
CD123 and More: How to Target the Cell Surface of Blastic Plasmacytoid Dendritic Cell Neoplasm

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
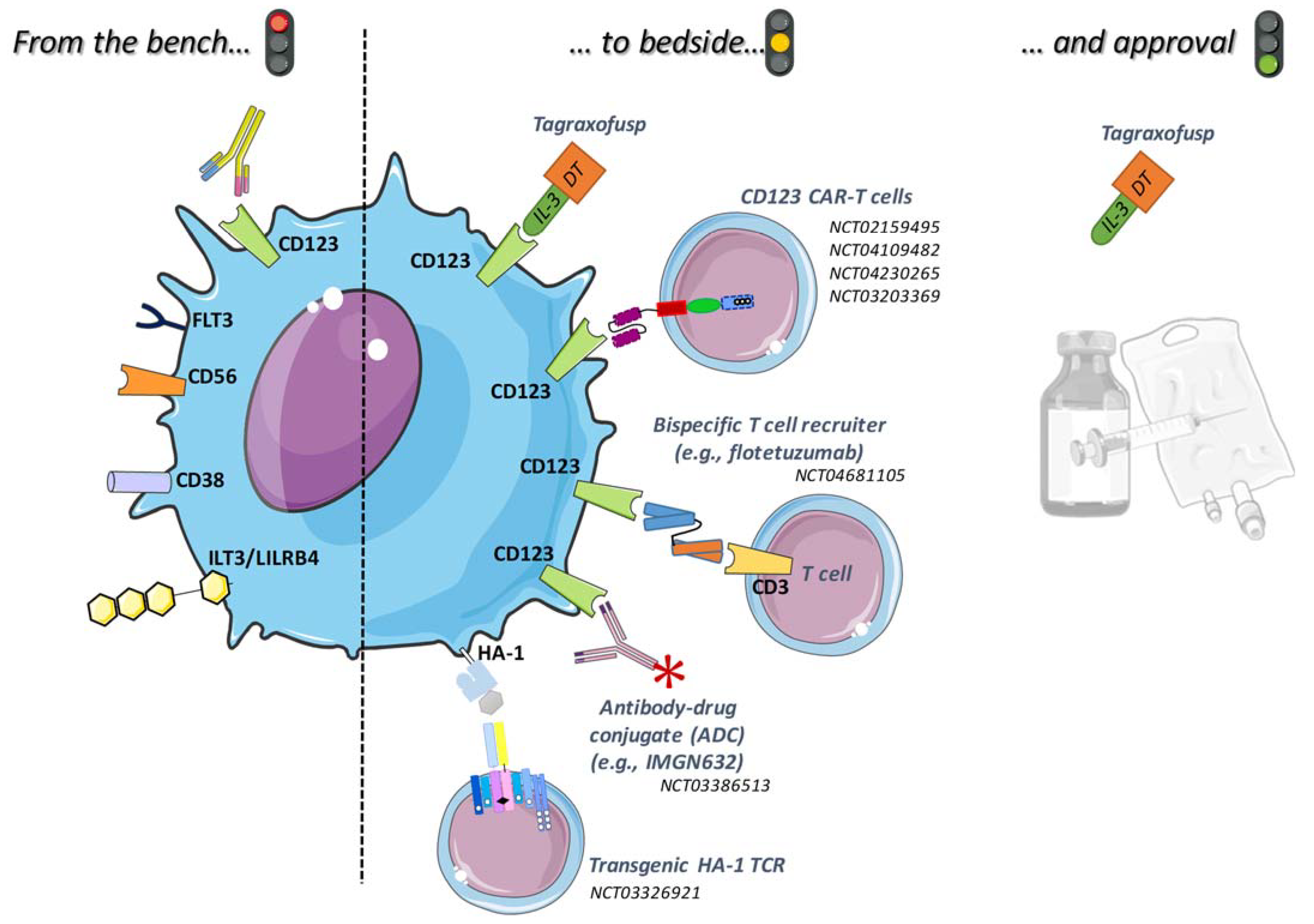
2. Targeted Therapies with Results or in Evaluation for BPDCN
2.1. Tagraxofusp
2.2. Monoclonal or Conjugated Antibodies
2.3. Bispecific Antibodies
2.4. CAR-T Cells
2.5. Promising New Targets
2.5.1. CD38
2.5.2. HA-1H
2.5.3. CD56
2.5.4. ILT3
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pemmaraju, N.; Lane, A.A.; Sweet, K.L.; Stein, A.S.; Vasu, S.; Blum, W.; Rizzieri, D.A.; Wang, E.S.; Duvic, M.; Sloan, J.M.; et al. Tagraxofusp in Blastic Plasmacytoid Dendritic-Cell Neoplasm. N. Engl. J. Med. 2019, 380, 1628–1637. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.; Stephansky, J.; Cai, T.; Griffin, G.K.; Cabal-Hierro, L.; Togami, K.; Hogdal, L.J.; Galinsky, I.; Morgan, E.A.; Aster, J.C.; et al. Blastic Plasmacytoid Dendritic Cell Neoplasm Is Dependent on BCL2 and Sensitive to Venetoclax. Cancer Discov. 2017, 7, 156–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philippe, L.; Ceroi, A.; Bôle-Richard, E.; Jenvrin, A.; Biichle, S.; Perrin, S.; Limat, S.; Bonnefoy, F.; Deconinck, E.; Saas, P.; et al. Bortezomib as a New Therapeutic Approach for Blastic Plasmacytoid Dendritic Cell Neoplasm. Haematologica 2017, 102, 1861–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marmouset, V.; Joris, M.; Merlusca, L.; Beaumont, M.; Charbonnier, A.; Marolleau, J.-P.; Gruson, B. The Lenalidomide/Bortezomib/Dexamethasone Regimen for the Treatment of Blastic Plasmacytoid Dendritic Cell Neoplasm. Hematol. Oncol. 2019, 37, 487–489. [Google Scholar] [CrossRef] [PubMed]
- Sapienza, M.R.; Fuligni, F.; Agostinelli, C.; Tripodo, C.; Righi, S.; Laginestra, M.A.; Pileri, A.; Mancini, M.; Rossi, M.; Ricci, F.; et al. Molecular Profiling of Blastic Plasmacytoid Dendritic Cell Neoplasm Reveals a Unique Pattern and Suggests Selective Sensitivity to NF-KB Pathway Inhibition. Leukemia 2014, 28, 1606–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garnache-Ottou, F.; Vidal, C.; Biichlé, S.; Renosi, F.; Poret, E.; Pagadoy, M.; Desmarets, M.; Roggy, A.; Seilles, E.; Soret, L.; et al. How Should We Diagnose and Treat Blastic Plasmacytoid Dendritic Cell Neoplasm Patients? Blood Adv. 2019, 3, 4238–4251. [Google Scholar] [CrossRef] [Green Version]
- Dalle, S.; Beylot-Barry, M.; Bagot, M.; Lipsker, D.; Machet, L.; Joly, P.; Dompmartin, A.; D’Incan, M.; Maubec, E.; Grange, F.; et al. Blastic Plasmacytoid Dendritic Cell Neoplasm: Is Transplantation the Treatment of Choice? Br. J. Dermatol. 2010, 162, 74–79. [Google Scholar] [CrossRef]
