
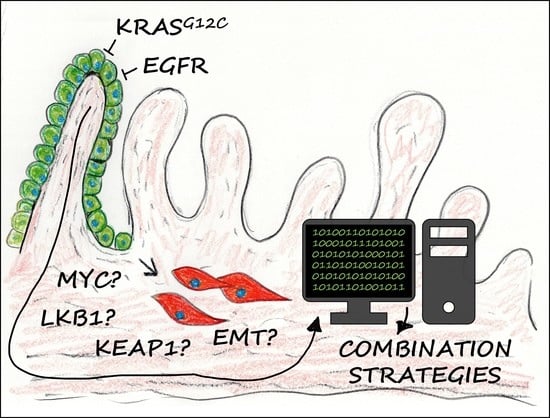
EMT, Stemness, and Drug Resistance in Biological Context: A 3D Tumor Tissue/In Silico Platform for Analysis of Combinatorial Treatment in NSCLC with Aggressive KRAS-Biomarker Signatures

 ,
,  , ,
, ,  ,
,
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Matrix Preparation
2.2. Cells
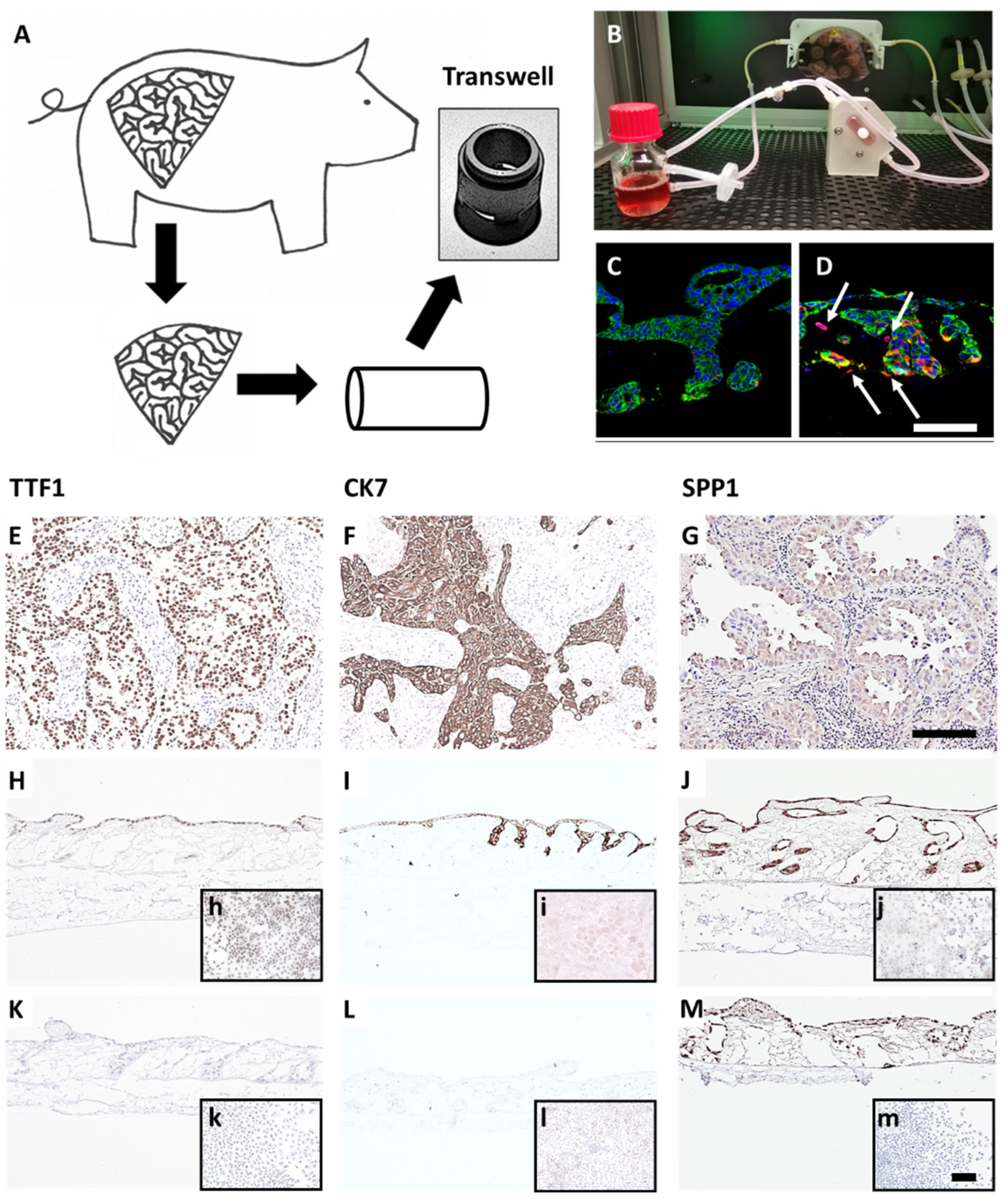
2.3. Preparation of Tumor Models
2.4. Patient Tumor Samples
2.5. Treatment of Cells in 2D and 3D
2.6. Stimulation of Cells with hTGF-β1
2.7. Immuno/Histochemical Stainings
2.8. Quantification of Proliferation and Cell Invasion
2.9. M30 ELISA
2.10. Viability Assays (MTT-Test and CellTiter-Glo®)
2.11. Western Blotting
2.12. Statistics
2.13. Lung Cancer PCR Array
2.14. Ultrastructural Analysis
2.15. In Silico 3D Tissue Simulations/Bioinformatics
3. Results
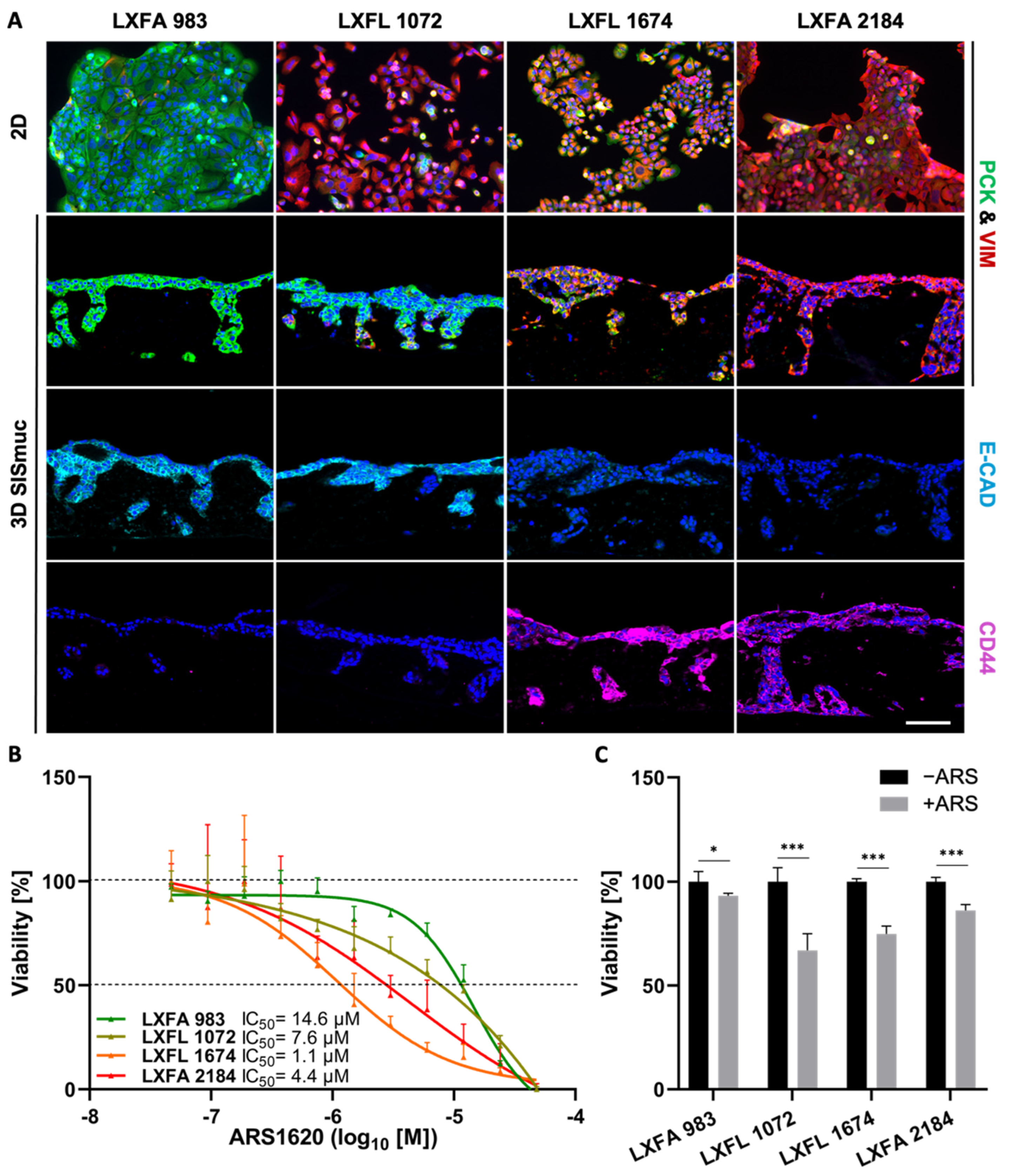
3.1. Generation and Characterization of the 3D Lung Cancer Tissue Model
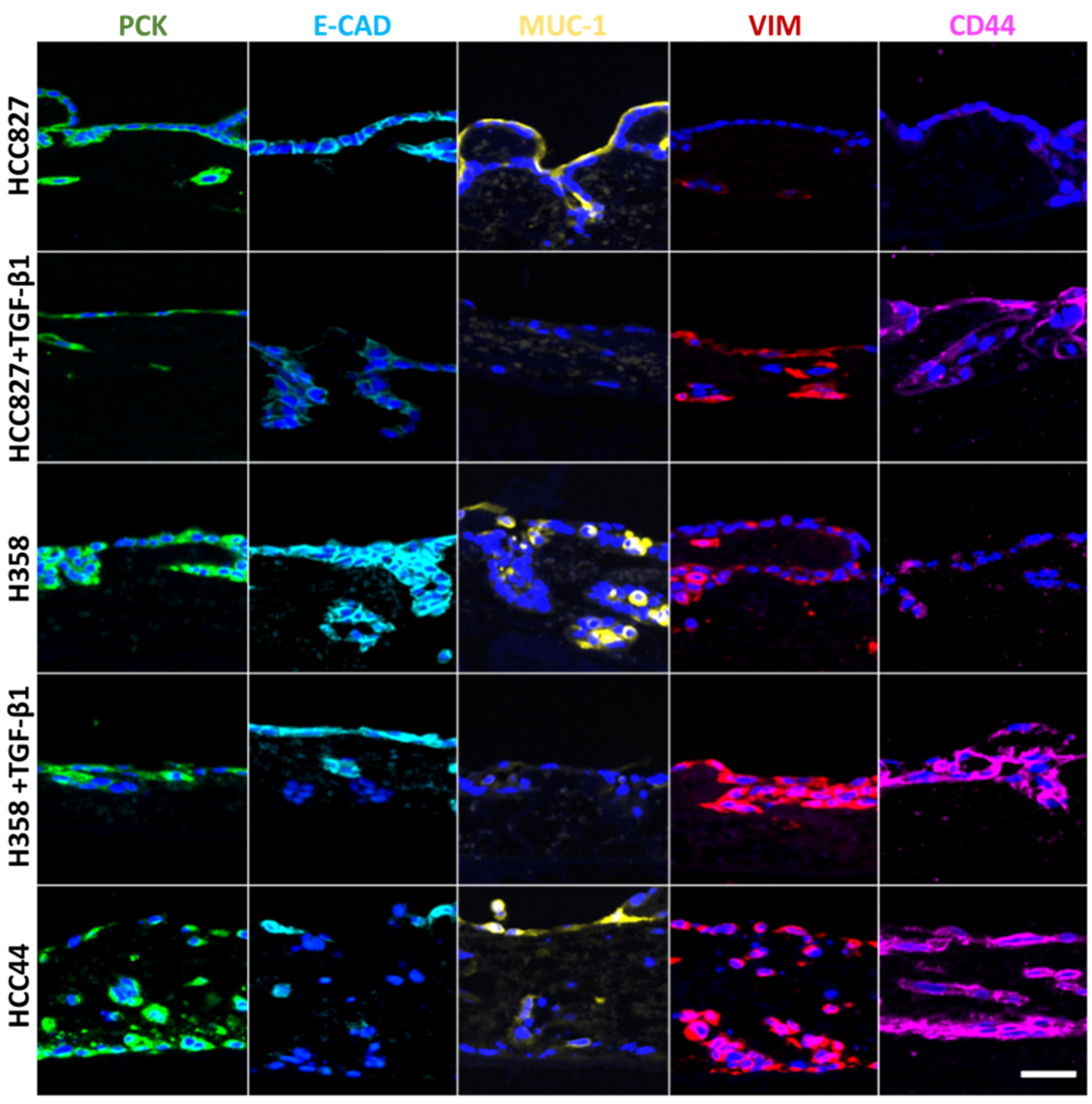
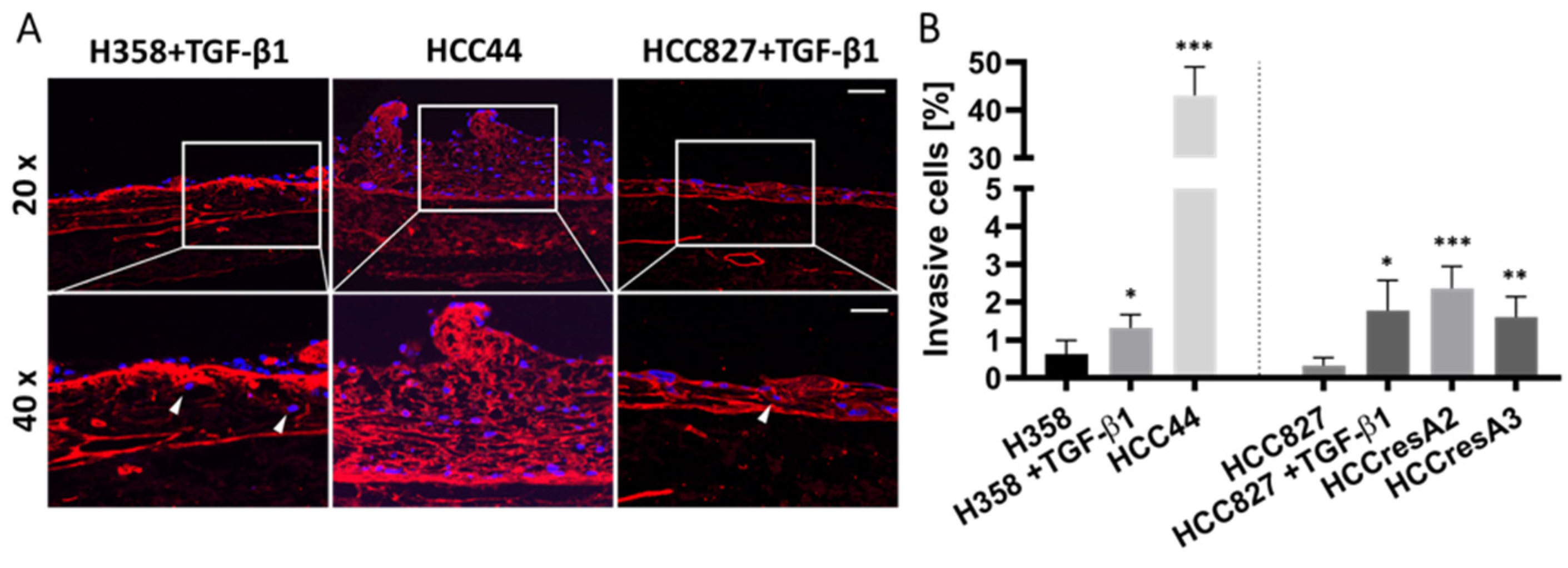
3.2. Assessment of EMT, Differentiation, Stemness, and Invasion
3.3. EMT Correlation with Drug Response
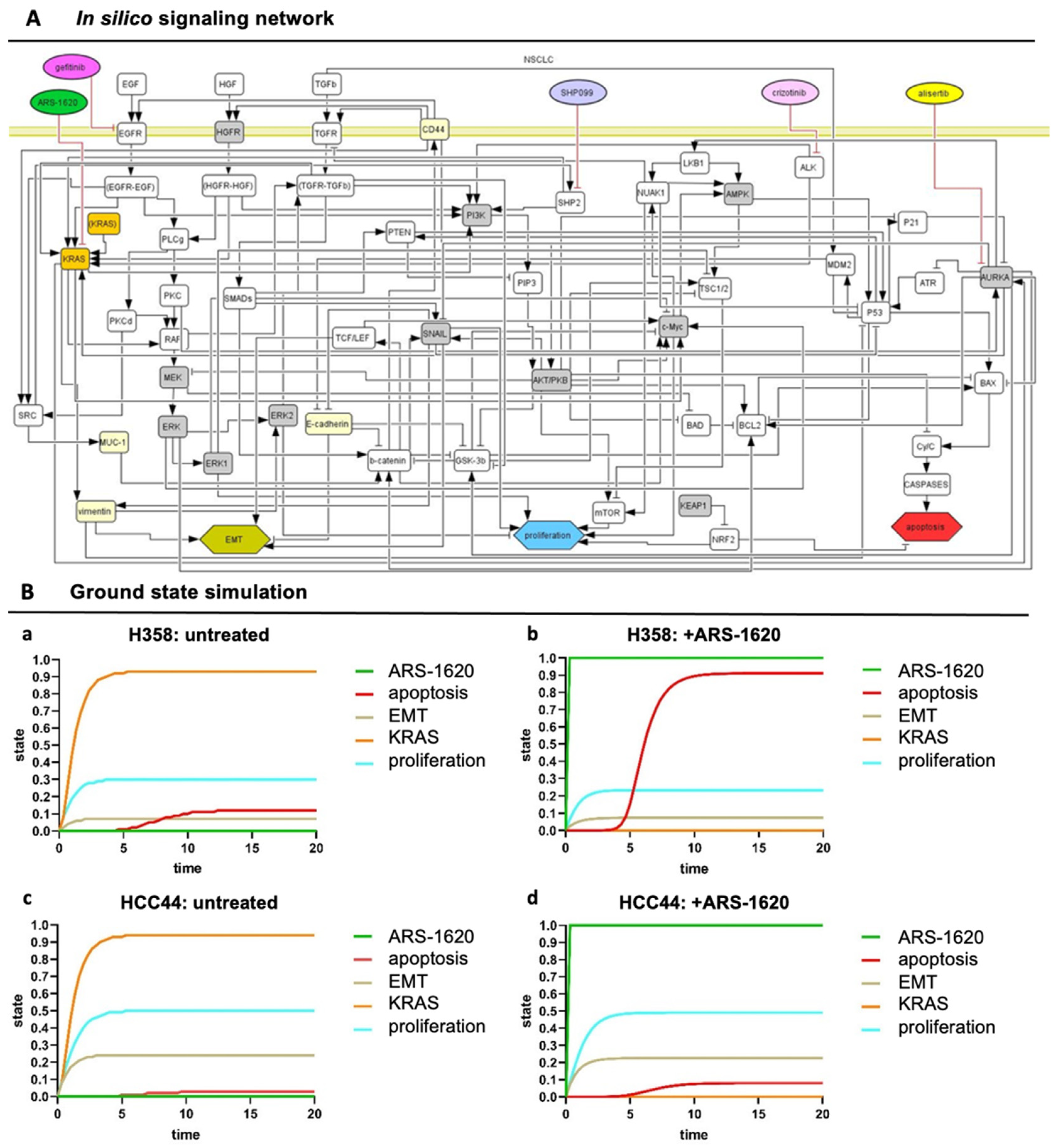
3.4. Set-Up of Combined In Vitro/In Silico Models with KRAS Signatures
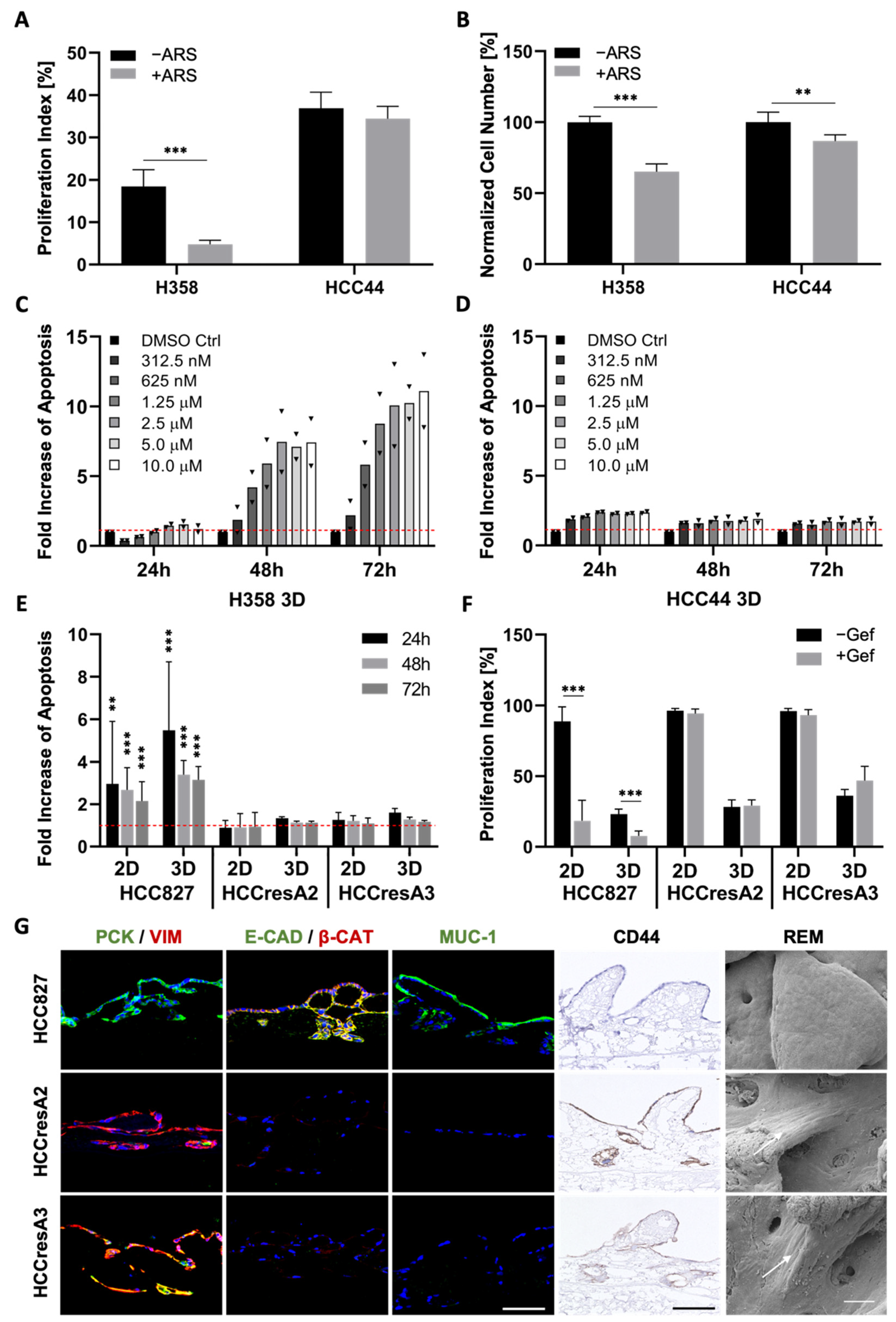
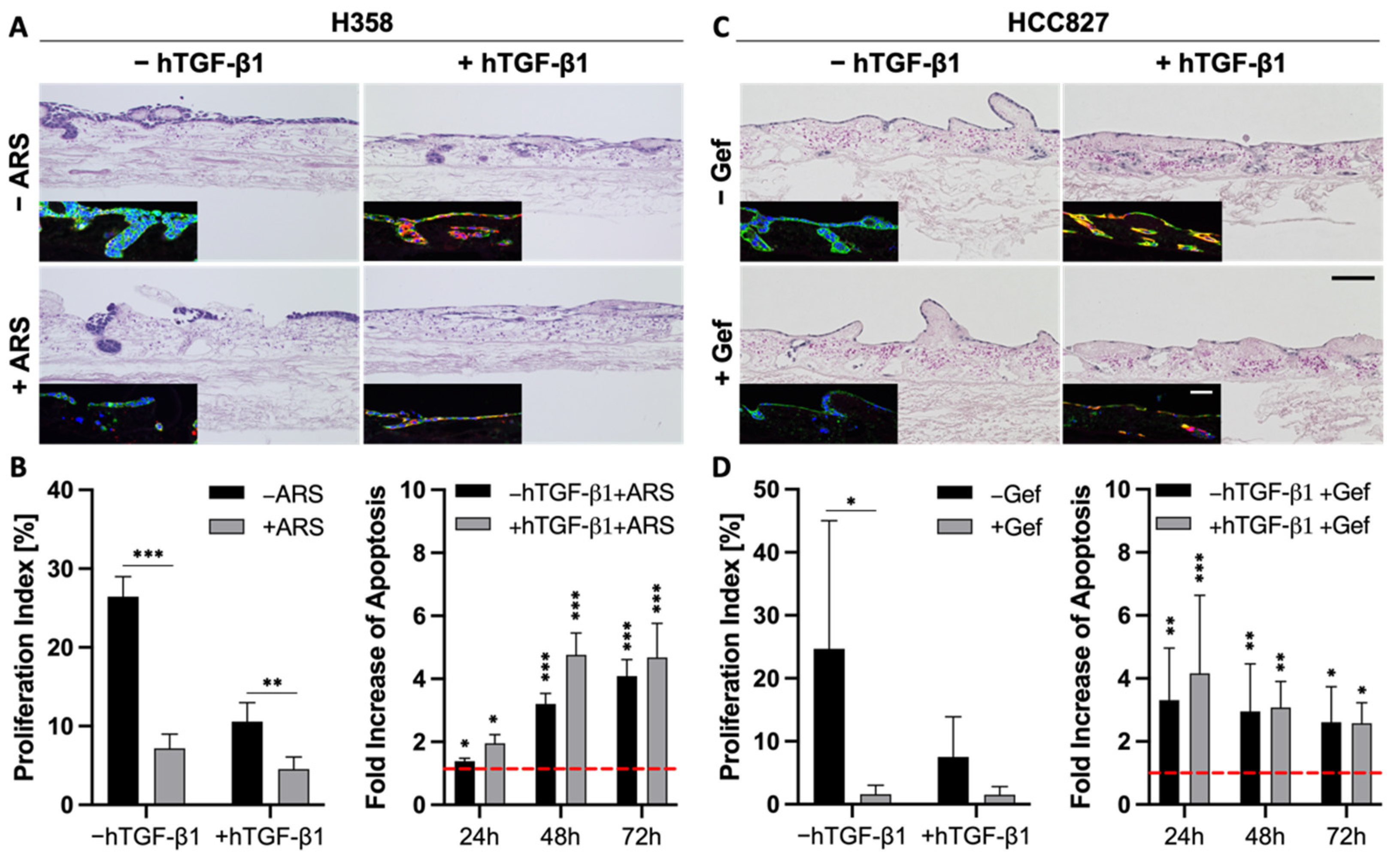
3.5. TGF-β1-Induced EMT Does Not Mediate Resistance toward Targeted Therapies
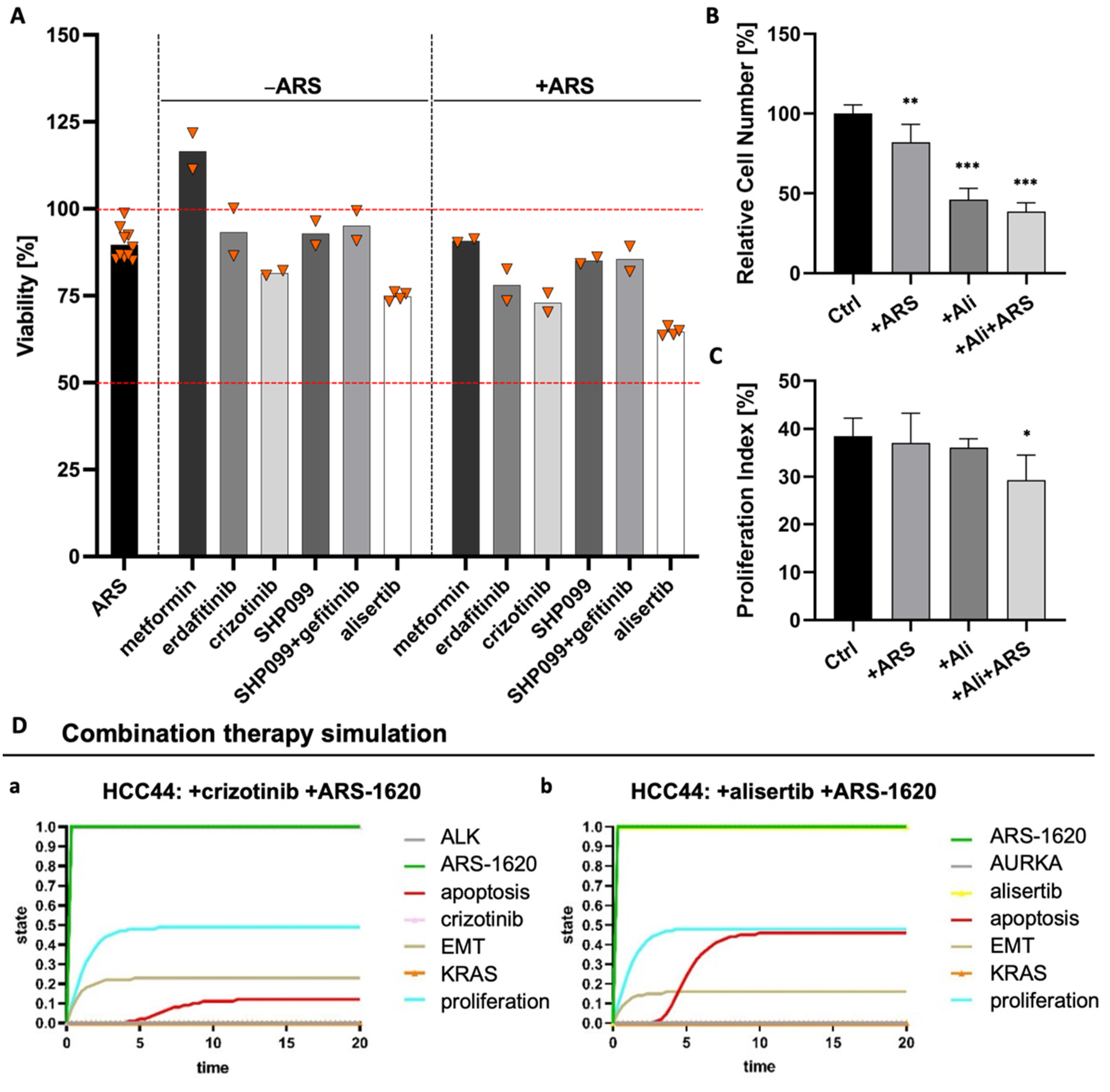
3.6. EMT Status and CD44 Expression Are No Predictors of Drug Response in PDX Cell Lines
3.7. Combination Strategies to Overcome Resistance in HCC44 Tumor Models
4. Discussion
4.1. D Tissue Models for More Realistic Preclinical Testing
4.2. EMT Correlation to Drug Resistance and Invasion
4.3. An In Vivo/In Silico Platform for Testing Targeted Therapies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.; Weinberg, R.A. Epithelial-to-mesenchymal transition in cancer: Complexity and opportunities. Front. Med. 2018, 12, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Brunen, D.; Willems, S.M.; Kellner, U.; Midgley, R.; Simon, I.; Bernards, R. Tgf-beta: An emerging player in drug resistance. Cell Cycle 2013, 12, 2960–2968. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Shin, J.H.; Stuelten, C.H.; Zhang, Y.E. Tgf-beta-induced alternative splicing of tak1 promotes emt and drug resistance. Oncogene 2019, 38, 3185–3200. [Google Scholar] [CrossRef] [PubMed]
- Voon, D.C.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. The emt spectrum and therapeutic opportunities. Mol. Oncol. 2017, 11, 878–891. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Hlubek, F.; Spaderna, S.; Schmalhofer, O.; Hiendlmeyer, E.; Jung, A.; Kirchner, T. Invasion and metastasis in colorectal cancer: Epithelial-mesenchymal transition, mesenchymal-epithelial transition, stem cells and beta-catenin. Cells Tissues Organs 2005, 179, 56–65. [Google Scholar] [CrossRef]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. Egfr mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. Kras(g12c) inhibition with sotorasib in advanced solid tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ct2/show/NCT04699188 (accessed on 1 December 2021).
- Goebel, L.; Muller, M.P.; Goody, R.S.; Rauh, D. Krasg12c inhibitors in clinical trials: A short historical perspective. RSC Med. Chem. 2020, 11, 760–770. [Google Scholar] [CrossRef]
- Scheffler, M.; Ihle, M.A.; Hein, R.; Merkelbach-Bruse, S.; Scheel, A.H.; Siemanowski, J.; Bragelmann, J.; Kron, A.; Abedpour, N.; Ueckeroth, F.; et al. K-ras mutation subtypes in nsclc and associated co-occuring mutations in other oncogenic pathways. J. Thorac. Oncol. 2019, 14, 606–616. [Google Scholar] [CrossRef]
- Bhattacharjee, Y. Biomedicine. Pharma firms push for sharing of cancer trial data. Science 2012, 338, 29. [Google Scholar] [CrossRef]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistics 2019, 20, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Kuhnemundt, J.; Leifeld, H.; Scherg, F.; Schmitt, M.; Nelke, L.C.; Schmitt, T.; Baur, F.; Gottlich, C.; Fuchs, M.; Kunz, M.; et al. Modular micro-physiological human tumor/tissue