
Molecular Profiles of Advanced Urological Cancers in the PERMED-01 Precision Medicine Clinical Trial

 , , , , ,
, , , , ,  ,
,  , and add
Show full author list
, and add
Show full author list
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Inclusion Criteria
2.3. Genome Analysis
2.4. Multidisciplinary Molecular Tumor Board
2.5. Objectives, Endpoints and Statistical Analysis
3. Results
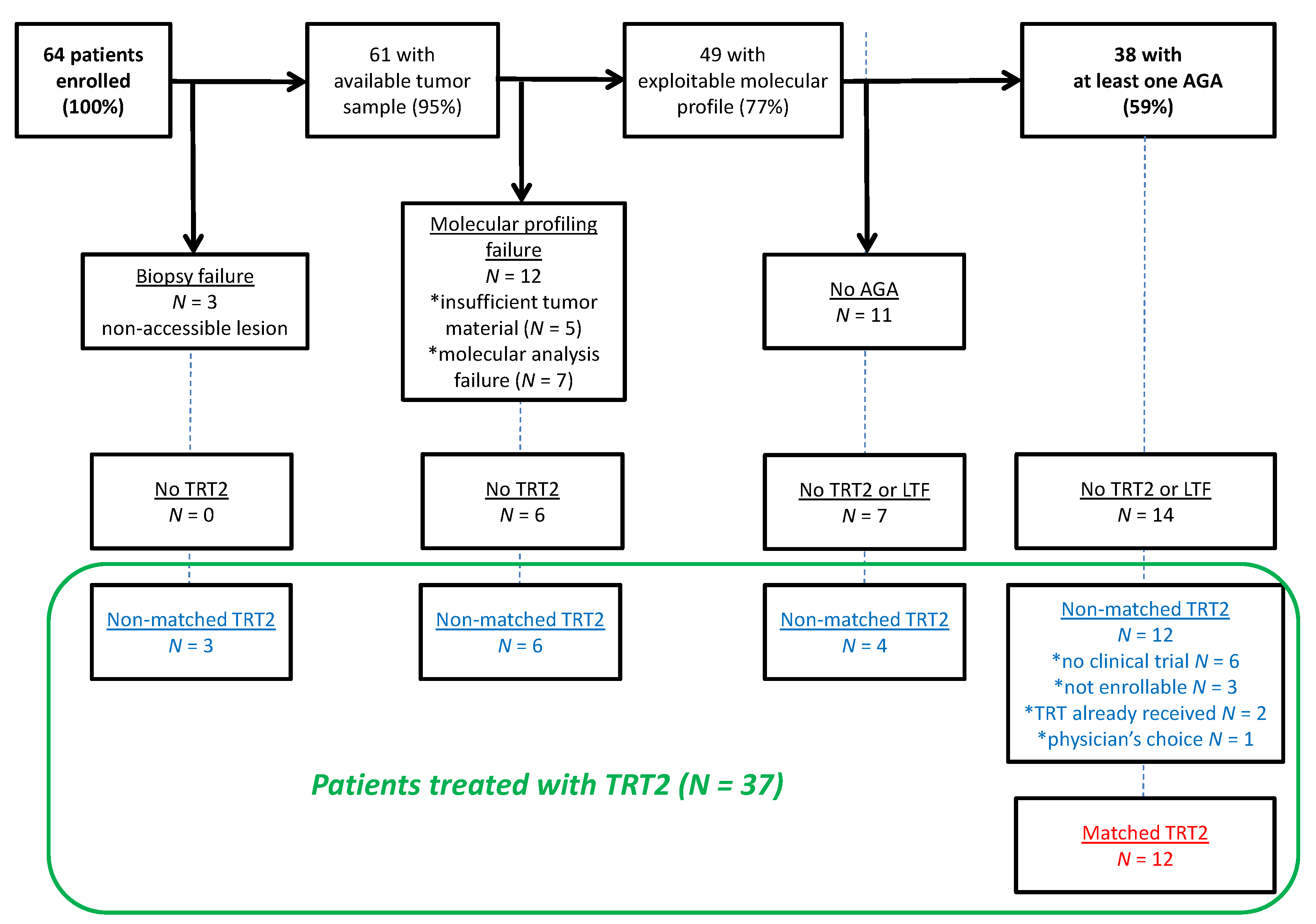
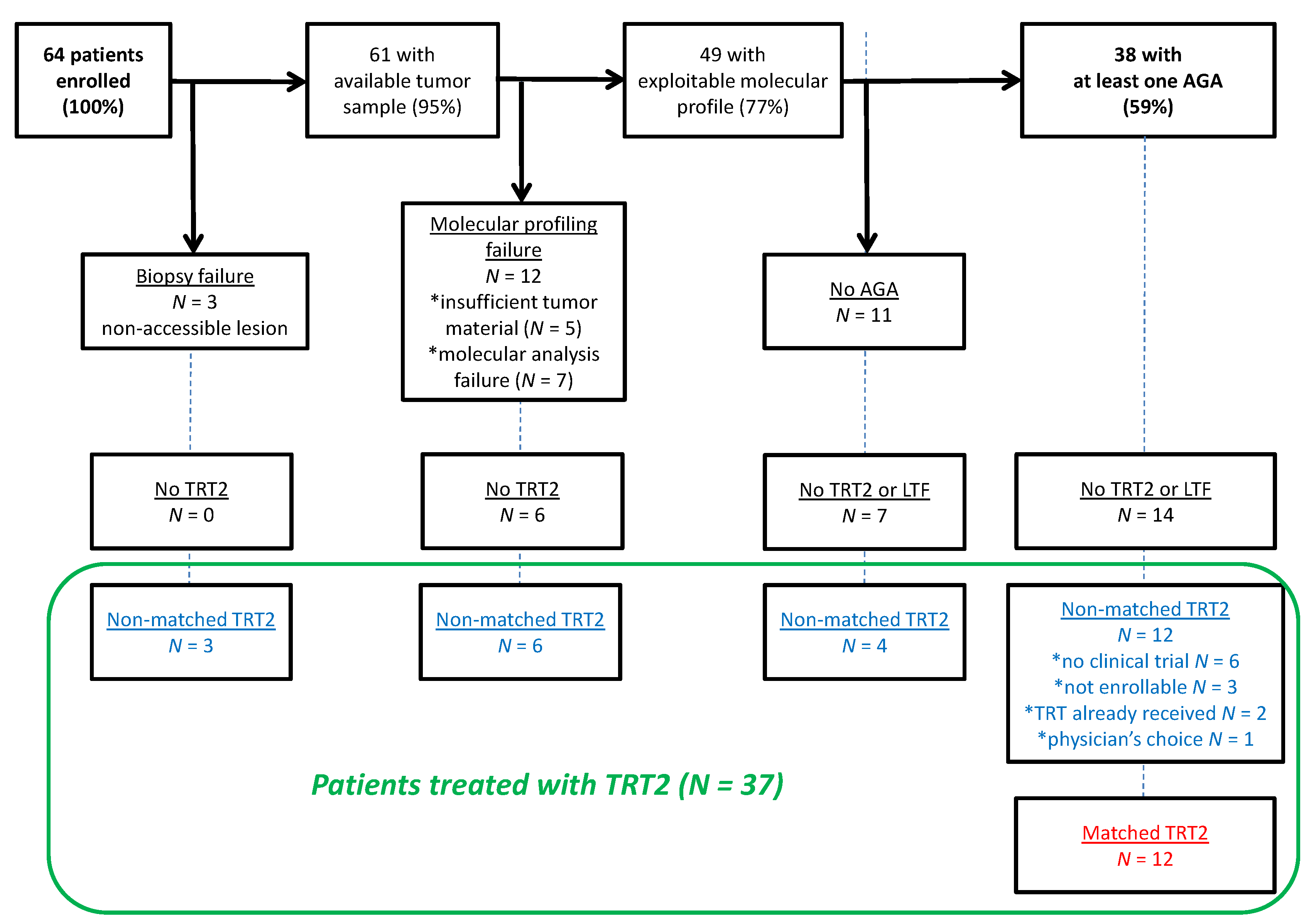
3.1. Patients’ Characteristics
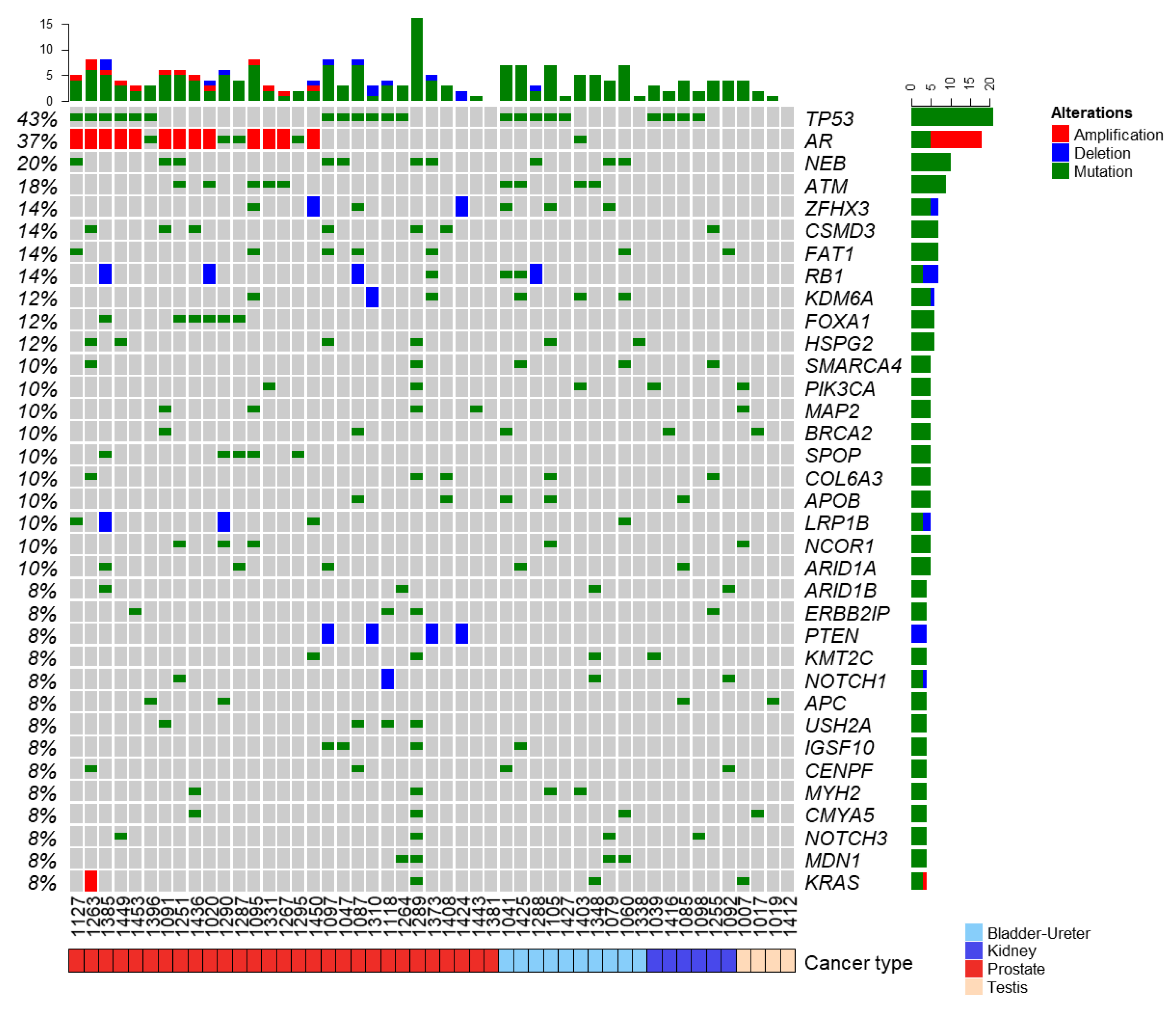
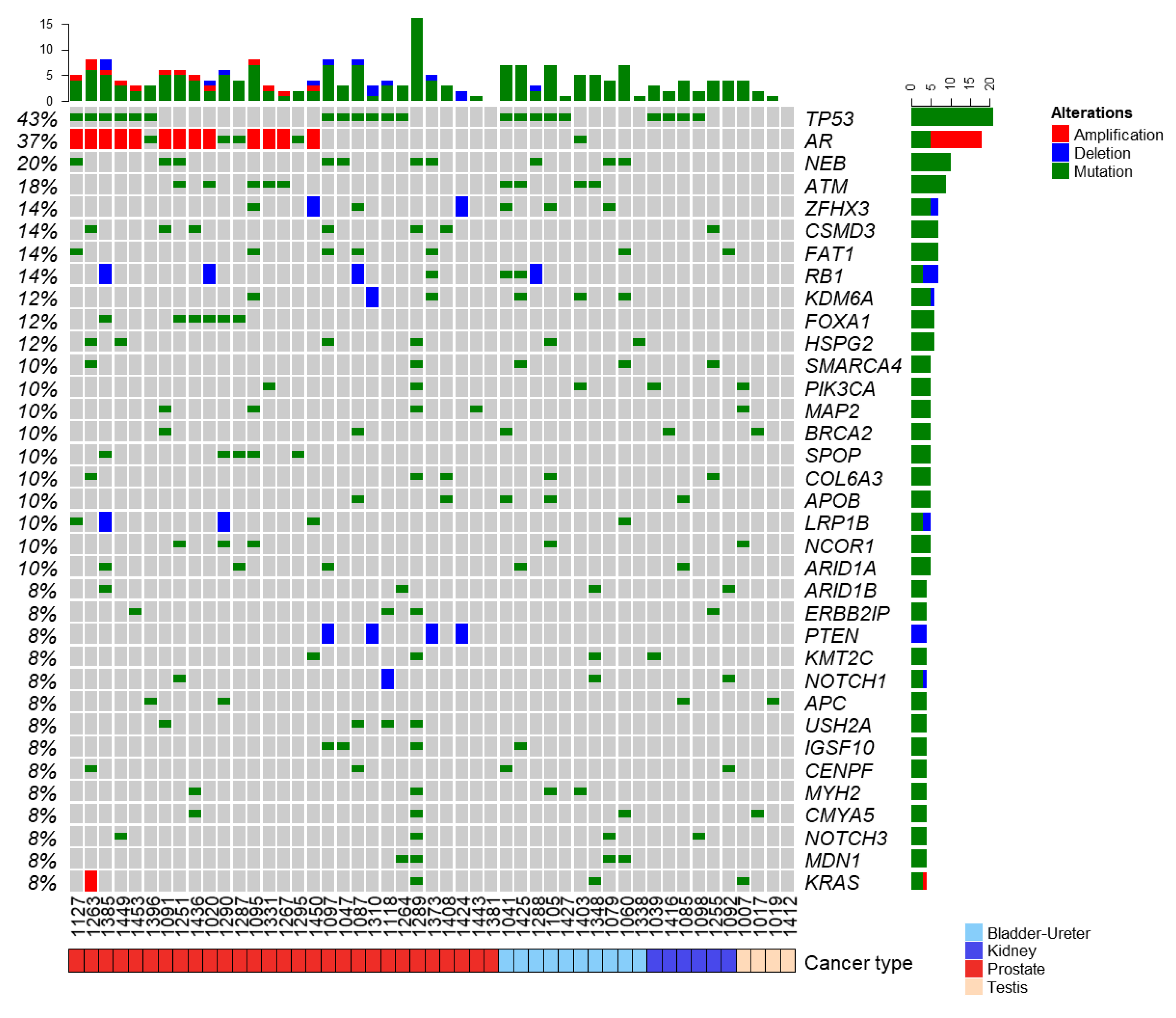
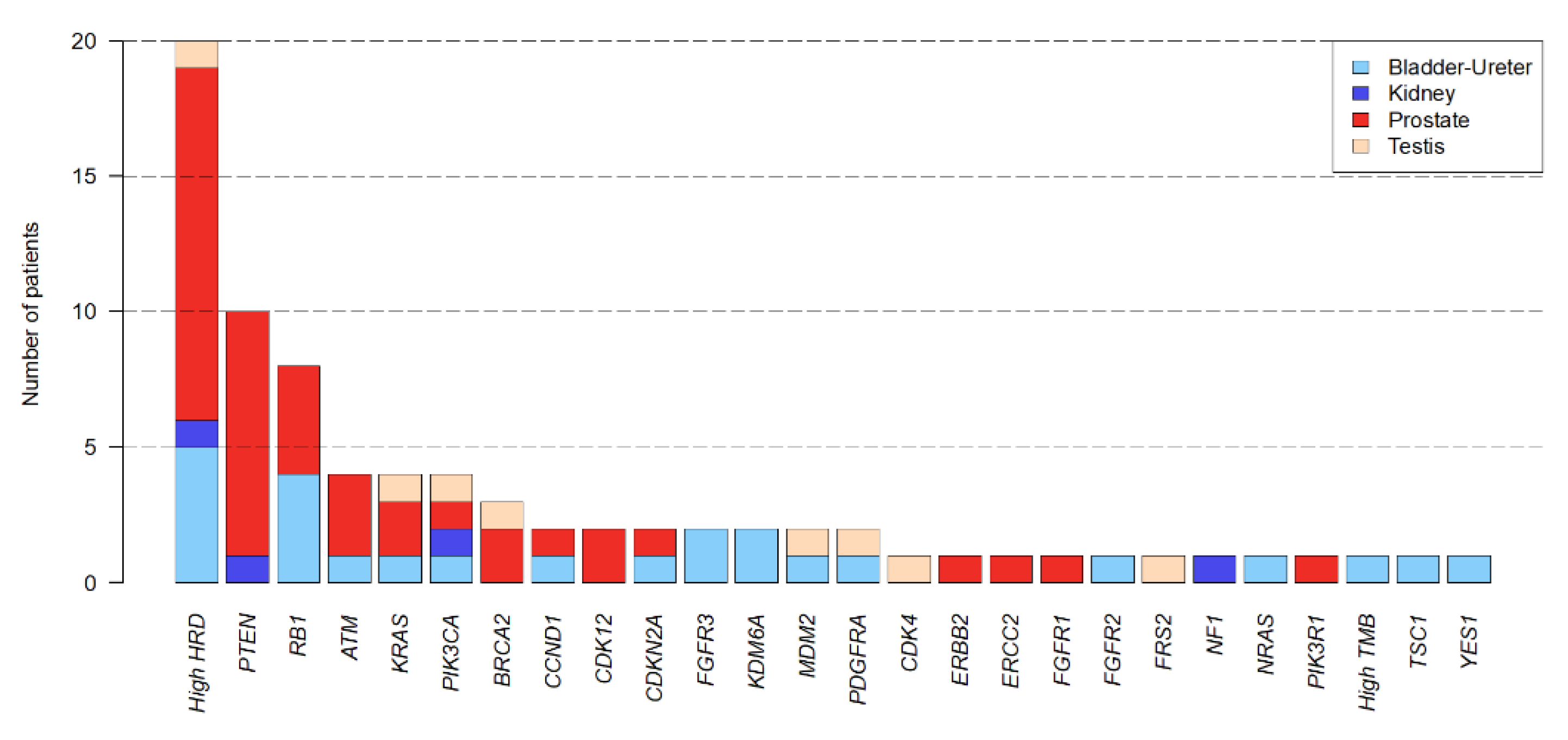
3.2. Landscape of Somatic and Germline Molecular Alterations
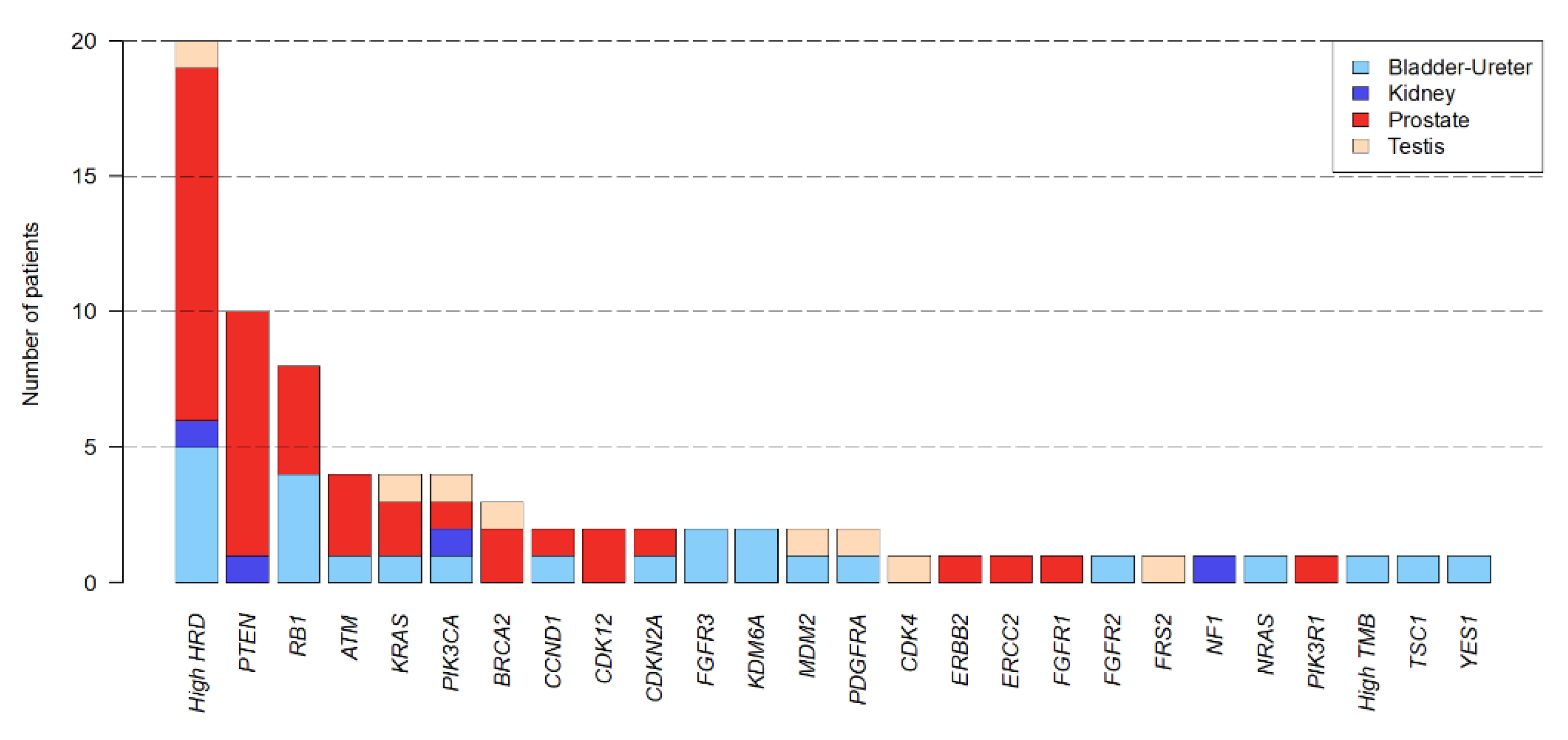
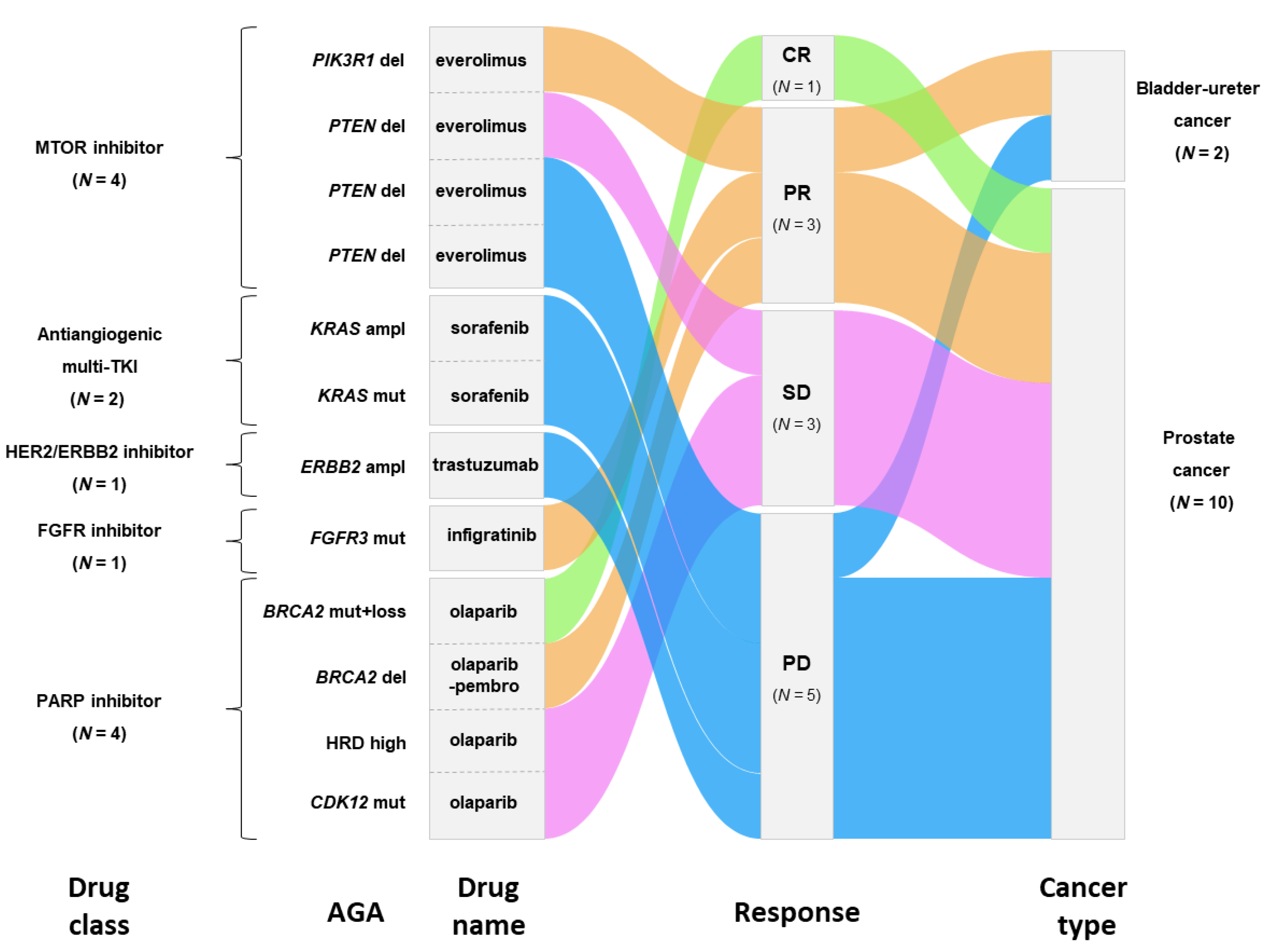
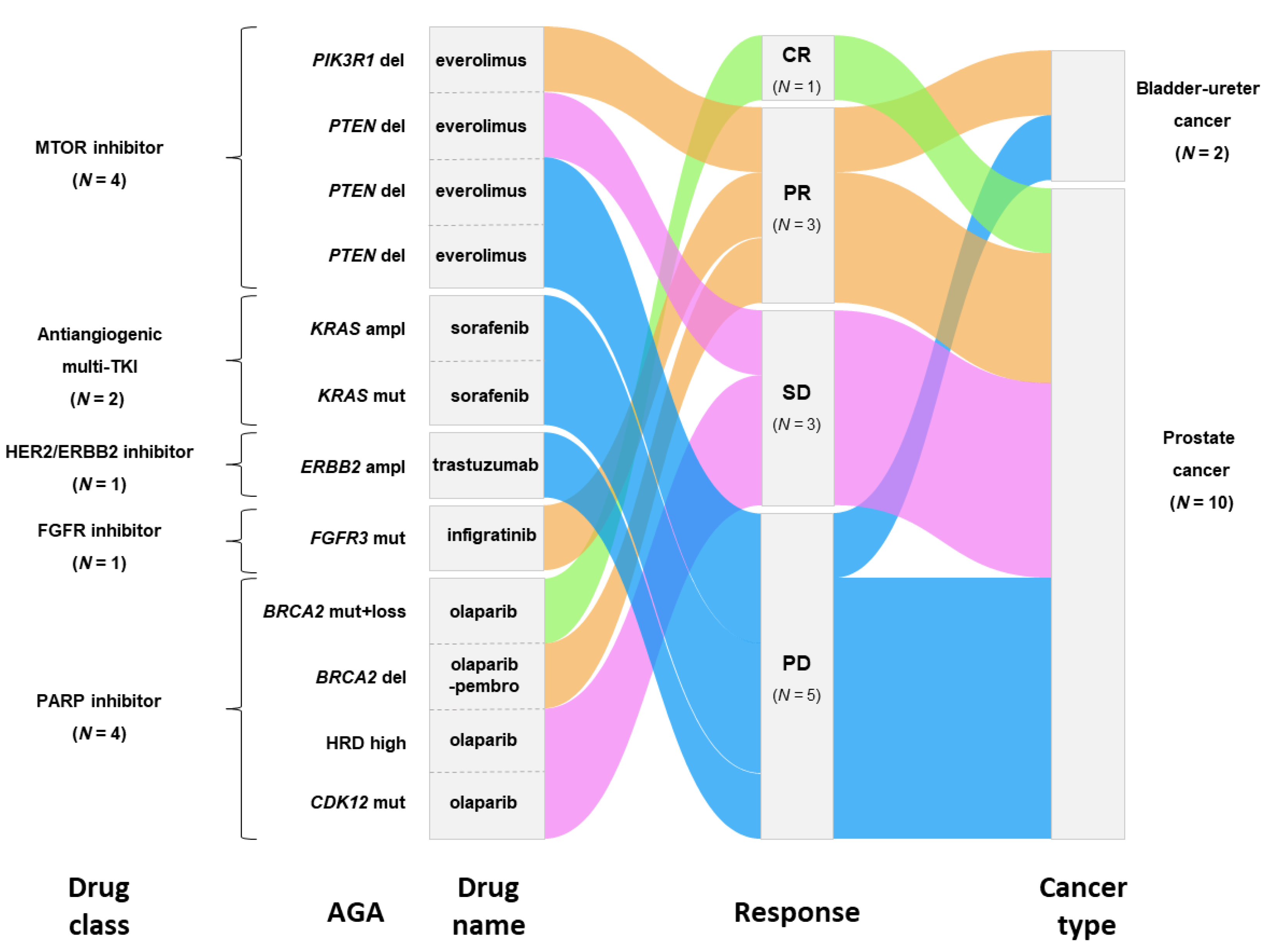
3.3. Actionable Genetic Alterations and Matched Therapy
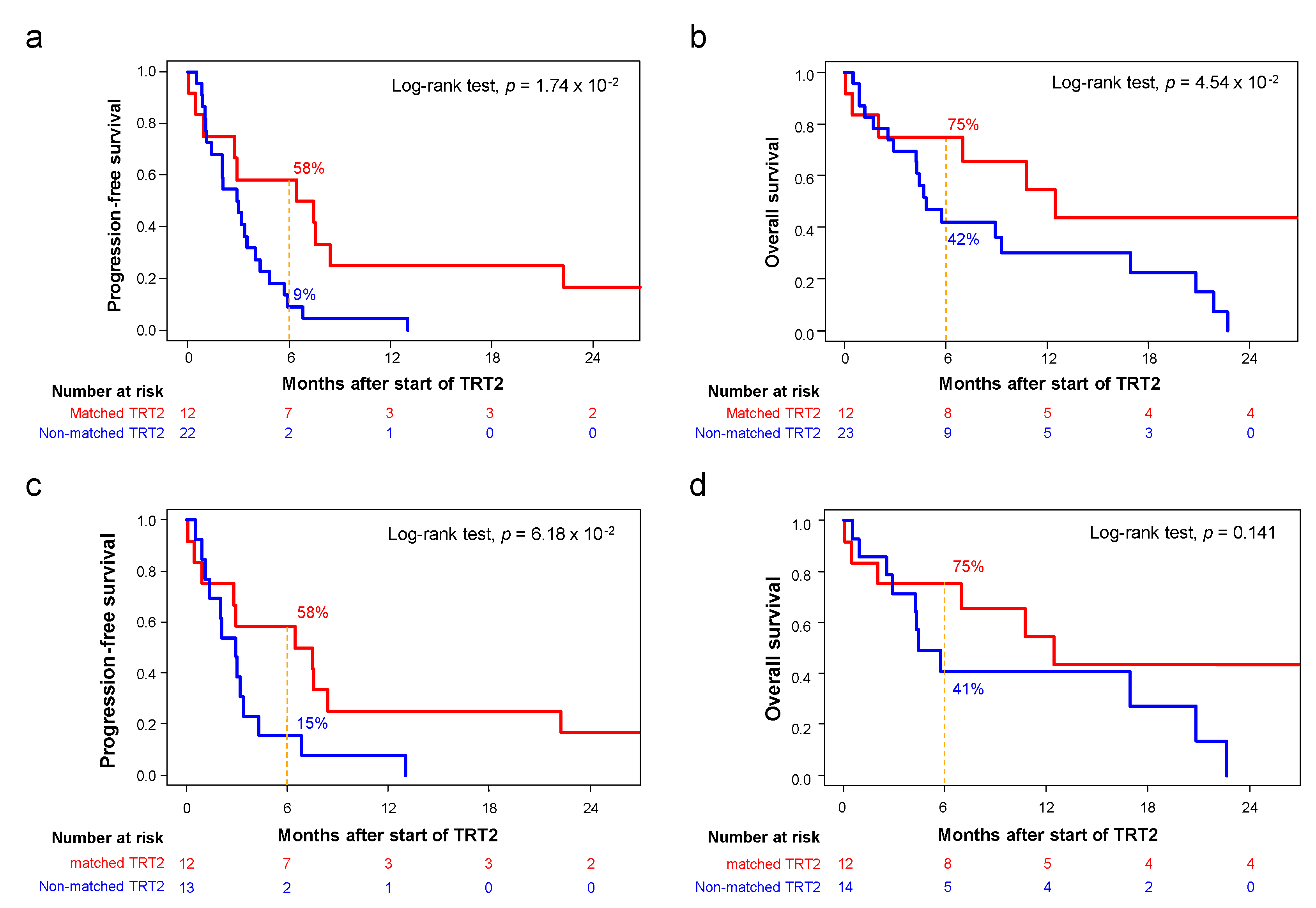
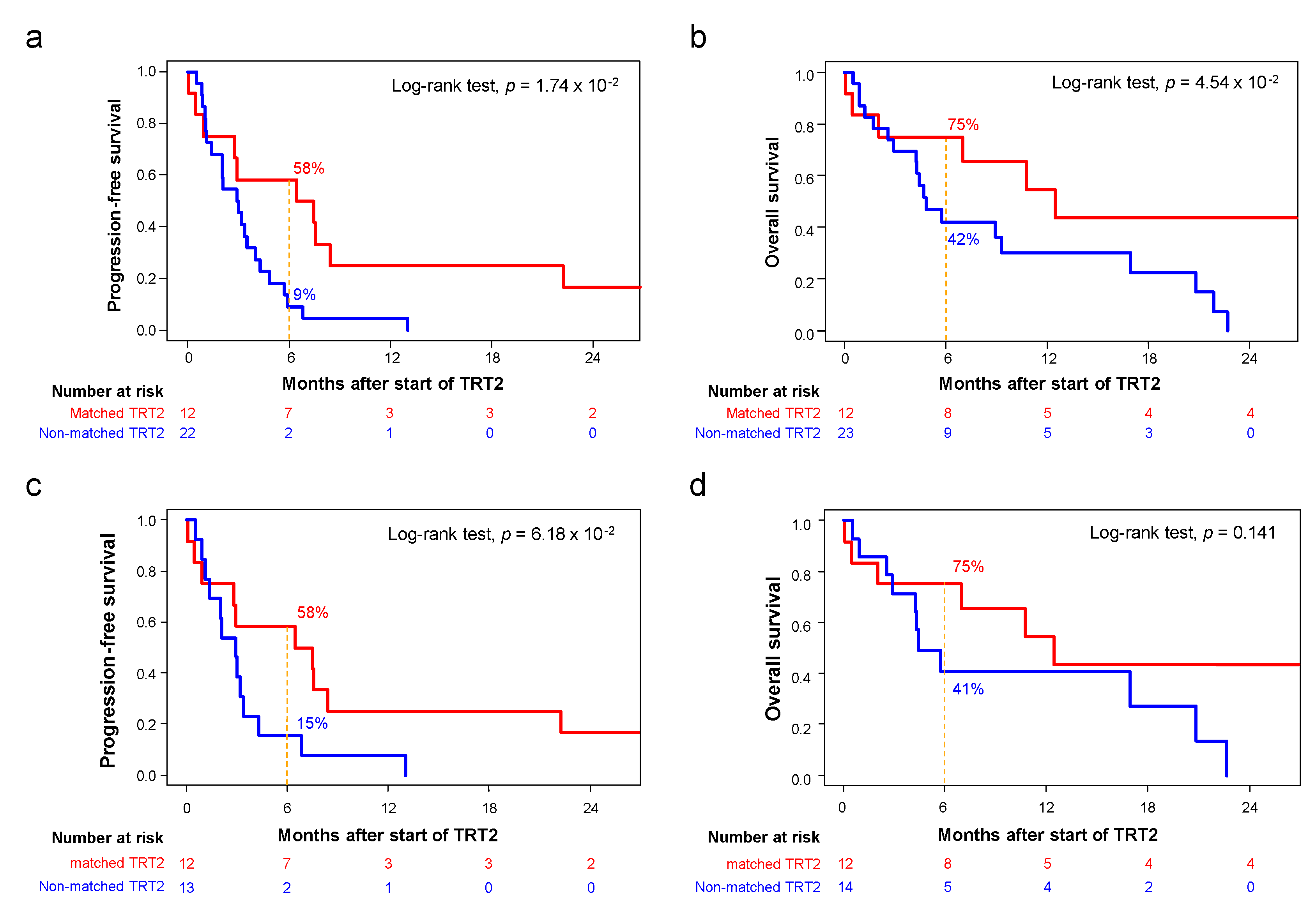
3.4. Outcome of Patients Treated after Delivery of the Molecular Report
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoffman, A.; Half, E.E. Update on Screening for Urological Malignancies. Rambam. Maimonides Med. J. 2017, 8, e0041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Gastaldo, A.; Kempf, E.; del Alba, A.G.; Duran, I. Systemic Treatment of Renal Cell Cancer: A Comprehensive Review. Cancer Treat. Rev. 2017, 60, 77–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Özgüroğlu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone Acetate plus Prednisone in Patients with Newly Diagnosed High-Risk Metastatic Castration-Sensitive Prostate Cancer (LATITUDE): Final Overall Survival Analysis of a Randomised, Double-Blind, Phase 3 Trial. Lancet Oncol. 2019, 20, 686–700. [Google Scholar] [CrossRef]