- Pagano, L.; Valentini, C.G.; Grammatico, S.; Pulsoni, A. Blastic Plasmacytoid Dendritic Cell Neoplasm: Diagnostic Criteria and Therapeutical Approaches. Br. J. Haematol. 2016, 174, 188–202. [Google Scholar] [CrossRef]
- Roos-Weil, D.; Dietrich, S.; Boumendil, A.; Polge, E.; Bron, D.; Carreras, E.; Iriondo Atienza, A.; Arcese, W.; Beelen, D.W.; Cornelissen, J.J.; et al. Stem Cell Transplantation Can Provide Durable Disease Control in Blastic Plasmacytoid Dendritic Cell Neoplasm: A Retrospective Study from the European Group for Blood and Marrow Transplantation. Blood 2013, 121, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Bashir, Q.; Milton, D.R.; Popat, U.R.; Kebriaei, P.; Hosing, C.; Khouri, I.F.; Rezvani, K.; Nieto, Y.; Oran, B.; Srour, S.A.; et al. Allogeneic Hematopoietic Cell Transplantation for Patients with Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN). Bone Marrow Transpl. 2021, 7, 51–56. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couzin-Frankel, J. Cancer Immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridgeman, J.S.; Hawkins, R.E.; Bagley, S.; Blaylock, M.; Holland, M.; Gilham, D.E. The Optimal Antigen Response of Chimeric Antigen Receptors Harboring the CD3ζ Transmembrane Domain Is Dependent upon Incorporation of the Receptor into the Endogenous TCR/CD3 Complex. J. Immunol. 2010, 184, 6938–6949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezzanzanica, D.; Canevari, S.; Mazzoni, A.; Figini, M.; Colnaghi, M.I.; Waks, T.; Schindler, D.G.; Eshhar, Z. Transfer of Chimeric Receptor Gene Made of Variable Regions of Tumor-Specific Antibody Confers Anticarbohydrate Specificity on T Cells. Cancer Gene Ther. 1998, 5, 401–407. [Google Scholar]
- Becker, M.L.B.; Near, R.; Mudgett-Hunter, M.; Margolies, M.N.; Kubo, R.T.; Kaye, J.; Hedrick, S.M. Expression of a Hybrid Immunoglobulin-T Cell Receptor Protein in Transgenic Mice. Cell 1989, 58, 911–921. [Google Scholar] [CrossRef]
- Goverman, J.; Gomez, S.M.; Segesman, K.D.; Hunkapiller, T.; Laug, W.E.; Hood, L. Chimeric Immunoglobulin-T Cell Receptor Proteins Form Functional Receptors: Implications for T Cell Receptor Complex Formation and Activation. Cell 1990, 60, 929–939. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of Immunoglobulin-T-Cell Receptor Chimeric Molecules as Functional Receptors with Antibody-Type Specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [Green Version]