models based on decellularized tissue for improved preclinical testing. ALTEX 2020, 38, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Baur, F.; Nietzer, S.L.; Kunz, M.; Saal, F.; Jeromin, J.; Matschos, S.; Linnebacher, M.; Walles, H.; Dandekar, T.; Dandekar, G. Connecting cancer pathways to tumor engines: A stratification tool for colorectal cancer combining human in vitro tissue models with boolean in silico models. Cancers 2019, 12, 28. [Google Scholar] [CrossRef]
- Gottlich, C.; Kunz, M.; Zapp, C.; Nietzer, S.L.; Walles, H.; Dandekar, T.; Dandekar, G. A combined tissue-engineered/in silico signature tool patient stratification in lung cancer. Mol. Oncol. 2018, 12, 1264–1285. [Google Scholar] [CrossRef]
- Gottlich, C.; Muller, L.C.; Kunz, M.; Schmitt, F.; Walles, H.; Walles, T.; Dandekar, T.; Dandekar, G.; Nietzer, S.L. A combined 3d tissue engineered in vitro/in silico lung tumor model for predicting drug effectiveness in specific mutational backgrounds. J. Vis. Exp. 2016, 110, e53885. [Google Scholar] [CrossRef]
- Stratmann, A.T.; Fecher, D.; Wangorsch, G.; Gottlich, C.; Walles, T.; Walles, H.; Dandekar, T.; Dandekar, G.; Nietzer, S.L. Establishment of a human 3d lung cancer model based on a biological tissue matrix combined with a boolean in silico model. Mol. Oncol. 2014, 8, 351–365. [Google Scholar] [CrossRef]
- Jannasch, M.; Groeber, F.; Brattig, N.W.; Unger, C.; Walles, H.; Hansmann, J. Development and application of three-dimensional skin equivalents for the investigation of percutaneous worm invasion. Exp. Parasitol. 2015, 150, 22–30. [Google Scholar] [CrossRef]
- Linke, K.; Schanz, J.; Hansmann, J.; Walles, T.; Brunner, H.; Mertsching, H. Engineered liver-like tissue on a capillarized matrix for applied research. Tissue Eng. 2007, 13, 2699–2707. [Google Scholar] [CrossRef]
- Schanz, J.; Pusch, J.; Hansmann, J.; Walles, H. Vascularised human tissue models: A new approach for the refinement of biomedical research. J. Biotechnol. 2010, 148, 56–63. [Google Scholar] [CrossRef]