- Powles, T.; Rosenberg, J.E.; Sonpavde, G.P.; Loriot, Y.; Durán, I.; Lee, J.-L.; Matsubara, N.; Vulsteke, C.; Castellano, D.; Wu, C.; et al. Enfortumab Vedotin in Previously Treated Advanced Urothelial Carcinoma. N. Engl. J. Med. 2021, 384, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive Molecular Characterization of Urothelial Bladder Carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Tsimberidou, A.M.; Fountzilas, E.; Nikanjam, M.; Kurzrock, R. Review of Precision Cancer Medicine: Evolution of the Treatment Paradigm. Cancer Treat. Rev. 2020, 86, 102019. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Stephenson, J.J.; Rosen, P.; Loesch, D.M.; Borad, M.J.; Anthony, S.; Jameson, G.; Brown, S.; Cantafio, N.; Richards, D.A.; et al. Pilot Study Using Molecular Profiling of Patients’ Tumors to Find Potential Targets and Select Treatments for Their Refractory Cancers. J. Clin. Oncol. 2010, 28, 4877–4883. [Google Scholar] [CrossRef]
- Tsimberidou, A.-M.; Wen, S.; Hong, D.S.; Wheler, J.J.; Falchook, G.S.; Fu, S.; Piha-Paul, S.; Naing, A.; Janku, F.; Aldape, K.; et al. Personalized Medicine for Patients with Advanced Cancer in the Phase I Program at MD Anderson: Validation and Landmark Analyses. Clin. Cancer Res. 2014, 20, 4827–4836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kris, M.G.; Johnson, B.E.; Berry, L.D.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Varella-Garcia, M.; Franklin, W.A.; Aronson, S.L.; Su, P.-F.; et al. Using Multiplexed Assays of Oncogenic Drivers in Lung Cancers to Select Targeted Drugs. JAMA 2014, 311, 1998. [Google Scholar] [CrossRef] [PubMed]
