
Optical Genome Mapping for Comprehensive Assessment of Chromosomal Aberrations and Discovery of New Fusion Genes in Pediatric B-Acute Lymphoblastic Leukemia

 , , and
, , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. UHMW DNA Isolation, Quantification and Labeling for Optical Genome Mapping
2.3. Structural Variant Calling and Variant Filtering
2.4. Comparison of Clinically Significant SVs/CNVs Identified by Conventional Testing
2.5. Confirmation of Additional SVs with Whole-Genome Sequencing
2.6. Verification of LMNB1::PPP2R2B and TMEM272::KDM4B Putative Fusion Genes
2.7. Statistics
3. Results
3.1. Clinical Characteristics of Patients and Technical Characteristics of OGM
3.1.1. Patient Characteristics
3.1.2. Raw Data Quality and SV/CNV Callings in OGM
3.2. Concordance between OGM and Conventional Cytogenetic Results
3.2.1. OGM Reaches 100% True Positive Rate for Known Aberrations, except for Specific Gene Regions
3.2.2. Refinement of Abnormal Karyotypes and Resolution of Complex Genome by OGM
3.2.3. Identification of Novel Chromosomal Alterations or Gene Fusions by OGM
3.3. Clinical Values of OGM Detection
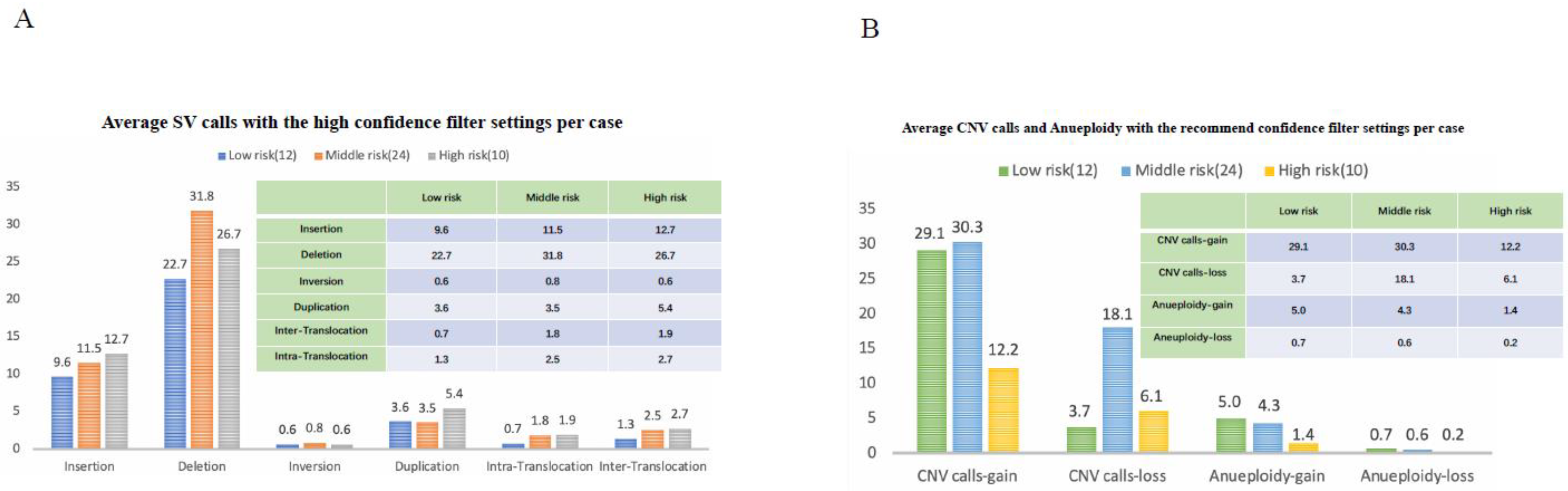
Difference in SV Numbers among the Three Risk Groups
3.4. Successful Validation of New SVs with Combination of OGM and NGS
3.4.1. NGS Validation of SVs Detected by OGM
3.4.2. Determination of LMNB1::PPP2R2B Fusion mRNA in Another Cohort of B-ALL Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

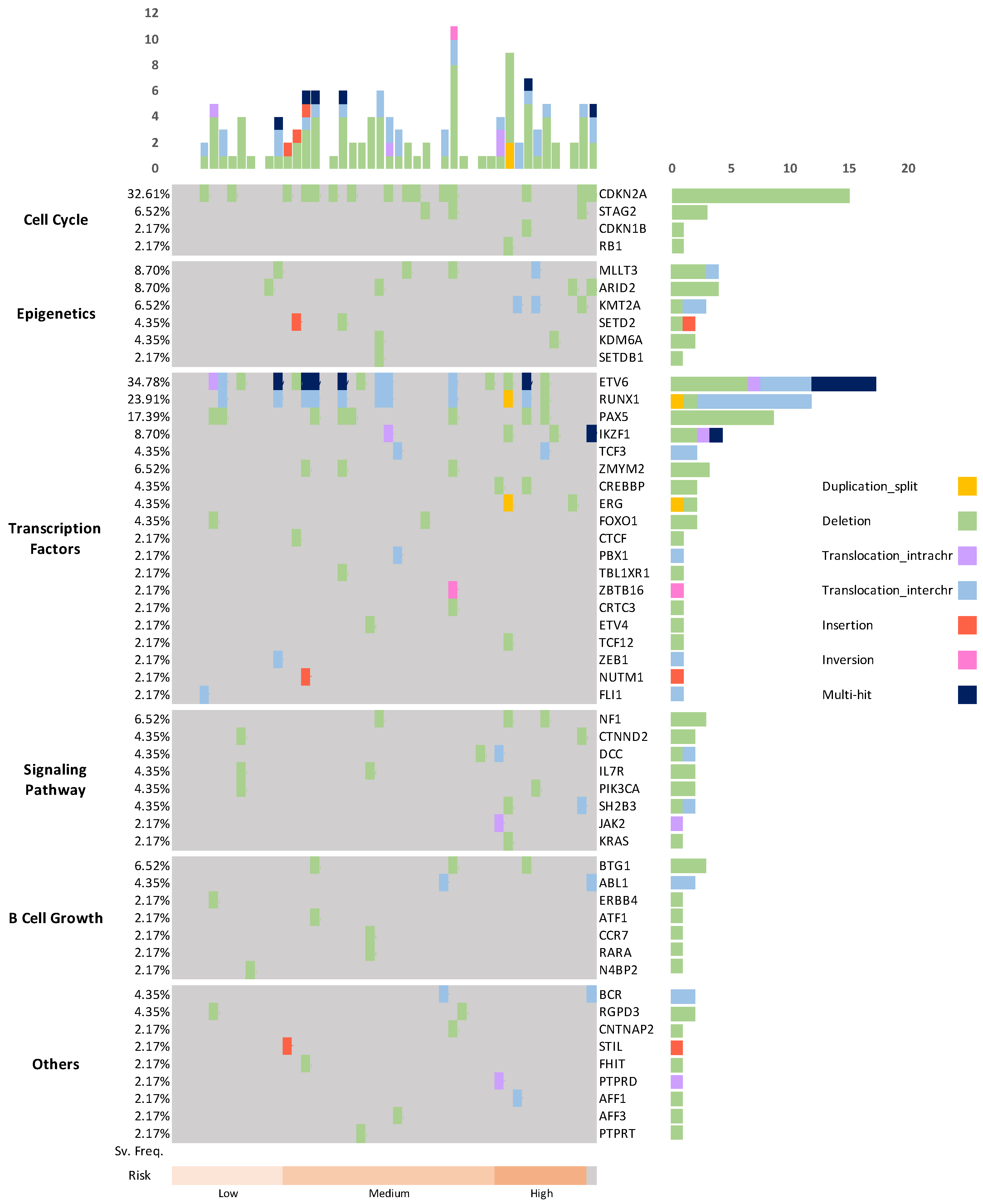
Appendix B. Genes and Cellular Biological Processes Affected by Chromosomal SVs
References
- Hunger, S.P.; Mullighan, C.G. Acute lymphoblastic leukemia in children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, I.; Mullighan, C.G. Genetic basis of acute lymphoblastic leukemia. J. Clin. Oncol. 2017, 35, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-F.; Wang, B.-Y.; Zhang, W.-N.; Huang, J.-Y.; Li, B.-S.; Zhang, M.; Jiang, L.; Li, J.-F.; Wang, M.-J.; Dai, Y.-J.; et al. Genomic profiling of adult and pediatric B-cell acute lymphoblastic leukemia. EBioMedicine 2016, 8, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Suttorp, J.; Lühmann, J.L.; Behrens, Y.L.; Göhring, G.; Steinemann, D.; Reinhardt, D.; Neuhoff, N.V.; Schneider, M. Optical Genome Mapping as a Diagnostic Tool in Pediatric Acute Myeloid Leukemia. Cancers 2022, 14, 2058. [Google Scholar] [CrossRef] [PubMed]
- Lafage-Pochitaloff, M.; Baranger, L.; Hunault, M.; Cuccuini, W.; Lefebvre, C.; Bidet, A.; Tigaud, I.; Eclache, V.; Delabesse, E.; Bilhou-Nabéra, C.; et al. Impact of cytoge- netic abnormalities in adults with Ph-negative B-cell precursor acute lymphoblastic leukemia. Blood 2017, 130, 1832–1844. [Google Scholar] [CrossRef]
- Chan, S.; Lam, E.; Saghbini, M.; Bocklandt, S.; Hastie, A.; Cao, H.; Holmlin, E.; Borodkin, M. Structural Variation Detection and Analysis Using Bionano Optical Mapping. Methods Mol. Biol. 2018, 1833, 193–203. [Google Scholar]
- Chan, E.K.; Cameron, D.L.; Petersen, D.C.; Lyons, R.J.; Baldi, B.F.; Papenfuss, A.T.; Thomas, D.M.; Hayes, V.M. Optical mapping reveals a higher level of genomic architecture of chained fusions in cancer. Genome Res. 2018, 28, 26–738. [Google Scholar] [CrossRef]
- Eisfeldt, J.; Pettersson, M.; Vezzi, F.; Wincent, J.; Käller, M.; Gruselius, J.; Nilsson, D.; Lundberg, E.S.; Carvalho, C.M.B.; Lindstrand, A. Comprehensive structural variation genome map of individuals carrying complex chromosomal rearrangements. PLoS Genet. 2019, 15, e1007858. [Google Scholar] [CrossRef]