- Noro, R.; Gemma, A.; Kosaihira, S.; Kokubo, Y.; Chen, M.; Seike, M.; Kataoka, K.; Matsuda, K.; Okano, T.; Minegishi, Y.; et al. Gefitinib (iressa) sensitive lung cancer cell lines show phosphorylation of akt without ligand stimulation. BMC Cancer 2006, 6, 277. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Funahashi, A.; Ghosh, S.; Kitano, H. Modeling and simulation using celldesigner. Methods Mol. Biol. 2014, 1164, 121–145. [Google Scholar] [PubMed]
- Di Cara, A.; Garg, A.; De Micheli, G.; Xenarios, I.; Mendoza, L. Dynamic simulation of regulatory networks using squad. BMC Bioinform. 2007, 8, 462. [Google Scholar] [CrossRef]
- Lee, C.J.; An, H.J.; Cho, E.S.; Kang, H.C.; Lee, J.Y.; Lee, H.S.; Cho, Y.Y. Stat2 stability regulation: An intersection between immunity and carcinogenesis. Exp. Mol. Med. 2020, 52, 1526–1536. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, S.; Ivanova, E.; Guo, S.; Yoshida, R.; Campisi, M.; Sundararaman, S.K.; Tange, S.; Mitsuishi, Y.; Thai, T.C.; Masuda, S.; et al. Suppression of sting associated with lkb1 loss in kras-driven lung cancer. Cancer Discov. 2019, 9, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Iwakawa, R.; Takahashi, K.; Kohno, T.; Nakanishi, Y.; Matsuno, Y.; Suzuki, K.; Nakamoto, M.; Shimizu, E.; Minna, J.D.; et al. Prevalence and specificity of lkb1 genetic alterations in lung cancers. Oncogene 2007, 26, 5911–5918. [Google Scholar] [CrossRef] [PubMed]
- Elkin, S.R.; Bendris, N.; Reis, C.R.; Zhou, Y.; Xie, Y.; Huffman, K.E.; Minna, J.D.; Schmid, S.L. A systematic analysis reveals heterogeneous changes in the endocytic activities of cancer cells. Cancer Res. 2015, 75, 4640–4650. [Google Scholar] [CrossRef] [PubMed]
- Lou, K.; Steri, V.; Ge, A.Y.; Hwang, Y.C.; Yogodzinski, C.H.; Shkedi, A.R.; Choi, A.L.M.; Mitchell, D.C.; Swaney, D.L.; Hann, B.; et al. Kras(g12c) inhibition produces a driver-limited state revealing collateral dependencies. Sci. Signal. 2019, 12, eaaw9450. [Google Scholar] [CrossRef]