- Bachelot, T.; Filleron, T.; Bieche, I.; Arnedos, M.; Campone, M.; Dalenc, F.; Coussy, F.; Sablin, M.-P.; Debled, M.; Lefeuvre-Plesse, C.; et al. Durvalumab Compared to Maintenance Chemotherapy in Metastatic Breast Cancer: The Randomized Phase II SAFIR02-BREAST IMMUNO Trial. Nat. Med. 2021, 27, 250–255. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Powles, T.; Carroll, D.; Chowdhury, S.; Gravis, G.; Joly, F.; Carles, J.; Fléchon, A.; Maroto, P.; Petrylak, D.; Rolland, F.; et al. An Adaptive, Biomarker-Directed Platform Study of Durvalumab in Combination with Targeted Therapies in Advanced Urothelial Cancer. Nat. Med. 2021, 27, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, F.; Gonçalves, A.; Guille, A.; Adelaïde, J.; Garnier, S.; Carbuccia, N.; Billon, E.; Finetti, P.; Sfumato, P.; Monneur, A.; et al. Prospective High-Throughput Genome Profiling of Advanced Cancers: Results of the PERMED-01 Clinical Trial. Genome Med. 2021, 13, 87. [Google Scholar] [CrossRef]
- Bertucci, F.; Finetti, P.; Guille, A.; Adélaïde, J.; Garnier, S.; Carbuccia, N.; Monneur, A.; Charafe-Jauffret, E.; Goncalves, A.; Viens, P.; et al. Comparative Genomic Analysis of Primary Tumors and Metastases in Breast Cancer. Oncotarget 2016, 7, 27208–27219. [Google Scholar] [CrossRef] [Green Version]