- Hematology Group, Scientific Society, Chinese Medical Association. Recommendations for Diagnosis and Treatment of pediatric Acute lymphoblastic Leukemia (Fourth Revision). Chin. J. Pediatr. 2014, 52, 641–644. [Google Scholar]
- Dixon, J.R.; Xu, J.; Dileep, V.; Zhan, Y.; Song, F.; Le, V.T.; Yardımcı, G.G.; Chakraborty, A.; Bann, D.V.; Wang, Y.; et al. Integrative detection and analysis of structural variation in cancer genomes. Nat. Genet. 2018, 50, 1388–1398. [Google Scholar] [CrossRef]
- Broach, J.R.; Xu, J.; Schleicher, E.; Pool, C.; Hennessy, M.; Sheldon, K.; Annageldiyev, C.; Sharma, A.; Chang, Y.; Hastie, A.; et al. An integrated framework for genome analysis reveals numerous previously unrecognizable structural variants in leukemia patients’ samples. Cancer Res. 2019, 79, 1708. [Google Scholar] [CrossRef]
- Kumar, K.R.; Cowley, M.J.; Davis, R.L. Next-Generation Sequencing and Emerging Technologies. Semin. Thromb. Hemost. 2019, 45, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Liu, M.; Huang, T.; Liao, W.; Xu, M.; Gu, J. GeneFuse: Detection and visualization of target gene fusions from DNA sequencing data. Int. J. Biol. Sci. 2018, 14, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wu, L.; Wang, Q.; Li, Y.; Wang, X. KDM4B promotes epithelial-mesenchymal transition through up-regulation of ZEB1 in pancreatic cancer. Acta Biochim. Biophys. Sin. 2015, 47, 997–1004. [Google Scholar] [CrossRef]
- Gerding, W.M.; Tembrink, M.; Nilius-Eliliwi, V.; Mika, T.; Dimopoulos, F.; Ladigan-Badura, S.; Eckhardt, M.; Pohl, M.; Wünnenberg, M.; Farshi, P.; et al. Optical genome mapping reveals additional prognostic information compared to conventional cytogenetics in AML/MDS patients. Int. J. Cancer 2022, 150, 1998–2011. [Google Scholar] [CrossRef]
- Moorman, A.V.; Richards, S.M.; Robinson, H.M.; Strefford, J.C.; Gibson, B.E.S.; Kinsey, S.E.; Eden, T.O.B.; Vora, A.J.; Mitchell, C.D.; Harrison, C.J.; et al. Prognosis of children with acute lymphoblastic leukemia (ALL) and intrachromosomal amplification of chromosome 21 (iAMP21). Blood 2006, 109, 2327–2330. [Google Scholar] [CrossRef]
- Strefford, J.C.; van Delft, F.W.; Robinson, H.M.; Worley, H.; Yiannikouris, O.; Selzer, R.; Richmond, T.; Hann, I.; Bellotti, T.; Raghavan, M.; et al. Complex genomic alterations and gene expression in acute lymphoblastic leukemia with intrachromosomal amplification of chromosome 21. Proc. Natl. Acad. Sci. USA 2006, 103, 8167–8172. [Google Scholar] [CrossRef]
- Sinclair, P.B.; Parker, H.; An, Q.; Rand, V.; Ensor, H.; Harrison, C.J.; Strefford, J.C. Analysis of a breakpoint cluster reveals insight into the mechanism of intrachromosomal amplification in a lymphoid malignancy. Hum. Mol. Genet. 2011, 20, 2591–2602. [Google Scholar] [CrossRef]
- McClintock, B. The stability of broken ends of chromosomes in Zea Mays. Genetics 1941, 26, 234–282. [Google Scholar] [CrossRef]
- Rand, V.; Parker, H.; Russell, L.J.; Schwab, C.; Ensor, H.; Irving, J.; Jones, L.; Masic, D.; Minto, L.; Morrison, H.; et al. Genomic characterization implicates iAMP21 as a likely primary genetic event in childhood B-cell pre-cursor acute lymphoblastic leukemia. Blood 2001, 117, 6848–6855. [Google Scholar] [CrossRef]