- Fedele, C.; Li, S.; Teng, K.W.; Foster, C.J.R.; Peng, D.; Ran, H.; Mita, P.; Geer, M.J.; Hattori, T.; Koide, A.; et al. Shp2 inhibition diminishes krasg12c cycling and promotes tumor microenvironment remodeling. J. Exp. Med. 2021, 218, e20201414. [Google Scholar] [CrossRef]
- Xue, J.Y.; Zhao, Y.; Aronowitz, J.; Mai, T.T.; Vides, A.; Qeriqi, B.; Kim, D.; Li, C.; de Stanchina, E.; Mazutis, L.; et al. Rapid non-uniform adaptation to conformation-specific kras(g12c) inhibition. Nature 2020, 577, 421–425. [Google Scholar] [CrossRef]
- Jiang, L.; Xu, W.; Chen, Y.; Zhang, Y. Shp2 inhibitor specifically suppresses the stemness of kras-mutant non-small cell lung cancer cells. Artif. Cells Nanomed. Biotechnol. 2019, 47, 3231–3238. [Google Scholar] [CrossRef]
- Kortlever, R.M.; Sodir, N.M.; Wilson, C.H.; Burkhart, D.L.; Pellegrinet, L.; Brown Swigart, L.; Littlewood, T.D.; Evan, G.I. Myc cooperates with ras by programming inflammation and immune suppression. Cell 2017, 171, 1301–1315.e14. [Google Scholar] [CrossRef]
- Klijn, C.; Durinck, S.; Stawiski, E.W.; Haverty, P.M.; Jiang, Z.; Liu, H.; Degenhardt, J.; Mayba, O.; Gnad, F.; Liu, J.; et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat. Biotechnol. 2015, 33, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, X.; Pu, J.; Yang, Q.; Guan, H.; Ji, M.; Shi, B.; Chen, M.; Hou, P. C-myc is a major determinant for antitumor activity of aurora a kinase inhibitor mln8237 in thyroid cancer. Thyroid 2018, 28, 1642–1654. [Google Scholar] [CrossRef] [PubMed]
- Middleton, G.; Yang, Y.; Campbell, C.D.; Andre, T.; Atreya, C.E.; Schellens, J.H.M.; Yoshino, T.; Bendell, J.C.; Hollebecque, A.; McRee, A.J.; et al. Braf-mutant transcriptional subtypes predict outcome of combined braf, mek, and egfr blockade with dabrafenib, trametinib, and panitumumab in patients with colorectal cancer. Clin. Cancer Res. 2020, 26, 2466–2476. [Google Scholar] [CrossRef]
- Lubtow, M.M.; Nelke, L.C.; Seifert, J.; Kuhnemundt, J.; Sahay, G.; Dandekar, G.; Nietzer, S.L.; Luxenhofer, R. Drug induced micellization into ultra-high capacity and stable curcumin nanoformulations: Physico-chemical characterization and evaluation in 2d and 3d in vitro models. J. Control Release 2019, 303, 162–180. [Google Scholar] [CrossRef]