- Kuwana, Y.; Asakura, Y.; Utsunomiya, N.; Nakanishi, M.; Arata, Y.; Itoh, S.; Nagase, F.; Kurosawa, Y. Expression of Chimeric Receptor Composed of Immunoglobulin-Derived V Regions and T-Cell Receptor-Derived C Regions. Biochem. Biophys. Res. Commun. 1987, 149, 960–968. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific Activation and Targeting of Cytotoxic Lymphocytes through Chimeric Single Chains Consisting of Antibody-Binding Domains and the Gamma or Zeta Subunits of the Immunoglobulin and T-Cell Receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Eshhar, Z.; Bach, N.; Fitzer-Attas, C.J.; Grosse, G.; Lustgarten, J.; Waks, T.; Schindler, D.G. The T-Body Approach: Potential for Cancer Immunotherapy. Springer Semin. Immunopathol. 1996, 18, 199–209. [Google Scholar] [CrossRef]
- Hwu, P.; Yang, J.C.; Cowherd, R.; Treisman, J.; Shafer, G.E.; Eshhar, Z.; Rosenberg, S.A. In Vivo Antitumor Activity of T Cells Redirected with Chimeric Antibody/T-Cell Receptor Genes. Cancer Res. 1995, 55, 3369–3373. [Google Scholar] [PubMed]
- Ramos, C.A.; Dotti, G. Chimeric Antigen Receptor (CAR)-Engineered Lymphocytes for Cancer Therapy. Expert Opin. Biol. Ther. 2011, 11, 855–873. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Brentjens, R.; Riviere, I. The Basic Principles of Chimeric Antigen Receptor (CAR) Design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Jensen, M.; Lin, Y.; Sui, X.; Chen, E.; Lindgren, C.G.; Till, B.; Raubitschek, A.; Forman, S.J.; Qian, X.; et al. Optimizing Adoptive Polyclonal T Cell Immunotherapy of Lymphomas, Using a Chimeric T Cell Receptor Possessing CD28 and CD137 Costimulatory Domains. Hum. Gene Ther. 2007, 18, 712–725. [Google Scholar] [CrossRef]
- Stoiber, S.; Cadilha, B.L.; Benmebarek, M.-R.; Lesch, S.; Endres, S.; Kobold, S. Limitations in the Design of Chimeric Antigen Receptors for Cancer Therapy. Cells 2019, 8, 472. [Google Scholar] [CrossRef] [Green Version]
- Gomes-Silva, D.; Mukherjee, M.; Srinivasan, M.; Krenciute, G.; Dakhova, O.; Zheng, Y.; Cabral, J.M.S.; Rooney, C.M.; Orange, J.S.; Brenner, M.K.; et al. Tonic 4-1BB Costimulation in Chimeric Antigen Receptors Impedes T Cell Survival and Is Vector Dependent. Cell Rep. 2017, 21, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Angelot-Delettre, F.; Roggy, A.; Frankel, A.E.; Lamarthee, B.; Seilles, E.; Biichle, S.; Royer, B.; Deconinck, E.; Rowinsky, E.K.; Brooks, C.; et al. In Vivo and in Vitro Sensitivity of Blastic Plasmacytoid Dendritic Cell Neoplasm to SL-401, an Interleukin-3 Receptor Targeted Biologic Agent. Haematologica 2015, 100, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Garnache-Ottou, F.; Feuillard, J.; Ferrand, C.; Biichle, S.; Trimoreau, F.; Seilles, E.; Salaun, V.; Garand, R.; Lepelley, P.; Maynadié, M.; et al. Extended Diagnostic Criteria for Plasmacytoid Dendritic Cell Leukaemia. Br. J. Haematol. 2009, 145, 624–636. [Google Scholar] [CrossRef]