- Stanulla, M.; Dagdan, E.; Zaliova, M.; Möricke, A.; Palmi, C.; Cazzaniga, G.; Eckert, C.; Te Kronnie, G.; Bourquin, J.P.; Bornhauser, B.; et al. International BFM Study Group. IKZF1plus defines a new minimal residual disease-dependent very-poor prognostic profile in pediatric B-Cell precursor acute lymphoblastic leukemia. J. Clin. Oncol. 2018, 36, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.-L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Gu, Z.; Payne-Turner, D.; McCastlain, K.; Harvey, R.; Chen, I.-M.; Pei, D.; Iacobucci, I.; Valentine, M.; Pounds, S.B.; et al. High frequency and poor outcome of Philadelphia chromosome-like acute lymphoblastic leukemia in adults. J. Clin. Oncol. 2017, 35, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, I.; Li, Y.; Roberts, K.G.; Dobson, S.M.; Kim, J.C.; Payne-Turner, D.; Harvey, R.C.; Valentine, M.; McCastlain, K.; Easton, J.; et al. Truncating erythropoietin receptor rearrangements in acute lymphoblastic leukemia. Cancer Cell 2016, 29, 186–200. [Google Scholar] [CrossRef]
- Schwab, C.; Ryan, S.L.; Chilton, L.; Elliott, A.; Murray, J.; Richardson, S.; Wragg, C.; Moppett, J.; Cummins, M.; Tunstall, O.; et al. EBF1-PDGFRB fusion in pediatric B-cell precursor acute lymphoblastic leukemia (BCP-ALL): Genetic profile and clinical implications. Blood 2016, 127, 2214–2218. [Google Scholar] [CrossRef]
- Yang, H.; Garcia-Manero, G.; Sasaki, K.; Montalban-Bravo, G.; Tang, Z.; Wei, Y.; Kadia, T.; Chien, K.; Rush, D.; Nguyen, H.; et al. High-resolution structural variant profiling of myelodysplastic syndromes by optical genome mapping uncovers cryptic aberrations of prognostic and therapeutic significance. Leukemia 2022, 36, 2306–2316. [Google Scholar] [CrossRef]
- Balducci, E.; Kaltenbach, S.; Villarese, P.; Duroyon, E.; Zalmai, L.; Friedrich, C.; Suarez, F.; Marcais, A.; Bouscary, D.; Decroocq, J.; et al. Optical genome mapping refines cytogenetic diagnostics, prognostic stratification and provides new molecular insights in adult MDS/AML patients. Blood Cancer J. 2022, 12, 126. [Google Scholar] [CrossRef]
- Finlay, D.; Murad, R.; Hong, K.; Lee, J.; Pang, A.; Lai, C.; Burian, C.; Mason, J.R.; Hastie, A.; Yin, J.; et al. Optical mapping uncovers multiple novel genomic structural variants in patient leukemias. Blood 2020, 136 (Suppl. S1), 33–34. [Google Scholar] [CrossRef]
- Levy, B.; Baughn, L.B.; Chartrand, S.; LaBarge, B.; Claxton, D.; Lennon, A.; Akkari, Y.; Cujar, C.; Kolhe, R.; Kroeger, K.; et al. A national multicenter evaluation of the clinical utility of optical genome mapping for assessment of genomic aberrations in acute myeloid leukemia. medRxiv 2020. [Google Scholar] [CrossRef]
- Hastie, A.R.; Lam, E.T.; Chun Pang, A.W.; Zhang, X.; Andrews, W.; Lee, J.; Liang, T.Y.; Wang, J.; Zhou, X.; Zhu, Z.; et al. Rapid Automated Large Structural Variation Detection in a Diploid Genome by NanoChannel Based Nest-Generation Mapping. bioRxiv 2017. [CrossRef]
- Elfassihi, L.; Giroux, S.; Bureau, A.; Laflamme, N.; Cole, D.E.; Rousseau, F. Association with replication between estrogen-related receptor gamma (ESRRgamma) polymorphisms and bone phenotypes in women of European ancestry. J. Bone Miner. Res. 2010, 25, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Jubb, A.M.; Pike, L.; Buffa, F.M.; Turley, H.; Baban, D.; Leek, R.; Gatter, K.C.; Ragoussis, J.; Harris, A.L. The histone demethylase JMJD2B is regulated by estrogen receptor alpha and hypoxia, and is a key mediator of estrogen induced growth. Cancer Res. 2010, 70, 6456–6466. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Chen, L.; Yang, J.; Ye, T.; Chen, Y.; Fang, J. HIF-1alpha-induced histone demethylase JMJD2B contributes to the malignant phenotype of colorectal cancer cells via an epigenetic mechanism. Carcinogenesis 2012, 33, 1664–1673. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Qiu, L.; Hong, Y.; Karnik, T.; Tadros, G.; Mau, B.; Ma, T.; Mu, Y.; New, J.; Louie, R.J.; et al. The histone demethylase KDM4B regulates peritoneal seeding of ovarian cancer. Oncogene 2017, 36, 2565–2576. [Google Scholar] [CrossRef]