- Bertucci, F.; Ng, C.K.Y.; Patsouris, A.; Droin, N.; Piscuoglio, S.; Carbuccia, N.; Soria, J.C.; Dien, A.T.; Adnani, Y.; Kamal, M.; et al. Genomic Characterization of Metastatic Breast Cancers. Nature 2019, 569, 560–564. [Google Scholar] [CrossRef]
- Niu, B.; Ye, K.; Zhang, Q.; Lu, C.; Xie, M.; McLellan, M.D.; Wendl, M.C.; Ding, L. MSIsensor: Microsatellite Instability Detection Using Paired Tumor-Normal Sequence Data. Bioinformatics 2014, 30, 1015–1016. [Google Scholar] [CrossRef] [Green Version]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of Genomic Loss of Heterozygosity Predict Homologous Recombination Repair Defects in Epithelial Ovarian Cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef] [Green Version]
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017, PO.17.00011. [Google Scholar] [CrossRef]
- Wu, Y.-M.; Cieślik, M.; Lonigro, R.J.; Vats, P.; Reimers, M.A.; Cao, X.; Ning, Y.; Wang, L.; Kunju, L.P.; de Sarkar, N.; et al. Inactivation of CDK12 Delineates a Distinct Immunogenic Class of Advanced Prostate Cancer. Cell 2018, 173, 1770–1782.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajrami, I.; Frankum, J.R.; Konde, A.; Miller, R.E.; Rehman, F.L.; Brough, R.; Campbell, J.; Sims, D.; Rafiq, R.; Hooper, S.; et al. Genome-Wide Profiling of Genetic Synthetic Lethality Identifies CDK12 as a Novel Determinant of PARP1/2 Inhibitor Sensitivity. Cancer Res. 2014, 74, 287–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, D.R.; Wu, Y.-M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative Clinical Genomics of Metastatic Cancer. Nature 2017, 548, 297–303. [Google Scholar] [CrossRef]