- Testa, U.; Pelosi, E.; Frankel, A. CD 123 Is a Membrane Biomarker and a Therapeutic Target in Hematologic Malignancies. Biomark Res. 2014, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Frankel, A.; Liu, J.-S.; Rizzieri, D.; Hogge, D. Phase I Clinical Study of Diphtheria Toxin-Interleukin 3 Fusion Protein in Patients with Acute Myeloid Leukemia and Myelodysplasia. Leuk. Lymphoma 2008, 49, 543–553. [Google Scholar] [CrossRef]
- FitzGerald, D.J. Targeted Diphtheria Toxin to Treat BPDCN. Blood 2014, 124, 310–312. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.E.; Woo, J.H.; Ahn, C.; Pemmaraju, N.; Medeiros, B.C.; Carraway, H.E.; Frankfurt, O.; Forman, S.J.; Yang, X.A.; Konopleva, M.; et al. Activity of SL-401, a Targeted Therapy Directed to Interleukin-3 Receptor, in Blastic Plasmacytoid Dendritic Cell Neoplasm Patients. Blood 2014, 124, 385–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pemmaraju, N.; Konopleva, M. Approval of Tagraxofusp-Erzs for Blastic Plasmacytoid Dendritic Cell Neoplasm. Blood Adv. 2020, 4, 4020–4027. [Google Scholar] [CrossRef]
- Wilson, N.R.; Konopleva, M.; Khoury, J.D.; Pemmaraju, N. Novel Therapeutic Approaches in Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN): Era of Targeted Therapy. Clin. Lymphoma Myeloma Leuk. 2021, 21, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, M.M.; Mughal, T.I.; Brooks, C.; Lindsay, R.; Pemmaraju, N. Targeting CD123 in Hematologic Malignancies: Identifying Suitable Patients for Targeted Therapy. Leuk. Lymphoma 2021, 62, 2568–2586. [Google Scholar] [CrossRef]
- Lane, A.A. Targeting CD123 in AML. Clin. Lymphoma Myeloma Leuk. 2020, 20 (Suppl. 1), S67–S68. [Google Scholar] [CrossRef]
- Lane, A.A.; Stein, A.S.; Garcia, J.S.; Garzon, J.L.; Galinsky, I.; Luskin, M.R.; Stone, R.M.; Winer, E.S.; Leonard, R.; Mughal, T.I.; et al. Safety and Efficacy of Combining Tagraxofusp (SL-401) with Azacitidine or Azacitidine and Venetoclax in a Phase 1b Study for CD123 Positive AML, MDS, or BPDCN. Blood 2021, 138, 2346. [Google Scholar] [CrossRef]
- He, S.Z.; Busfield, S.; Ritchie, D.S.; Hertzberg, M.S.; Durrant, S.; Lewis, I.D.; Marlton, P.; McLachlan, A.J.; Kerridge, I.; Bradstock, K.F.; et al. A Phase 1 Study of the Safety, Pharmacokinetics and Anti-Leukemic Activity of the Anti-CD123 Monoclonal Antibody CSL360 in Relapsed, Refractory or High-Risk Acute Myeloid Leukemia. Leuk. Lymphoma 2015, 56, 1406–1415. [Google Scholar] [CrossRef]
- Li, F.; Sutherland, M.K.; Yu, C.; Walter, R.B.; Westendorf, L.; Valliere-Douglass, J.; Pan, L.; Cronkite, A.; Sussman, D.; Klussman, K.; et al. Characterization of SGN-CD123A, A Potent CD123-Directed Antibody–Drug Conjugate for Acute Myeloid Leukemia. Mol. Cancer Ther. 2018, 17, 554–564. [Google Scholar] [CrossRef] [Green Version]
- Archer, K.E.; Reid, E.E.; Shizuka, M.; Woods, J.; Harris, L.; Maloney, E.K.; Bartle, L.M.; Ab, O.; Wilhelm, A.; Setiady, Y.; et al. Synthesis of Highly Potent N-10 Amino-Linked DNA-Alkylating Indolinobenzodiazepine Antibody–Drug Conjugates (ADCs). ACS Med. Chem. Lett. 2019, 10, 1211–1215. [Google Scholar] [CrossRef]