- Li, W.; Zhao, L.; Zang, W.; Liu, Z.; Chen, L.; Liu, T.; Xu, D.; Jia, J. Histone demethylase JMJD2B is required for tumor cell proliferation and survival and is over-expressed in gastric cancer. Biochem. Biophys. Res. Commun. 2011, 416, 372–378. [Google Scholar] [CrossRef]
- Slee, R.B.; Steiner, C.M.; Herbert, B.S.; Vance, G.H.; Hickey, R.J.; Schwarz, T.; Christan, S.; Radovich, M.; Schneider, B.P.; Schindelhauer, D.; et al. Cancer-associated alteration of pericentromeric heterochromatin may contribute to chromosome instability. Oncogene 2012, 31, 3244–3253. [Google Scholar] [CrossRef]
- Tan, J.; Lee, P.L.; Li, Z.; Jiang, X.; Lim, Y.C.; Hooi, S.C.; Yu, Q. B55β-associated PP2A complex controls PDK1-directed MYC signaling and modulates rapamycin sensitivity in colorectal cancer. Cancer Cell 2010, 18, 459–471. [Google Scholar] [CrossRef]
- Li, Z.; Li, Y.; Wang, X.; Yang, Q. PPP2R2B downregulation is associated with immune evasion and predicts poor clinical outcomes in triple-negative breast cancer. Cancer Cell Int. 2021, 21, 13. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sum in All Samples | Average per Sample | |
|---|---|---|
| SV calls using the recommend confidence filter settings | ||
| Insertion | 30,895 | 671.6 |
| Deletion | 29,099 | 632.6 |
| Inversion | 3105 | 67.5 |
| Duplication | 8256 | 179.5 |
| Translocation inter-chromosomal | 72 | 1.6 |
| Translocation intrachromosomal | 104 | 2.3 |
| Total | 71,531 | 1555.1 |
| SV calls using the high confidence filter settings | ||
| Insertion | 517 | 11.2 |
| Deletion | 1301 | 28.3 |
| Inversion | 33 | 0.7 |
| Duplication | 180 | 3.1 |
| Translocation inter-chromosomal | 70 | 1.5 |
| Translocation intra-chromosomal | 103 | 2.2 |
| Total | 2204 | 47.0 |
| CNV calls (non-masked only) | ||
| Gain (called duplication in the file) | 1189 | 25.8 |
| Loss (called deletion in the file) | 403 | 8.7 |
| Total | 1592 | 34.5 |
| Aneuploidy (non-masked only) | ||
| Aneuploidy Gain | 177 | 3.8 |
| Aneuploidy Loss | 23 | 0.5 |
| Total | 200 | 4.3 |
| Sample ID | Karyotype | FISH/PCR | OGM[GRCh38] | Karyotype Predicted by OGM | Overlapping Genes | Additional Findings |
|---|---|---|---|---|---|---|
| AL-41 | 46,XX[16] | RUNX1,IGH and CRLF2 copy number gain | (4)×3,(5)×3,(6)×3,(9p24.3-9q11)(14566-43279152)×3,(9q21.11-9q34.3)(68310629-138334464)×3,(10)×3,(14q11.2-14q32.33)(19761872-105833056)×3,(17)×3,(18)×3, (21q11.2-21q22.3)(13120706-45845512)×4, (23)×3 | 53,XX,+X,+4,+5,+6, dup(9)(p24.3q11), dup(9)(q21.11q34.3),+10, dup(14) (q11.2q32.33), +17,+18, dup(21)(q11.2q22.3) | RUNX1, IGH, CRLF2 | Hyperdiploid |
| AL-45 | 45,XX,del(9)(p13) [10] | MEF2D::HNRNPUL1, CRLF2 copy number gain | t(1q22;19q13.2)(156553387;41431238), t(9p24.1;9p21.2)(5025357;27143532), (9p24.2-p24.1)(3552238-8731962)×1,(9p22.3-9p21.3)(15980358-23496538)×1,(9p21.2-9p21.1)(96703784-96724287)×1,(12p13.33-12p12.33)(14568-16540369)×1,(Xp11.23-Xp22.2)(4170788-56416870)×3 | 46,XX,t(1;19)(q22;q13.2),der(9) t(9;9)(p24.1;p21.2),del(9)(p24.2p24.1),del(9)(p22.3p21.3), del(9)(p21.2p21.1), del(12)(p13.33p12.33),dup(X)(p11.23p22.2) | MEF2D::HNRNPUL1, CRLF2,JAK2::TEK | t(9;9)(p24.1;p21.2) JAK2::TEK |
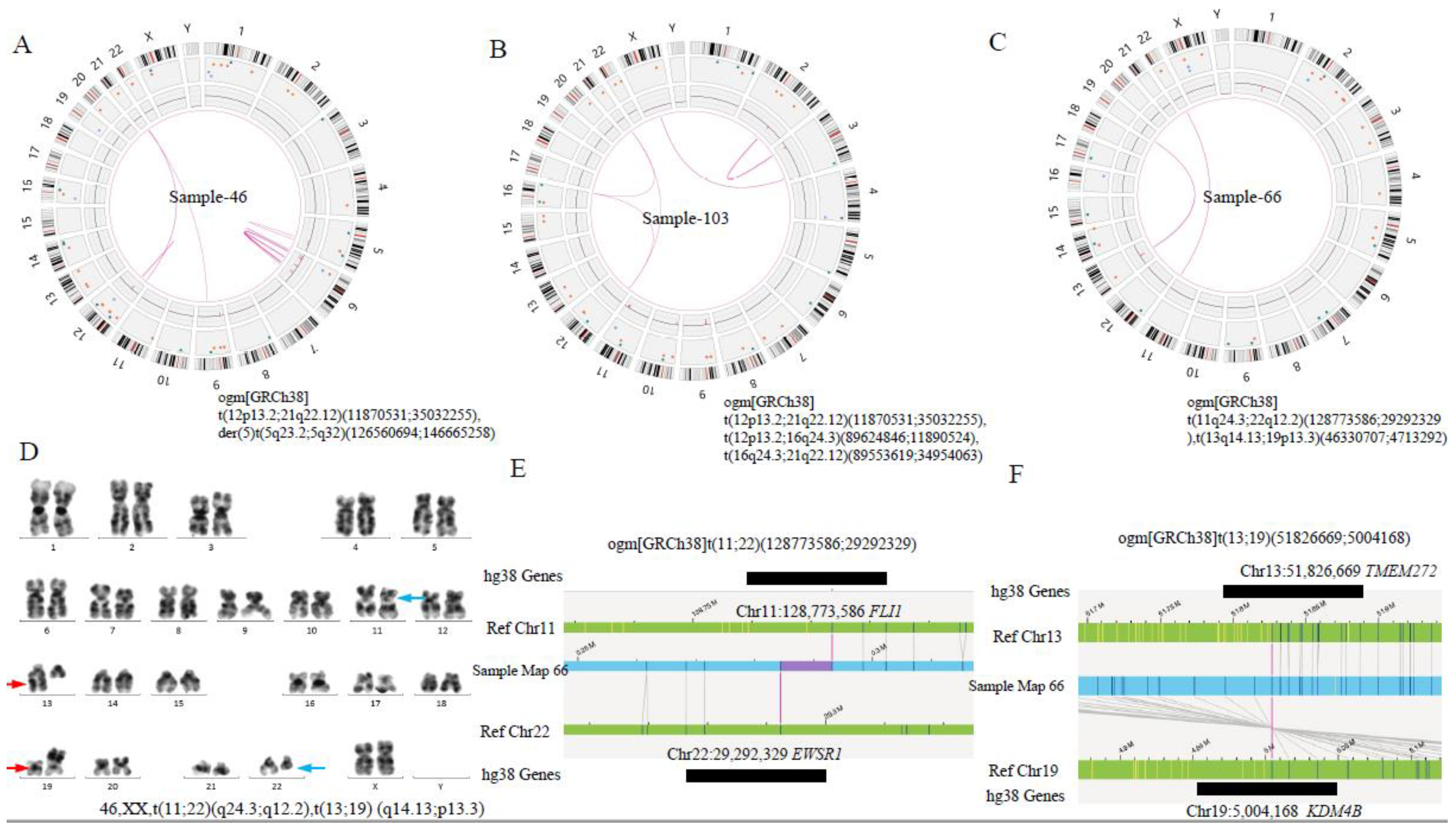
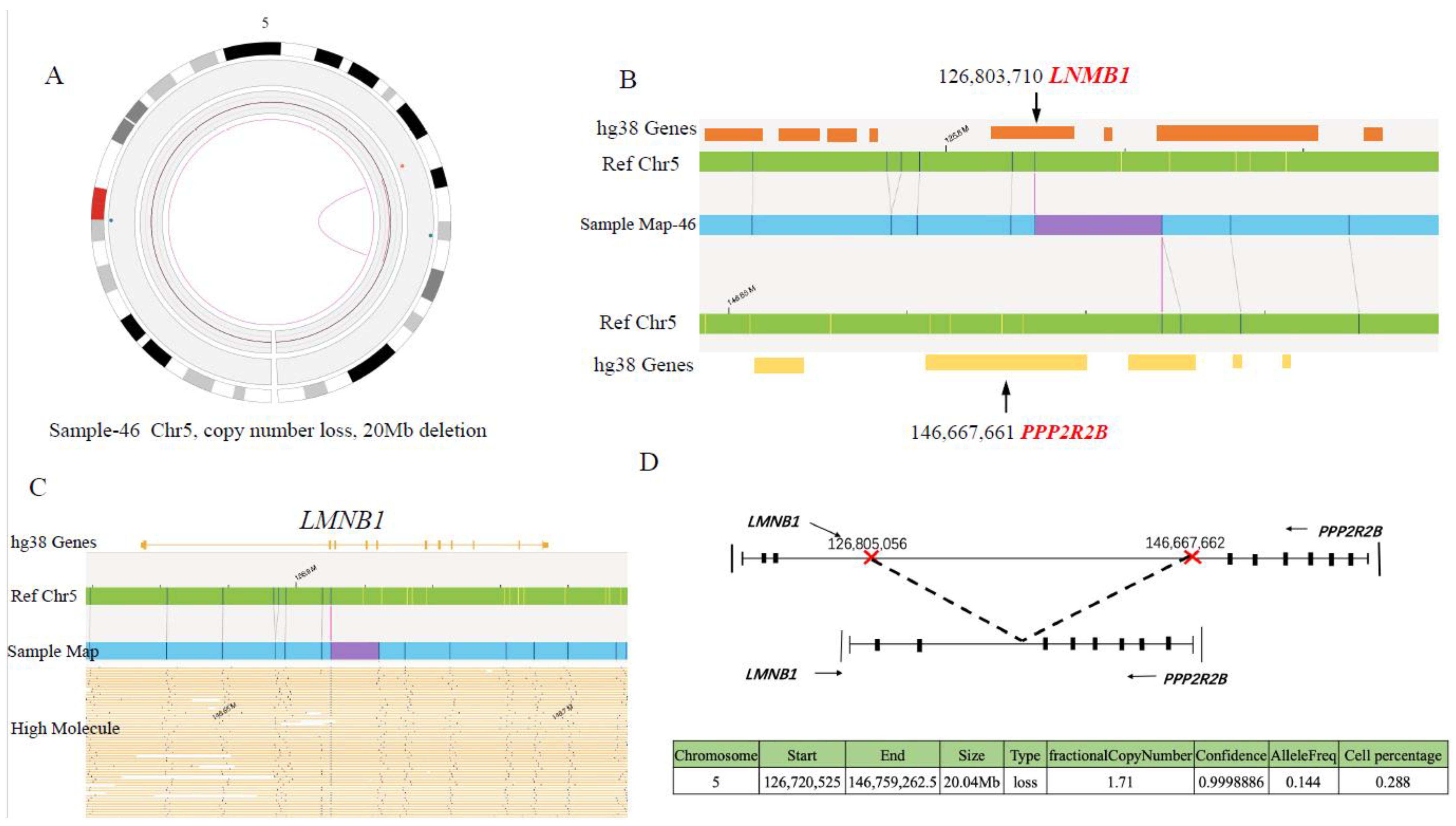
| AL-46 | 46,XX[9] | ETV6::RUNX1 | t(12p13.2;21q22.12)(11870531;35032255),der(5),t(5q23.2;5q32)(126560694;146665258) | 46,XX,t(12;21)(p13.2;q22.12), der(5),t(5;5)(q23.2q32) | ETV6::RUNX1 LMNB1::PPP2R2B | t(5;5)(q23.2q32) LMNB1::PPP2R2B |