- Tsimberidou, A.-M.; Iskander, N.G.; Hong, D.S.; Wheler, J.J.; Falchook, G.S.; Fu, S.; Piha-Paul, S.; Naing, A.; Janku, F.; Luthra, R.; et al. Personalized Medicine in a Phase I Clinical Trials Program: The MD Anderson Cancer Center Initiative. Clin. Cancer Res. 2012, 18, 6373–6383. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.B.; Dahlman, K.H.; Knol, J.; Gilbert, J.; Puzanov, I.; Means-Powell, J.; Balko, J.M.; Lovly, C.M.; Murphy, B.A.; Goff, L.W.; et al. Enabling a Genetically Informed Approach to Cancer Medicine: A Retrospective Evaluation of the Impact of Comprehensive Tumor Profiling Using a Targeted next-Generation Sequencing Panel. Oncologist 2014, 19, 616–622. [Google Scholar] [CrossRef] [Green Version]
- Stockley, T.L.; Oza, A.M.; Berman, H.K.; Leighl, N.B.; Knox, J.J.; Shepherd, F.A.; Chen, E.X.; Krzyzanowska, M.K.; Dhani, N.; Joshua, A.M.; et al. Molecular Profiling of Advanced Solid Tumors and Patient Outcomes with Genotype-Matched Clinical Trials: The Princess Margaret IMPACT/COMPACT Trial. Genome Med. 2016, 8, 109. [Google Scholar] [CrossRef] [Green Version]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational Landscape of Metastatic Cancer Revealed from Prospective Clinical Sequencing of 10,000 Patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Trédan, O.; Wang, Q.; Pissaloux, D.; Cassier, P.; de la Fouchardière, A.; Fayette, J.; Desseigne, F.; Ray-Coquard, I.; de la Fouchardière, C.; Frappaz, D.; et al. Molecular Screening Program to Select Molecular-Based Recommended Therapies for Metastatic Cancer Patients: Analysis from the ProfiLER Trial. Ann. Oncol. 2019, 30, 757–765. [Google Scholar] [CrossRef]