- Kovtun, Y.; Noordhuis, P.; Whiteman, K.R.; Watkins, K.; Jones, G.E.; Harvey, L.; Lai, K.C.; Portwood, S.; Adams, S.; Sloss, C.M.; et al. IMGN779, a Novel CD33-Targeting Antibody–Drug Conjugate with DNA-Alkylating Activity, Exhibits Potent Antitumor Activity in Models of AML. Mol. Cancer Ther. 2018, 17, 1271–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovtun, Y.; Jones, G.E.; Adams, S.; Harvey, L.; Audette, C.A.; Wilhelm, A.; Bai, C.; Rui, L.; Laleau, R.; Liu, F.; et al. A CD123-Targeting Antibody-Drug Conjugate, IMGN632, Designed to Eradicate AML While Sparing Normal Bone Marrow Cells. Blood Adv. 2018, 2, 848–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pemmaraju, N.; Martinelli, G.; Todisco, E.; Lane, A.A.; Acuña-Cruz, E.; Deconinck, E.; Wang, E.S.; Sweet, K.L.; Rizzieri, D.A.; Mazzarella, L.; et al. Clinical Profile of IMGN632, a Novel CD123-Targeting Antibody-Drug Conjugate (ADC), in Patients with Relapsed/Refractory (R/R) Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN). Blood 2020, 136, 11–13. [Google Scholar] [CrossRef]
- Allen, C.; Zeidan, A.M.; Bewersdorf, J.P. BiTEs, DARTS, BiKEs and TriKEs—Are Antibody Based Therapies Changing the Future Treatment of AML? Life 2021, 11, 465. [Google Scholar] [CrossRef] [PubMed]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as Salvage Immunotherapy for Refractory Acute Myeloid Leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Ravandi, F.; Bashey, A.; Foran, J.M.; Stock, W.; Mawad, R.; Blum, W.; Saville, M.W.; Johnson, C.M.; Vanasse, K.G.J.; Ly, T.; et al. Complete Responses in Relapsed/Refractory Acute Myeloid Leukemia (AML) Patients on a Weekly Dosing Schedule of XmAb14045, a CD123 x CD3 T Cell-Engaging Bispecific Antibody: Initial Results of a Phase 1 Study. Blood 2018, 132, 763. [Google Scholar] [CrossRef]
- Watts, J.M.; Lin, T.; Wang, E.S.; Mims, A.S.; Cull, E.H.; Patel, P.A.; Shami, P.J.; Walter, R.B.; Cogle, C.R.; Chenault, R.A.; et al. Preliminary Results from a Phase 1 Study of APVO436, a Novel Anti-CD123 x Anti-CD3 Bispecific Molecule, in Relapsed/Refractory Acute Myeloid Leukemia and Myelodysplastic Syndrome. Blood 2020, 136, 11–12. [Google Scholar] [CrossRef]