| AL-66 | 46,XX,t(11;22)(q23;q11);t(13;19)(q14;p13)[10] | t(11q24.3;22q12.2)(128773586;29292329), t(13q14.13;19p13.3)(46330707;4713292) | 46,XX,t(11;22)(q24.3;q12.2),t(13;19) (q14.13;p13.3) | FLI1::EWSR1; TMEM272::KDM4B,AL162377.3::KDM4B | t(13;19) (q14.13;p13.3) AL162377.3::KDM4B | |
| AL-74 | 46,XY[9] | ZNF384 copy number loss | (12p13.311-12p12.1)(6670124-25189935) ×1, der(12) t(12p13.2;12p12.1)(11736073; 25175777), der(12)t(12p13.31;12p13.2) (6686961;11758879) | 46,XY,der(12)t(12;12)(p13.2;p12.1),der(12)t(12;12)(p13.31;p13.2) | ETV6::CASC1, ZNF384::ETV6, ZNF384 | t(12;12)(p13.2;p12.1) ETV6::CASC1 t(12;12)(p13.31;p13.2) ZNF384::ETV6 |
| AL-77 | 46,XX[9] | RUNX1 and CRLF2 copy number gain | (4)×3,(5)×3,(6)×3,(10)×3,(14)×3,(18)×3,(21)×3,(22)×3,(23)×3,(1q21.3)(151751846-15379814)×3,1q23.1q43(156616741_240089410)×3, (12p11.22-12q24.33)(27982715_132081687)×3, 17q11.1q25.3(26692353_81102800)×3, t(5;5)(q21.1;q33.2)(99,141,474;155,917,152), t(5;5)(q23.1;q34)(118,853,883;168,502,903), t(11;11)(q14.3;q24.1)(91,671,788;122,517,878), t(11;11)(q14.1;q25)(84,542,296;132,777,096) | 54,XX, +4,+5,+6, +10,+14,+18,+21, +22, +X, dup(1q21.3),dup(1q23.1q43), dup(12)(p11.2q24.33),dup(17)(q11.1q25.3), der(5)t(5;5)(q23.1;q34), der(5)t(5;5)(q21.1;q33.2), der(11)t(11;11)(q14.3;q24.1), der(11)t(11;11)(q14.1;q25) | RUNX1, CRLF2, DTWD2::RARS | Hyperdiploid; t(5;5)(q23.1;q34) DTWD2::RARS |
| AL-80 | 46, XX[1] | MEF2D,RUNX1,IGH,CRLF2 and ETV6 copy number gain | (1q21.1-1q44)(144085065_248943333)×3,(3)×3,(5)×3,(6)×3,(8)×3, (10)×4,(11)×3,(12p12.3-12p11.1) (19087028-34717936)×3, (12p13.31p12.3) (6245718-15306812)×1, t(12;16)(p13.31;p13.13)(6,245,718;11,607,398) t(9;12)(p24.1;p12.3)(19,087,028;5,761,495), (14)×4,(16p13.13-16 p11.2)(11584915-31987698)×3,(17q12-17q25.3)(38731593_82564467)×3,t(17;17)(q21.33;q22)(51,672,152;59,147,333), (18)×4,(21)×4,(23)×4 | 56,XX,+X,dup(1)(q21.1q44),+3,+5,+6,+8,+10,+11,dup(12)(p12.3p11.1),del(12)(p13.31p12.3),+14,dup(16)(p13.13p11.2),dup(17)(q12q25.3),+18,+21, t(12;16)(p13.31;p13.13) t(9;12)(p24.1;p12.3) t(17;17)(q21.33;q22) | RUNX1, IGH, CRLF2, ETV6 MEF2D | Hyperdiploid; |
| AL-97 | 46,XX[20] | RUNX1,IGH and CRLF2 copy number gain | (4)×3,(6)×3,(9)×3,(10)×3,(14)×3,(18)×3,(21)×3,(X)×3 | 54,XX,+X,+4,+6,+9+10,+14,+18,+21 | RUNX1, IGH, CRLF2 | Hyperdiploid |
| AL-101 | 46,XY[8] | RUNX1 copy number gain, IKZF1 deletion | (7)×1,21q22.11(32926920-33533692)×3, 21q22.3(41288580-45259300)×3 | 45,XY,−7,dup(21)(q22.11),dup(21)(q22.3) | RUNX1, IKZF1 | chr7 CN loss aenuploidy |
| AL-103 | 46,XY[20] | ETV6::RUNX1 | t(12p13.2;21q22.12)(11870531;35032255),t(12p13.2;16q24.3)(89624846;11890524),t(16q24.3;21q22.12)(89553619;34954063) | 46,XY,t(12;21)(p13.2;q22.12), t(12;16)(p13.2;q24.3),t(16;21)(q24.3;q22.12) | ETV6::RUNX1, ETV6::DPEP1, SPG7::RUNX1 | three-way trans; t(12;16)ETV6::DPEP1; t(16;21)SPG7::RUNX1 |
| AL-109 | 46,XX[9] | RUNX1, IGH and CRLF2 copy number gain | (1q21.1-1q41)(145439805-215837313) ×3,(4)×3,(6)×3,(10)×3,(14)×3,(17)×3, (18)×3,(21)×4,(23)×4 | 54,XX,+X,+X,dup(1)(q21.1q41),+4,+6,+14,+17,+18,+21 | RUNX1, IGH, CRLF2 | Hyperdiploid |
| Sample ID | Karyotype | FISH/PCR | OGM[GRCh38] | Karyotype Predicted by OGM | Overlapping Genes |
|---|---|---|---|---|---|
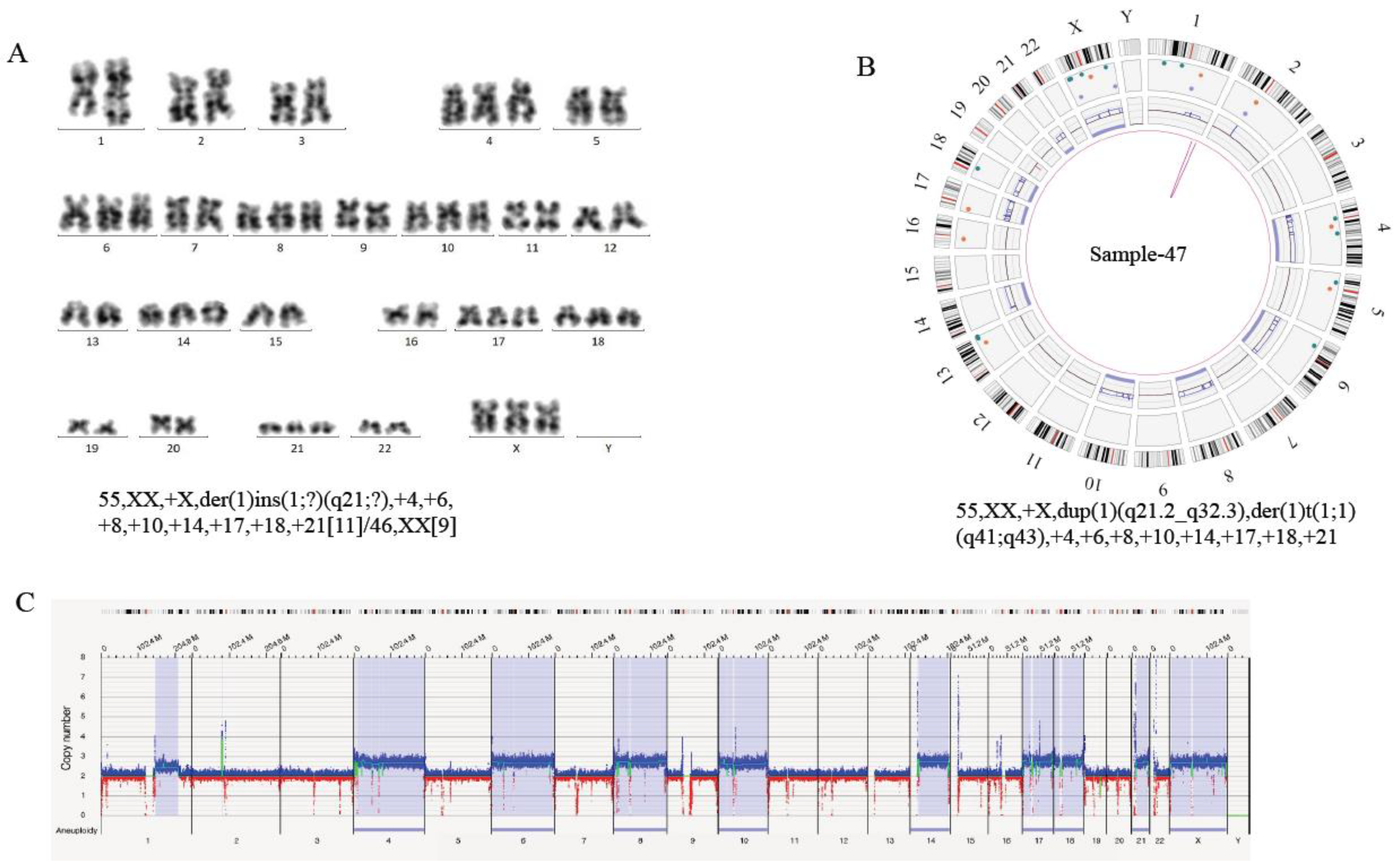
| AL-47 | 55,XX,+X,der(1) ins(1;?)(q21;?), +4,+6,+8,+10, +14,+17,+18,+21 [11]/46,XX[9] | IGH and CRLF2 copy number gain | (1q21.2-1q32.3)(149910330-213101514)×3, der(1)t(1q41;1q43)(217001914;236988091), (4)×3,(6)×3,(8)×3,(10)×3,(14)×3,(17)×3,(18)×3, (21)×3,(23)×3 | 55,XX,+X,dup(1)(q21.2 q32.3),der(1)t(1;1)(q41;q43),+4,+6,+8,+10,+14,+17,+18,+21 | IGH, CRLF2, ESRRG |