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-Cancer Whole-Genome Analyses of Metastatic Solid Tumours. Nature 2019, 575, 210–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwaederle, M.; Parker, B.A.; Schwab, R.B.; Daniels, G.A.; Piccioni, D.E.; Kesari, S.; Helsten, T.L.; Bazhenova, L.A.; Romero, J.; Fanta, P.T.; et al. Precision Oncology: The UC San Diego Moores Cancer Center PREDICT Experience. Mol. Cancer 2016, 15, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massard, C.; Michiels, S.; Ferté, C.; Le Deley, M.-C.; Lacroix, L.; Hollebecque, A.; Verlingue, L.; Ileana, E.; Rosellini, S.; Ammari, S.; et al. High-Throughput Genomics and Clinical Outcome in Hard-to-Treat Advanced Cancers: Results of the MOSCATO 01 Trial. Cancer Discov. 2017, 7, 586–595. [Google Scholar] [CrossRef] [Green Version]
- Tsimberidou, A.M.; Fountzilas, E.; Bleris, L.; Kurzrock, R. Transcriptomics and Solid Tumors: The next Frontier in Precision Cancer Medicine. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, A.-M.; Hong, D.S.; Ye, Y.; Cartwright, C.; Wheler, J.J.; Falchook, G.S.; Naing, A.; Fu, S.; Piha-Paul, S.; Janku, F.; et al. Initiative for Molecular Profiling and Advanced Cancer Therapy (IMPACT): An MD Anderson Precision Medicine Study. JCO Precis. Oncol. 2017, 1–18. [Google Scholar] [CrossRef]
- Vano, Y.-A.; Elaidi, R.; Bennamoun, M.; Chevreau, C.; Borchiellini, D.; Pannier, D.; Maillet, D.; Gross-Goupil, M.; Tournigand, C.; Laguerre, B.; et al. Nivolumab, Nivolumab-Ipilimumab, and VEGFR-Tyrosine Kinase Inhibitors as First-Line Treatment for Metastatic Clear-Cell Renal Cell Carcinoma (BIONIKK): A Biomarker-Driven, Open-Label, Non-Comparative, Randomised, Phase 2 Trial. Lancet Oncol. 