- Brentjens, R.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef] [Green Version]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef] [Green Version]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. New Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [Green Version]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.T.; Carpenter, R.O.; Stetler-Stevenson, M.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-Refractory Diffuse Large B-Cell Lymphoma and Indolent B-Cell Malignancies Can Be Effectively Treated With Autologous T Cells Expressing an Anti-CD19 Chimeric Antigen Receptor. J. Clin. Oncol. 2015, 33, 540–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadelain, M. CAR Therapy: The CD19 Paradigm. J. Clin. Investig. 2015, 125, 3392–3400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Mardiros, A.; Dos Santos, C.; McDonald, T.; Brown, C.E.; Wang, X.; Budde, L.E.; Hoffman, L.; Aguilar, B.; Chang, W.-C.; Bretzlaff, W.; et al. T Cells Expressing CD123-Specific Chimeric Antigen Receptors Exhibit Specific Cytolytic Effector Functions and Antitumor Effects against Human Acute Myeloid Leukemia. Blood 2013, 122, 3138–3148. [Google Scholar] [CrossRef] [Green Version]
- Gill, S.; Tasian, S.K.; Ruella, M.; Shestova, O.; Li, Y.; Porter, D.L.; Carroll, M.; Danet-Desnoyers, G.; Scholler, J.; Grupp, S.A.; et al. Preclinical Targeting of Human Acute Myeloid Leukemia and Myeloablation Using Chimeric Antigen Receptor–Modified T Cells. Blood 2014, 123, 2343–2354. [Google Scholar] [CrossRef] [Green Version]
- Tasian, S.K.; Kenderian, S.S.; Shen, F.; Ruella, M.; Shestova, O.; Kozlowski, M.; Li, Y.; Schrank-Hacker, A.; Morrissette, J.J.D.; Carroll, M.; et al. Optimized Depletion of Chimeric Antigen Receptor T Cells in Murine Xenograft Models of Human Acute Myeloid Leukemia. Blood 2017, 129, 2395–2407. [Google Scholar] [CrossRef] [Green Version]
- Bôle-Richard, E.; Fredon, M.; Biichlé, S.; Anna, F.; Certoux, J.-M.; Renosi, F.; Tsé, F.; Molimard, C.; Valmary-Degano, S.; Jenvrin, A.; et al. CD28/4-1BB CD123 CAR T Cells in Blastic Plasmacytoid Dendritic Cell Neoplasm. Leukemia 2020, 34, 3228–3241. [Google Scholar] [CrossRef]
- Loff, S.; Dietrich, J.; Meyer, J.-E.; Riewaldt, J.; Spehr, J.; von Bonin, M.; Gründer, C.; Swayampakula, M.; Franke, K.; Feldmann, A.; et al. Rapidly Switchable Universal CAR-T Cells for Treatment of CD123-Positive Leukemia. Mol. Ther. Oncolytics. 2020, 17, 408–420. [Google Scholar] [CrossRef]
- Cai, T.; Galetto, R.; Gouble, A.; Smith, J.; Cavazos, A.; Konoplev, S.; Lane, A.A.; Guzman, M.L.; Kantarjian, H.M.; Pemmaraju, N.; et al. Pre-Clinical Studies of Anti-CD123 CAR-T Cells for the Treatment of Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN). Blood 2016, 128, 4039. [Google Scholar] [CrossRef]
- Riberdy, J.M.; Zhou, S.; Zheng, F.; Kim, Y.-I.; Moore, J.; Vaidya, A.; Throm, R.E.; Sykes, A.; Sahr, N.; Bonifant, C.L.; et al. The Art and Science of Selecting a CD123-Specific Chimeric Antigen Receptor for Clinical Testing. Mol. Ther.—Methods Clin. Dev. 2020, 18, 571–581. [Google Scholar] [CrossRef]