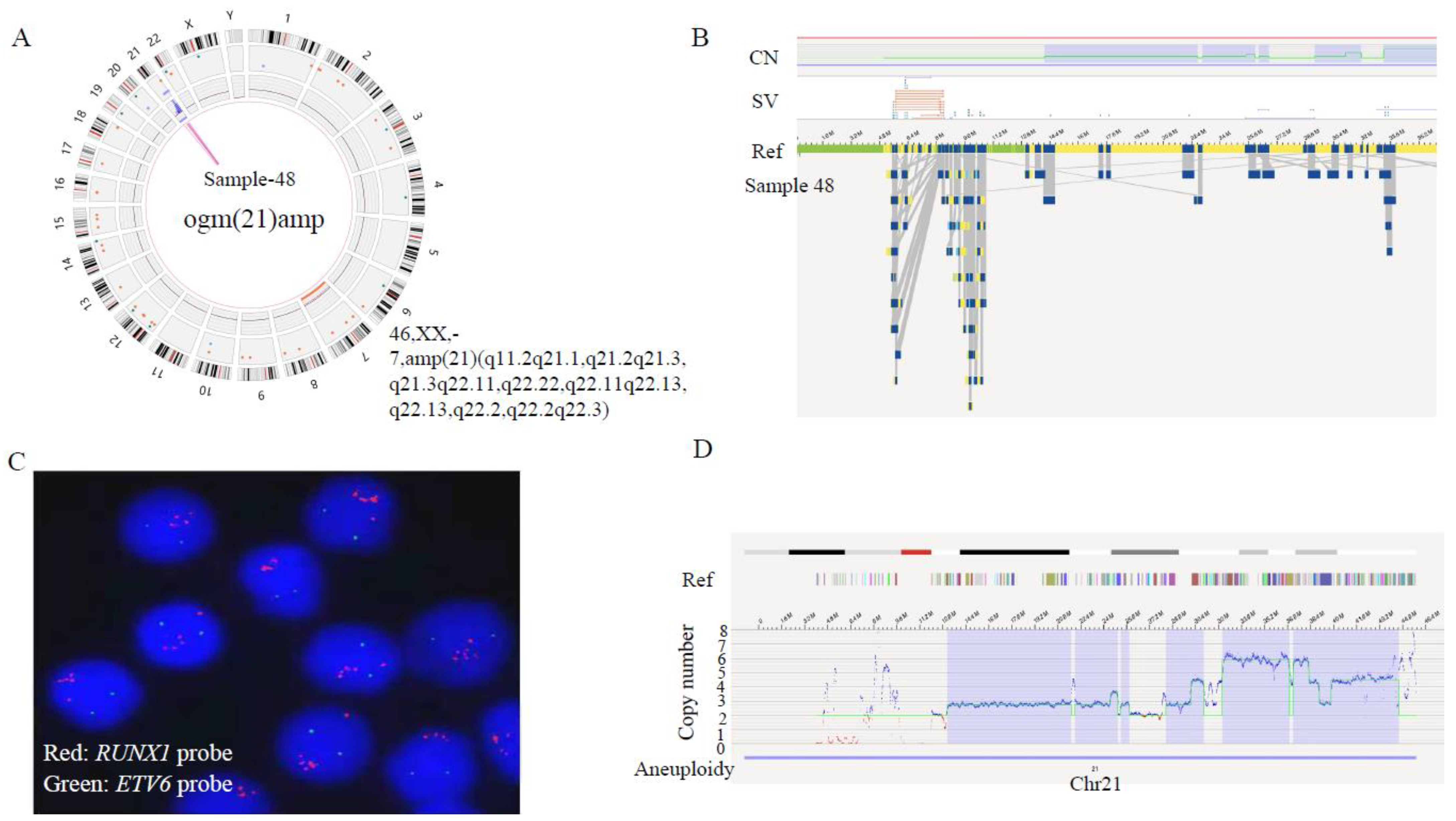
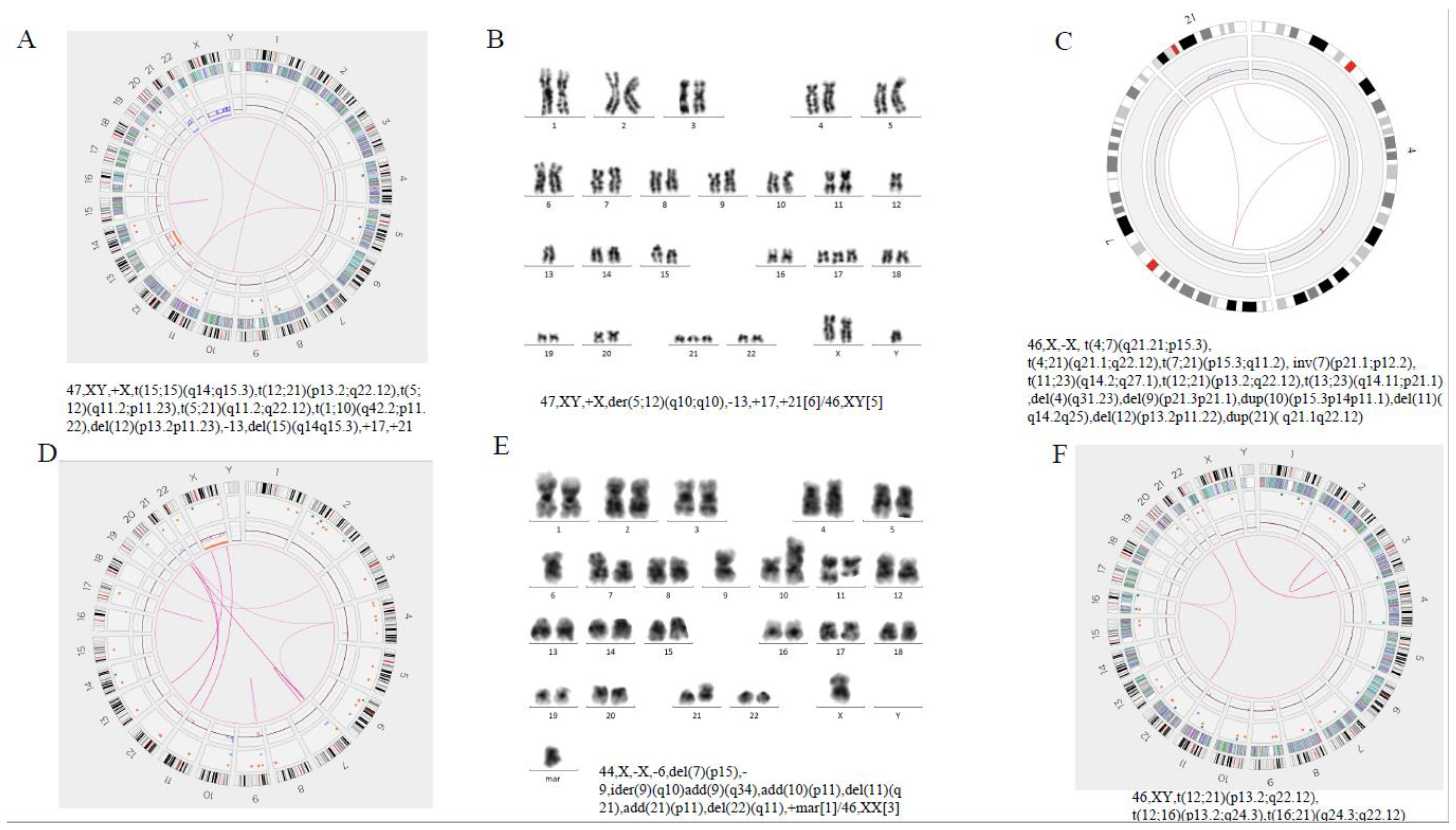
| AL-48 | 45 XX, −21, +mar[1]/45/idem,−7[14]/46, XX[5] | chr21(iAMP21),P2RY8::CRLF2,IKZF1 deletion | (7)×1,(21q22.3)(45427332-46402888) ×1,(21q11.2-21q21.1)(14097084-25448211)×3,(21q21.2-21q21.3)(25448806-25975600)×4,(21q21.3-21q22.11)(26177986-31070035)×3,(21q22.11)(31077495-31942346)amp,(21q22.11-21q22.13)(33213701-37921102)amp,(21q22.13-21q22.2)(38134198-39259099)amp,(21q22.2)(39267796-39966121)amp,(21q22.2-21q22.3)(40804931-45514719)amp | 46,XX,−7,+21,amp(21)(q11.2q22.3) | chr21(iAMP21),IKZF1, APP, BRWD1,ERG,EST2,GET1 |
| AL-71 | 48~49, XX, +X, t(2;12)(p13;q24), −6,add(6)(q23), −7,−17,−20,+21, +3~5mar[7]/46,XX[13] | RUNX1 and CRLF2 copy number gain | (8)×3,(10)×3,(23)×3, (21)×3, 6q15q22.1(91357467-115474254)×1, 20q11.21q13.33(31182877-61256295)×1,17p13.3p11.2(1342670-20101698)×1, 9p24.3p13.1(14566-38890429)×1, 7p14.3q11.21(31801042-66549041)×1, 7q11.21q11.22(67337248-72514593)×1, t(2;12)(p11.2;q24.12)(88827954;111430007), t(6;7)(q22.1;q11.21)(66546520;116497165), der(7)t(7;7)(p14.3;q11.22)(31802046;67525321),t(7;10)(q11.21;q21.1)(51434361;67432607),t(6;10)(q15;q11.23)(51098504;91367054) | 49, XX, +X,+8,+10,+21,−7,t(2;12)(p11.2;q24.12) del(6)(q15q22.1), del(20)(q11.21q13.33), del(17)(p13.3p11.2), del(9)(p24.3p13.1), t(6;7)(q22.1;q11.21), der(7),t(7;7)(p14.3;q11.22),t(7;10)(q11.21;q21.1), t(6;10)(q15;q11.23) | AC244205.1::SH2B3,RUNX1, CRLF2 |
| AL-78 | 46, XX[16] | ETV6::RUNX1 | t(12p13.2;21q22.12)(11870531;35032255), (12p13.33-12p12.3)(14568-17462436)×1,(22 q13.1-22q13.32)(38840431-48399879)×3, t(12;22)(p12.3;q13.1)(17474765;38809117) t(20;21)(p11.21;q22.12)(35029693;22329802) | 46,XX,t(12;21)(p13.2;q22.12),del(12)(p13.33p12.3),dup(22)(q13.1q13.32), t(12;22)(p12.3;q13.1), t(20;21)(p11.21;q22.12) | ETV6::RUNX1 |
| AL-85 | 46,XX,add(19)(p13)[2]/46,XX[5] | TCF3::PBX1 | t(1p23.3;19q13.3)(164783197;1638016), (1q23.3-1q44)(164773702-248458732)×3,2p25.3(743869-1959263)×3,10q21.1(54833448-55584392)×1 | 46,XX,t(1;19)(p23.3;q13.3),dup(1)(q23.3q44), dup(2)(p25.3),del(10)(q21.1) | TCF3::PBX1 |
| AL-107 | 55,XX,+X, der(1) ins(1;?)(q21;?), +4,+5,+6,+8,+10,+21, +21,+22[8]/55, idem, add(21) (q22)[2]/46,XX[10] | RUNXL1 and CRLF2 copy number gain | (1q21.1-1q32.3)(146397612-212907422) ×3,(4)×3,(5)×3,(6)×3,(8)×3,(10)×3,(21)×3, (22q11.21-22q13.1)(18746350-38096173)×3, (X)×3 | 53,XX,+X,dup(1)(q21.1q32.3),+4,+5,+6,+8,+10,+21,dup(22)(q11.21q13.1) | RUNX1, CRLF2 |
| Sample ID | Karyotype | FISH/PCR | OGM[GRCh38] | Karyotype Predicted by OGM | Overlapping Genes |
|---|---|---|---|---|---|
| AL-42 | / | ETV6::RUNX1 | t(12p13.2;21q22.12)(11870531;35032255) | 46,XY,t(12;21)(p13.2;q22.12) | ETV6::RUNX1 |
| AL-49 | 46,XX,t(4;11)(q21;q23)[12]/46,XX[2] | KMT2A::AF4 | t(4q21.3;11q23.3)(87082301;118477357) | 46,XX, t(4;11) (q21.3;q23.3) | KMT2A::AF4 |
| AL-58 | 46,XY,t(9;22)(q34;q11)[1]/45,sl,dic(7;9)(p12;p12)[17]/46,sdl,+der(22)t(9;22)[1]/46,XY[1] | BCR::ABL1,IKZF1 deletion | t(9q34.12;22q11.23)(130864214;23203247),(2q32.2)(194855238-195411332)×1,(7p22.3-7p14.1)(205606-38213349) ×1, (7p12.2)(49320190-50264542) ×1,(7p12.3-p12.2)(48462939-49316285) ×3,(9p24.3-9p12)(585489-39591818) ×1,(19p13.2)(10015151-10983318) ×1 | 46,XY,t(9;22)(q34.12;q11.23),del(2)(2q32.3),del(7)(p22.3p14.1),del(7)(p12.2),dup(7)(p12.3p12.2,del(9)(p24.3p12),del(19)(p13.2) | BCR::ABL1,IKZF1 |