- Nietzer, S.; Baur, F.; Sieber, S.; Hansmann, J.; Schwarz, T.; Stoffer, C.; Hafner, H.; Gasser, M.; Waaga-Gasser, A.M.; Walles, H.; et al. Mimicking metastases including tumor stroma: A new technique to generate a three-dimensional colorectal cancer model based on a biological decellularized intestinal scaffold. Tissue Eng. Part C Methods 2016, 22, 621–635. [Google Scholar] [CrossRef]
- Wallstabe, L.; Gottlich, C.; Nelke, L.C.; Kuhnemundt, J.; Schwarz, T.; Nerreter, T.; Einsele, H.; Walles, H.; Dandekar, G.; Nietzer, S.L.; et al. Ror1-car t cells are effective against lung and breast cancer in advanced microphysiologic 3d tumor models. JCI Insight 2019, 4, e126345. [Google Scholar] [CrossRef]
- Stuber, T.; Monjezi, R.; Wallstabe, L.; Kuhnemundt, J.; Nietzer, S.L.; Dandekar, G.; Wockel, A.; Einsele, H.; Wischhusen, J.; Hudecek, M. Inhibition of tgf-beta-receptor signaling augments the antitumor function of ror1-specific car t-cells against triple-negative breast cancer. J. Immunother. Cancer 2020, 8, e000676. [Google Scholar] [CrossRef]
- Rafaeva, M.; Erler, J.T. Framing cancer progression: Influence of the organ- and tumour-specific matrisome. FEBS J. 2020, 287, 1454–1477. [Google Scholar] [CrossRef]
- Bissell, M.J.; Radisky, D.C.; Rizki, A.; Weaver, V.M.; Petersen, O.W. The organizing principle: Microenvironmental influences in the normal and malignant breast. Differentiation 2002, 70, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Brugge, J.S. Modelling glandular epithelial cancers in three-dimensional cultures. Nat. Rev. Cancer 2005, 5, 675–688. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, L.E.; Zegers, M.M.; Mostov, K.E. Opinion: Building epithelial architecture: Insights from three-dimensional culture models. Nat. Rev. Mol. Cell Biol. 2002, 3, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Radisky, D.; Muschler, J.; Bissell, M.J. Order and disorder: The role of extracellular matrix in epithelial cancer. Cancer Investig. 