patients With Recurrent Glioblastoma Harboring Phosphatidylinositol 3-Kinase Pathway Activation: An Open-Label, Multicenter, Multi-Arm, Phase II Trial. J. Clin. Oncol. 2019, 37, 741–750. [Google Scholar] [CrossRef]
- Chang, S.M.; Wen, P.; Cloughesy, T.; Greenberg, H.; Schiff, D.; Conrad, C.; Fink, K.; Robins, H.I.; De Angelis, L.; Raizer, J.; et al. Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Investig. New Drugs 2005, 23, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Priego, N.; Zhu, L.; Monteiro, C.; Mulders, M.; Wasilewski, D.; Bindeman, W.; Doglio, L.; Martínez, L.; Martínez-Saez, E.; Ramón Y Cajal, S.; et al. STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat. Med. 2018, 24, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Wacquier, B.; Voorsluijs, V.; Combettes, L.; Dupont, G. Coding and decoding of oscillatory Ca2+ signals. Semin. Cell Dev. Biol. 2019, 94, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.; Burris, H.A.; Lewis, N.; Donehower, R.; Redman, J.; Friedman, S.; Scherle, P.; Fridman, J.; Li, J.; Emm, T. A multicenter phase Ib study of the safety, pharmacokinetics, biological activity and clinical efficacy of INCB7839, a potent and selective inhibitor of ADAM10 and ADAM17. In Breast Cancer Research and Treatment; Springer: New York, NY, USA, 2007; p. S269. [Google Scholar]
- Friedman, S.; Levy, R.; Garrett, W.; Doval, D.; Bondarde, S.; Sahoo, T.; Lokanatha, D.; Julka, P.; Shenoy, K.; Nagarkar, R. Clinical Benefit of INCB7839, a Potent and Selective Inhibitor of ADAM10 and ADAM17, in Combination with Trastuzumab in Metastatic HER2 Positive Breast Cancer Patients. Cancer Res. 2009, 69 (Suppl. S24), 5056. [Google Scholar] [CrossRef]
- Borghaei, H.; Gettinger, S.; Vokes, E.E.; Chow, L.Q.M.; Burgio, M.A.; de Castro Carpeno, J.; Pluzanski, A.; Arrieta, O.; Frontera, O.A.; Chiari, R.; et al. Five-Year Outcomes From the Randomized, Phase III Trials CheckMate 017 and 057: Nivolumab Versus Docetaxel in Previously Treated Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2021, 39, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Hellmann, M.D.; Paz-Ares, L.G.; Ramalingam, S.S.; Reck, M.; O’Byrne, K.J.; Bhagavatheeswaran, P.; Nathan, F.E.; Brahmer, J.R. Nivolumab (Nivo) + platinum-doublet chemotherapy (Chemo) vs chemo as first-line (1L) treatment (Tx) for advanced non-small cell lung cancer (NSCLC) with <1% tumor PD-L1 expression: Results from CheckMate 227. JCO 2018, 36, 9001. [Google Scholar] [CrossRef]
- Sim, H.-W.; Lwin, Z.; Barnes, E.; McDonald, K.; Yip, S.; Verhaak, R.; Heimberger, A.; Hall, M.; Wong, M.; Jennens, R.; et al. CTIM-24. Nutmeg: A Randomized Phase Ii Study of Nivolumab and Temozolomide Versus Temozolomide Alone in Newly Diagnosed Elderly Patients with Glioblastoma. Neuro Oncol. 2022, 24, vii65. [Google Scholar] [CrossRef]
- Stafford, J.H.; Hirai, T.; Deng, L.; Chernikova, S.B.; Urata, K.; West, B.L.; Brown, J.M. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro Oncol. 2016, 18, 797–806. [Google Scholar] [CrossRef]