| AL-65 | 46,XY[10] | ETV6::RUNX1 | t(12;21)(p13.2;q22.12)(11870531;35032255), (21q11.1-21q22.3)(12983105-45259300)×3 | 46,XY, t(12;21)(p13.2;q22.12),dup(21)(q11.1q22.3) | ETV6::RUNX1 |
| AL-82 | 46,XY[20] | ETV6::RUNX1 | t(12p13.2;21q22.12)(11870531;35032255),(4q26-4q35.2)(117373259-190202564)×3,(12p13.33-12p13.2)(14568-11843683)×3 | 46,XY,t(12;21)(p13.2;q22.12),dup(4)(q26q35.2),dup(12)(p13.33p13.2) | ETV6::RUNX1 |
| AL-83 | 44,X,-X,-6,del(7)(p15),-9,ider(9)(q10)add(9)(q34),add(10)(p11),del(11)(q21),add(21)(p11),del(22)(q11),+mar[1]/46,XX[3] | ETV6::RUNX1 | t(4;7)(q21.21;p15.3)(78918774;24807169),t(4;21)(q21.1;q22.12)(75639432;34981504),t(7;21)(p15.3;q11.2)(24843132;14996044),inv(7)(p21.1;p12.2)(16927511;50407985),t(11;23)(q14.2;q27.1)(87167189;139837627),t(12;21)(p13.2;q22.12)(11870531;34981504),t(12;21)(p13.2;q21.1)(11881907;15483164),t(13;23)(q14.11;p21.1)(40417525;33567746) (X)×1,4q31.23(148400189-148979475)×1, 9p21.3p21.1(21148269-29886519)×1,der(9)t(9;9)(p21.3;p21.1)(21155066;29880139), 10p15.3p14p11.1(2660922-38780901)×3,11q14.2q25(87162780-135069565)×1,12p13.2p11.22(11809511-29745022)×1, 21q21.1q22.12(19535402-34928264)×3, | 46,X,-X, t(4;7)(q21.21;p15.3), t(4;21)(q21.1;q22.12),t(7;21)(p15.3;q11.2), inv(7)(p21.1;p12.2), t(11;23)(q14.2;q27.1),t(12;21)(p13.2;q22.12),t(13;23)(q14.11;p21.1),del(4)(q31.23),del(9)(p21.3p21.1),dup(10)(p15.3p14p11.1),del(11)(q14.2q25),del(12)(p13.2p11.22),dup(21)(q21.1q22.12), | ETV6::RUNX1 BZW2::IKZF1 OSBPL3::NRIP1 PAQR3::OSBPL3 OSBPL3::AF127577.4 |
| AL-84 | 47,XY,+X,t(9;11)(p22;q23)[16]/46,XY[4] | KMT2A::MLLT3 | t(9p21.3;11q23.3)(20358621;118493942),(X)×3,22q11.22(22010337-22908320)×1, t(11;11)(q14.1;q21)(77603065;94621267) | 47,XY,+X,t(9;11)(p21.3;q23.3),inv(11)(q14.1;q21), del(22)q11.22 | KMT2A::MLLT3 |
| AL-89 | 46,XY[20] | IGH copy number gain | (14q11.2-14q32.33)(22525374-104169671)×3,(21q11.1-21q22.3)(12406577-43289581)×3 | 46,XY,dup(14)(q11.2q13.1q32.33),dup(21)(q11.1q22.3) | IGH |
| AL-95 | 45,XX,−7, add(9)(p13)[10]/46,idem,+mar[4]/46,XX[6] | TCF3::ZNF384; ZNF384 copy number gain | t(12p13.31;19p13.3)(1643841;6674678), (7)×1,(12p13.33-12p13.31)(377048-6670124) ×3 | 45,XX,−7,t(12;19)(p13.31;p13.3),dup(12)(p13.33p13.31) | TCF3::ZNF384, ZNF384 |
| AL-98 | 56,XX,+X,+2,+4,+6,t(9;22)(q34;q11),+10,+15,+18,+21,der(22)t(9;22),mar[4]/55,idem,-15,add(12)(q24)[7]/46,XX[9] | BCR::ABL1,RUNX1 copy number gain | t(9q34.12;22q11.23)(130732573;23244051),(2)×3,(4)×3,(6)×3,(10)×3,(12q24.21-1q24.33)(115355442-133263960)×1,(15)×3,(18)×3,(21)×3,(22q11.21-22q11.23)(18636137-23191585)×3,(X)×3 | 56,XX,+X,+2,+4,+6,t(9;22)(q34.12;q11.23),+10,del(12)(q24.21q24.33),+15,+18,+21,dup(22)(q11.21q11.23) | BCR::ABL1,RUNX1 |
| AL-114 | 47,XY,+X,der(5;12)(q10;q10),-13,+17,+21[6]/46,XY[5] | ETV6::RUNX1, CRLF2 copy number gain | der(15)t(15;15)(q14;q15.3)(38438035;43496447),t(12;21)(p13.2;q22.12)(11870531;34883313),t(5;12)(q11.2;p11.23)(27128273;58971017),t(5;21)(q11.2;q22.12)(34899705;58971017),t(1;10)(q42.2;p11.22)(31401031;233989381) (X)x2,(17)×3,(21)×3,(13)×1, 12p13.2p11.23(11845928-27123509)×1,15q14q15.3(38411329-43516043)×1, | 47,XY,+X,t(15;15)(q14;q15.3),t(12;21)(p13.2;q22.12),t(5;12)(q11.2;p11.23), t(5;21)(q11.2;q22.12),t(1;10)(q42.2;p11.22),del(12)(p13.2p11.23),-13,del(15)(q14q15.3),+17,+21 | ETV6::RUNX1,CRLF2 |
| Methods | Aneuploidies Only | Translocation and/or Aneuploidies | Negative Karyotype | Total |
|---|---|---|---|---|
| Conventional technologies | 18 * | 11 | 17 | 46 |
| OGM | 22 | 11 | 13 * | 46 |
| OGM concordance | 100% | 100% | 100% |
| Putative Gene Fusion | Clinical Features | Spearman ρ | p |
|---|---|---|---|