- Wermke, M.; Kraus, S.; Ehninger, A.; Bargou, R.C.; Goebeler, M.-E.; Middeke, J.M.; Kreissig, C.; von Bonin, M.; Koedam, J.; Pehl, M.; et al. Proof of Concept for a Rapidly Switchable Universal CAR-T Platform with UniCAR-T-CD123 in Relapsed/Refractory AML. Blood 2021, 137, 3145–3148. [Google Scholar] [CrossRef] [PubMed]
- Deotare, U.; Yee, K.W.L.; Le, L.W.; Porwit, A.; Tierens, A.; Musani, R.; Barth, D.; Torlakovic, E.; Schimmer, A.; Schuh, A.C.; et al. Blastic Plasmacytoid Dendritic Cell Neoplasm with Leukemic Presentation: 10-Color Flow Cytometry Diagnosis and HyperCVAD Therapy. Am. J. Hematol. 2016, 91, 283–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iversen, K.F.; Holdgaard, P.C.; Preiss, B.; Nyvold, C.G.; Plesner, T. Daratumumab for Treatment of Blastic Plasmacytoid Dendritic Cell Neoplasm. A Single-Case Report. Haematologica 2019, 104, e432–e433. [Google Scholar] [CrossRef] [PubMed]
- Mirgh, S.; Sharma, A.; Folbs, B.; Khushoo, V.; Kapoor, J.; Tejwani, N.; Ahmed, R.; Agrawal, N.; Choudhary, P.S.; Mehta, P.; et al. Daratumumab-Based Therapy after Prior Azacytidine-Venetoclax in an Octagenerian Female with BPDCN (Blastic Plasmacytoid Dendritic Cell Neoplasm)—A New Perspective. Leuk. Lymphoma 2021, 62, 3039–3042. [Google Scholar] [CrossRef]
- van Loenen, M.M.; de Boer, R.; Hagedoorn, R.S.; van Egmond, E.H.M.; Falkenburg, J.H.F.; Heemskerk, M.H.M. Optimization of the HA-1-Specific T-Cell Receptor for Gene Therapy of Hematologic Malignancies. Haematologica 2011, 96, 477–481. [Google Scholar] [CrossRef]
- van Balen, P.; Jedema, I.; van Loenen, M.M.; de Boer, R.; van Egmond, H.M.; Hagedoorn, R.S.; Hoogstaten, C.; Veld, S.A.J.; Hageman, L.; van Liempt, P.A.G.; et al. HA-1H T-Cell Receptor Gene Transfer to Redirect Virus-Specific T Cells for Treatment of Hematological Malignancies After Allogeneic Stem Cell Transplantation: A Phase 1 Clinical Study. Front. Immunol. 2020, 11, 1804. [Google Scholar] [CrossRef]
- Esnault, C.; Leblond, V.; Martin, C.; Desgranges, A.; Baltus, C.B.; Aubrey, N.; Lakhrif, Z.; Lajoie, L.; Lantier, L.; Clémenceau, B.; et al. Adcitmer®, a New CD56-Targeting Monomethyl Auristatin E-Conjugated Antibody, Is a Potential Therapeutic Approach in Merkel Cell Carcinoma. Br. J. Dermatol. 2021, 186, 295–306. [Google Scholar] [CrossRef]
- Crossland, D.L.; Denning, W.L.; Ang, S.; Olivares, S.; Mi, T.; Switzer, K.; Singh, H.; Huls, H.; Gold, K.S.; Glisson, B.S.; et al. Antitumor Activity of CD56-Chimeric Antigen Receptor T Cells in Neuroblastoma and SCLC Models. Oncogene 2018, 37, 3686–3697. [Google Scholar] [CrossRef]
- Vlad, G.; Chang, C.-C.; Colovai, A.I.; Berloco, P.; Cortesini, R.; Suciu-Foca, N. Immunoglobulin-like Transcript 3: A Crucial Regulator of Dendritic Cell Function. Hum. Immunol. 2009, 70, 340–344. [Google Scholar] [CrossRef]