2022, 23, 612–624. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | All Patients (N = 64) | Matched TRT2 (N = 12) | Non-Matched TRT2 (N = 25) | p-Value * | |
|---|---|---|---|---|---|
| Age, years | |||||
| Median (range) | 65.55 (28–83) | 70.5 (55–83) | 62.52 (28–81) | 3.2 × 10−2 | |
| Sex | 0.168 | ||||
| Male | 58 (91%) | 12 (100%) | 19 (76%) | ||
| Female | 6 (9%) | 0 (0%) | 6 (24%) | ||
| ECOG performance status | 0.890 | ||||
| 0 | 34 (57%) | 7 (58%) | 14 (61%) | ||
| 1 | 23 (38%) | 4 (33%) | 8 (35%) | ||
| 2 | 3 (5%) | 1 (8%) | 1 (4%) | ||
| Cancer type | 0.071 | ||||
| Prostate | 39 (61%) | 10 (83%) | 11 (44%) | ||
| Bladder-Ureter | 12 (19%) | 2 (17%) | 4 (16%) | ||
| Kidney | 9 (14%) | 0 (0%) | 7 (28%) | ||
| Testicular | 4 (6%) | 0 (0%) | 3 (12%) | ||
| Site of the biopsy | 0.493 | ||||
| Liver | 22 (36%) | 4 (33%) | 7 (32%) | ||
| Lymph node | 13 (21%) | 3 (25%) | 4 (18%) | ||
| Lung | 7 (11%) | 0 (0%) | 5 (23%) | ||
| Peritoneum | 2 (3%) | 0 (0%) | 1 (5%) | ||
| Prostate | 10 (16%) | 3 (25%) | 3 (14%) | ||
| Other | 7 (11%) | 2 (17%) | 2 (9%) | ||
| Pathological type | 0.341 | ||||
| Carcinoma | 59 (92%) | 12 (100%) | 21 (84%) | ||
| Germ cell tumor | 3 (5%) | 0 (0%) | 2 (8%) | ||
| Other | 2 (3%) | 0 (0%) | 2 (8%) | ||
| Extension stage | 1 | ||||
| Locally advanced | 1 (2%) | 0 (0%) | 1 (4%) | ||
| Metastatic | 63 (98%) | 12 (100%) | 24 (96%) | ||
| Number of metastatic sites | |||||
| Median (range) | 2.44 (0–4) | 2.17 (1–4) | 2.48 (0–4) | 0.433 | |