| BCR::ABL1 | WBC | 0.297 | 0.045 |
| GPN3::FAM216A | WBC | 0.313 | 0.034 |
| AC026202.2::EDEM1 | WBC | 0.305 | 0.039 |
| ARL8B::EDEM1 | WBC | 0.305 | 0.039 |
| MTAP::CDKN2B-AS1 | WBC | 0.322 | 0.029 |
| GRAPL::AC106017.1 | age | 0.378 | 0.01 |
| GRAPL::KYNUP3 | age | 0.378 | 0.01 |
| ETV6::AP000331.1 | d15 MRD | −0.307 | 0.04 |
| AC141586.1::KCTD5 | d33 MRD | 0.405 | 0.006 |
| ATP10A::AC016266.1 | d33 MRD | 0.329 | 0.029 |
| CALCOCO2::SUMO2P17 | d33 MRD | 0.350 | 0.020 |
| MIR4435.2HG::AC017002.5 | d33 MRD | 0.298 | 0.049 |
| AL034430.1::SLX4IP | d78 MRD | 0.318 | 0.038 |
| MKKS::SLX4IP | d78 MRD | 0.318 | 0.038 |
| AC141586.1::KCTD5 | Risk stratification | 0.318 | 0.033 |
| ATP10A::AC016266.1 | Risk stratification | 0.318 | 0.033 |
| CALCOCO2::SUMO2P17 | Risk stratification | 0.318 | 0.033 |
| PDCD6IPP1::AC138649.1 | Risk stratification | 0.305 | 0.041 |
| AC133919.2::LINC02193 | Percentage of blasts in PB at diagnosis | −0.388 | 0.008 |
| FAM157C::LINC02193 | Percentage of blasts in PB at diagnosis | −0.388 | 0.008 |
| GPN3::FAM216A | Percentage of blasts in PB at diagnosis | 0.297 | 0.045 |
| Genes | Gene Alteration Type | Clinical Features | Spearman ρ | p |
|---|---|---|---|---|
| BCR | Inter-trans | WBC | 0.297 | 0.045 |
| ABL2 | Inter-trans | WBC | 0.297 | 0.045 |
| TCF3 | Inter-trans | WBC | 0.321 | 0.030 |
| IKZF1 | Intra-trans, del | age | 0.361 | 0.014 |
| ERG | dup | d15MRD | 0.361 | 0.035 |
| NF1 | del | d15MRD | 0.302 | 0.044 |
| CREBBP | del | d33MRD | 0.340 | 0.024 |
| ERG | dup | d33MRD | 0.372 | 0.013 |
| IKZF1 | Intra-trans, del | d33MRD | 0.394 | 0.008 |
| NF1 | del | d33MRD | 0.283 | 0.042 |
| SH2B3 | Inter-trans, del | d33MRD | 0.329 | 0.029 |
| BTG1 | del | d78MRD | 0.383 | 0.011 |
| CREBBP | del | d78MRD | 0.488 | 0.001 |
| KMT2A | Inter-trans, del | d78MRD | 0.363 | 0.017 |
| PIK3CA | del | d78MRD | 0.463 | 0.002 |
| CREBBP | del | Risk stratification | 0.318 | 0.033 |
| ERG | dup | Risk stratification | 0.318 | 0.033 |
| KMT2A | Inter-trans, del | Risk stratification | 0.394 | 0.007 |
| SH2B3 | Inter-trans, del | Risk stratification | 0.318 | 0.033 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, H.; Xu, H.; Wang, C.; Cui, L.; Huang, X.; Li, W.; Yue, Z.; Tian, S.; Zhao, X.; Xue, T.; et al. Optical Genome Mapping for Comprehensive Assessment of Chromosomal Aberrations and Discovery of New Fusion Genes in Pediatric B-Acute Lymphoblastic Leukemia. Cancers 2023, 15, 35. https://doi.org/10.3390/cancers15010035
Gao H, Xu H, Wang C, Cui L, Huang X, Li W, Yue Z, Tian S, Zhao X, Xue T, et al. Optical Genome Mapping for Comprehensive Assessment of Chromosomal Aberrations and Discovery of New Fusion Genes in Pediatric B-Acute Lymphoblastic Leukemia. Cancers. 2023; 15(1):35. https://doi.org/10.3390/cancers15010035
Chicago/Turabian StyleGao, Huixia, Hanli Xu, Chanjuan Wang, Lei Cui, Xiaotong Huang, Weijing Li, Zhixia Yue, Shuo Tian, Xiaoxi Zhao, Tianlin Xue, and et al. 2023. "Optical Genome Mapping for Comprehensive Assessment of Chromosomal Aberrations and Discovery of New Fusion Genes in Pediatric B-Acute Lymphoblastic Leukemia" Cancers 15, no. 1: 35. https://doi.org/10.3390/cancers15010035
APA StyleGao, H., Xu, H., Wang, C., Cui, L., Huang, X., Li, W., Yue, Z., Tian, S., Zhao, X., Xue, T., Xing, T., Li, J., Wang, Y., Zhang, R., Li, Z., & Wang, T. (2023). Optical Genome Mapping for Comprehensive Assessment of Chromosomal Aberrations and Discovery of New Fusion Genes in Pediatric B-Acute Lymphoblastic Leukemia. Cancers, 15(1), 35. https://doi.org/10.3390/cancers15010035