- John, S.; Chen, H.; Deng, M.; Gui, X.; Wu, G.; Chen, W.; Li, Z.; Zhang, N.; An, Z.; Zhang, C.C. A Novel Anti-LILRB4 CAR-T Cell for the Treatment of Monocytic AML. Mol. Ther. 2018, 26, 2487–2495. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
| CD123 CAR T-Cell Trials | ||||||
|---|---|---|---|---|---|---|
| NCT | System | Safety Switch | Condition/Disease | Dose | Phase | Status |
| NCT04318678 | CD123-CAR CD28 TM-CD28-CD3z | CD20 | AML, B-ALL, T-ALL, BPDCN | 3 × 105, 1 × 106, 3 × 106, 1 × 107 cells/kg | 1 | Recruiting |
| NCT02159495 | CD123-CAR IgG4 TM-CD28-CD3z | EGFRt | CD123+ diseases | 1 | Recruiting | |
| NCT04109482 | CD123-CAR IgG4 TM-CD28-CD3z (MB-102) | EGFRt | BPDCN | Up to 600 × 106 cells | 1/2 | Recruiting |
| NCT02623582 | CD123-CAR 41BB-CD3z (RNA electroporated) | AML | Early phase 1 | Terminated | ||
| NCT03766126 | CD123-CAR 41BB-CD3z (lentiviral transduced) | AML | 1–5 × 106 cells/kg | 1 | Active, not recruiting | |
| NCT03203369 | Allogenic UCART123-41BB-CD3z | RQR8 | BPDCN | 6.25 × 105 –6.25 × 106 cells/kg | 1 | Terminated |
| NCT03190278 | Allogenic UCART123 v1.2 -41BB-CD3z | RQR8 | AML | 1 | Recruiting | |
| NCT04678336 | CD123-CAR 41BB-CD3z | Pediatric AML | 2 × 106 cells/kg | 1 | Recruiting | |
| Other active non-CAR-T cell BPDCN trials | ||||||
| NCT | Agent(s) | Condition/Disease | Phase | Status | ||
| NCT03113643 | SL-401, venetoclax, azacitidine | BPDCN, AML, MDS | 1 | Recruiting | ||
| NCT03386513 | IMGN632 | BPDCN | 1/2 | Recruiting | ||
| NCT04216524 | SL-401, venetoclax, Hyper-CVAD | BPDCN | 1 | Recruiting | ||
| NCT04317781 | SL-401 | BPDCN after stem cell transplant | 2 | Active, not recruiting | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bôle-Richard, E.; Pemmaraju, N.; Caël, B.; Daguindau, E.; Lane, A.A. CD123 and More: How to Target the Cell Surface of Blastic Plasmacytoid Dendritic Cell Neoplasm. Cancers 2022, 14, 2287. https://doi.org/10.3390/cancers14092287
Bôle-Richard E, Pemmaraju N, Caël B, Daguindau E, Lane AA. CD123 and More: How to Target the Cell Surface of Blastic Plasmacytoid Dendritic Cell Neoplasm. Cancers. 2022; 14(9):2287. https://doi.org/10.3390/cancers14092287
Chicago/Turabian StyleBôle-Richard, Elodie, Naveen Pemmaraju, Blandine Caël, Etienne Daguindau, and Andrew A. Lane. 2022. "CD123 and More: How to Target the Cell Surface of Blastic Plasmacytoid Dendritic Cell Neoplasm" Cancers 14, no. 9: 2287. https://doi.org/10.3390/cancers14092287
APA StyleBôle-Richard, E., Pemmaraju, N., Caël, B., Daguindau, E., & Lane, A. A. (2022). CD123 and More: How to Target the Cell Surface of Blastic Plasmacytoid Dendritic Cell Neoplasm. Cancers, 14(9), 2287. https://doi.org/10.3390/cancers14092287