| Number of previous treatment lines for advanced disease | |||||
| Median (range) | 3.50 (0–11) | 3.50 (1–9) | 3.0 (0–8) | 0.229 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Billon, E.; Gravis, G.; Guille, A.; Carbuccia, N.; Adelaide, J.; Garnier, S.; Finetti, P.; Denicolaï, E.; Sfumato, P.; Brunelle, S.; et al. Molecular Profiles of Advanced Urological Cancers in the PERMED-01 Precision Medicine Clinical Trial. Cancers 2022, 14, 2275. https://doi.org/10.3390/cancers14092275
Billon E, Gravis G, Guille A, Carbuccia N, Adelaide J, Garnier S, Finetti P, Denicolaï E, Sfumato P, Brunelle S, et al. Molecular Profiles of Advanced Urological Cancers in the PERMED-01 Precision Medicine Clinical Trial. Cancers. 2022; 14(9):2275. https://doi.org/10.3390/cancers14092275
Chicago/Turabian StyleBillon, Emilien, Gwenaelle Gravis, Arnaud Guille, Nadine Carbuccia, Jose Adelaide, Séverine Garnier, Pascal Finetti, Emilie Denicolaï, Patrick Sfumato, Serge Brunelle, and et al. 2022. "Molecular Profiles of Advanced Urological Cancers in the PERMED-01 Precision Medicine Clinical Trial" Cancers 14, no. 9: 2275. https://doi.org/10.3390/cancers14092275
APA StyleBillon, E., Gravis, G., Guille, A., Carbuccia, N., Adelaide, J., Garnier, S., Finetti, P., Denicolaï, E., Sfumato, P., Brunelle, S., Thomassin-Piana, J., Pignot, G., Walz, J., Chabannon, C., Pakradouni, J., Sabatier, R., Vicier, C., Popovici, C., Mamessier, E., ... Bertucci, F. (2022). Molecular Profiles of Advanced Urological Cancers in the PERMED-01 Precision Medicine Clinical Trial. Cancers, 14(9), 2275. https://doi.org/10.3390/cancers14092275