- Falchook, G.S.; Peeters, M.; Rottey, S.; Dirix, L.Y.; Obermannova, R.; Cohen, J.E.; Perets, R.; Frommer, R.S.; Bauer, T.M.; Wang, J.S.; et al. A phase 1a/1b trial of CSF-1R inhibitor LY3022855 in combination with durvalumab or tremelimumab in patients with advanced solid tumors. Investig. New Drugs 2021, 39, 1284–1297. [Google Scholar] [CrossRef]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol. 2016, 18, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Amulic, B.; Aziz, M.; Barnes, B.J.; Eruslanov, E.; Fridlender, Z.G.; Goodridge, H.S.; Granot, Z.; Hidalgo, A.; Huttenlocher, A.; et al. Neutrophil phenotypes and functions in cancer: A consensus statement. J. Exp. Med. 2022, 219, e20220011. [Google Scholar] [CrossRef] [PubMed]
- Batich, K.A.; Mitchell, D.A.; Healy, P.; Herndon, J.E., 2nd; Sampson, J.H. Once, Twice, Three Times a Finding: Reproducibility of Dendritic Cell Vaccine Trials Targeting Cytomegalovirus in Glioblastoma. Clin. Cancer Res. 2020, 26, 5297–5303. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.A.; Batich, K.A.; Gunn, M.D.; Huang, M.-N.; Sanchez-Perez, L.; Nair, S.K.; Congdon, K.L.; Reap, E.A.; Archer, G.E.; Desjardins, A.; et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 2015, 519, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Ashkan, K.; Brem, S.; Campian, J.L.; Trusheim, J.E.; Iwamoto, F.M.; Tran, D.D.; Ansstas, G.; Cobbs, C.S.; Heth, J.A.; et al. Association of Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination With Extension of Survival Among Patients With Newly Diagnosed and Recurrent Glioblastoma: A Phase 3 Prospective Externally Controlled Cohort Trial. JAMA Oncol. 2023, 9, 112–121. [Google Scholar] [CrossRef]
- Preusser, M.; van den Bent, M.J. Autologous tumor lysate-loaded dendritic cell vaccination (DCVax-L) in glioblastoma: Breakthrough or fata morgana? Neuro Oncol. 2023, 25, 631–634. [Google Scholar] [CrossRef]
- Sheykhhasan, M.; Manoochehri, H.; Dama, P. Use of CAR T-cell for acute lymphoblastic leukemia (ALL) treatment: A review study. Cancer Gene 2022, 29, 1080–1096. [Google Scholar] [CrossRef]
- Ma, R.; Lu, T.; Li, Z.; Teng, K.-Y.; Mansour, A.G.; Yu, M.; Tian, L.; Xu, B.; Ma, S.; Zhang, J.; et al. An oncolytic virus expressing IL-15/IL-15Rα combined with off-the-shelf EGFR-CAR NK cells targets glioblastoma. Cancer Res. 2021, 81, 3635–3648. [Google Scholar] [CrossRef]
- Hosseinalizadeh, H.; Habibi Roudkenar, M.; Mohammadi Roushandeh, A.; Kuwahara, Y.; Tomita, K.; Sato, T. Natural killer cell immunotherapy in glioblastoma. Discov. Oncol. 2022, 13, 113. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.-C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Rα2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuile, A.; Wei, J.Q.; Mohan, A.A.; Hotchkiss, K.M.; Khasraw, M. Interdependencies of the Neuronal, Immune and Tumor Microenvironment in Gliomas. Cancers 2023, 15, 2856. https://doi.org/10.3390/cancers15102856
Yuile A, Wei JQ, Mohan AA, Hotchkiss KM, Khasraw M. Interdependencies of the Neuronal, Immune and Tumor Microenvironment in Gliomas. Cancers. 2023; 15(10):2856. https://doi.org/10.3390/cancers15102856
Chicago/Turabian StyleYuile, Alexander, Joe Q. Wei, Aditya A. Mohan, Kelly M. Hotchkiss, and Mustafa Khasraw. 2023. "Interdependencies of the Neuronal, Immune and Tumor Microenvironment in Gliomas" Cancers 15, no. 10: 2856. https://doi.org/10.3390/cancers15102856
APA StyleYuile, A., Wei, J. Q., Mohan, A. A., Hotchkiss, K. M., & Khasraw, M. (2023). Interdependencies of the Neuronal, Immune and Tumor Microenvironment in Gliomas. Cancers, 15(10), 2856. https://doi.org/10.3390/cancers15102856