2002, 20, 139–153. [Google Scholar] [CrossRef]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.M.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 world health organization classification of lung tumors: Impact of genetic, clinical and radiologic advances since the 2004 classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef]
- Bergholz, J.; Xiao, Z.X. Role of p63 in development, tumorigenesis and cancer progression. Cancer Microenviron. 2012, 5, 311–322. [Google Scholar] [CrossRef]
- Steurer, S.; Riemann, C.; Buscheck, F.; Luebke, A.M.; Kluth, M.; Hube-Magg, C.; Hinsch, A.; Hoflmayer, D.; Weidemann, S.; Fraune, C.; et al. P63 expression in human tumors and normal tissues: A tissue microarray study on 10,200 tumors. Biomark Res. 2021, 9, 7. [Google Scholar] [CrossRef]
- Chang, Y.S.; Kim, H.J.; Chang, J.; Ahn, C.M.; Kim, S.K.; Kim, S.K. Elevated circulating level of osteopontin is associated with advanced disease state of non-small cell lung cancer. Lung Cancer 2007, 57, 373–380. [Google Scholar] [CrossRef]
- Donati, V.; Boldrini, L.; Dell’Omodarme, M.; Prati, M.C.; Faviana, P.; Camacci, T.; Lucchi, M.; Mussi, A.; Santoro, M.; Basolo, F.; et al. Osteopontin expression and prognostic significance in non-small cell lung cancer. Clin. Cancer Res. 2005, 11, 6459–6465. [Google Scholar] [CrossRef]
- Mack, P.C.; Redman, M.W.; Chansky, K.; Williamson, S.K.; Farneth, N.C.; Lara, P.N., Jr.; Franklin, W.A.; Le, Q.T.; Crowley, J.J.; Gandara, D.R.; et al. Lower osteopontin plasma levels are associated with superior outcomes in advanced non-small-cell lung cancer patients receiving platinum-based chemotherapy: Swog study s0003. J. Clin. Oncol. 2008, 26, 4771–4776. [Google Scholar] [CrossRef]
- Wai, P.Y.; Kuo, P.C. The role of osteopontin in tumor metastasis. J. Surg. Res. 2004, 121, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Han, S.S.; Lee, S.J.; Kim, W.J.; Ryu, D.R.; Won, J.Y.; Park, S.; Cheon, M.J. Plasma osteopontin is a useful diagnostic biomarker for advanced non-small cell lung cancer. Tuberc. Respir. Dis. 2013, 75, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Lin, D.; Yuan, J.; Xiao, T.; Zhang, H.; Sun, W.; Han, N.; Ma, Y.; Di, X.; Gao, M.; et al. Overexpression of osteopontin is associated with more aggressive phenotypes in human non-small cell lung cancer. Clin. Cancer Res. 2005, 11, 4646–4652. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Liang, Y.; Chen, R.; Liu, G.; Wang, H.; Tang, M.; Zhou, X.; Wang, H.; Yang, Y.; Wei, H.; et al. Osteopontin facilitates tumor metastasis by regulating epithelial-mesenchymal plasticity. Cell Death Dis. 2016, 7, e2564. [Google Scholar] [CrossRef]
- Kothari, A.N.; Arffa, M.L.; Chang, V.; Blackwell, R.H.; Syn, W.K.; Zhang, J.; Mi, Z.; Kuo, P.C. Osteopontin-a master regulator of epithelial-mesenchymal transition. J. Clin. Med. 2016, 5, 39. [Google Scholar] [CrossRef]
- Weber, C.E.; Li, N.Y.; Wai, P.Y.; Kuo, P.C. Epithelial-mesenchymal transition, tgf-beta, and osteopontin in wound healing and tissue remodeling after injury. J. Burn Care Res. 2012, 33, 311–318. [Google Scholar] [CrossRef]
- Zang, M.; Zhang, B.; Zhang, Y.; Li, J.; Su, L.; Zhu, Z.; Gu, Q.; Liu, B.; Yan, M. Ceacam6 promotes gastric cancer invasion and metastasis by inducing epithelial-mesenchymal transition via pi3k/akt signaling pathway. PLoS ONE 2014, 9, e112908. [Google Scholar] [CrossRef]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. Emt in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Weinberg, R.A. Emt and cancer: More than meets the eye. Dev. Cell 2019, 49, 313–316. [Google Scholar] [CrossRef]
- Li, C.H.; Hsu, T.I.; Chang, Y.C.; Chan, M.H.; Lu, P.J.; Hsiao, M. Stationed or relocating: The seesawing emt/met determinants from embryonic development to cancer metastasis. Biomedicines 2021, 9, 1265. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; LeBleu, V.S.; Carstens, J.L.; Sugimoto, H.; Zheng, X.; Malasi, S.; Saur, D.; Kalluri, R. Dual reporter genetic mouse models of pancreatic cancer identify an epithelial-to-mesenchymal transition-independent metastasis program. EMBO Mol. Med. 2018, 10, e9085. [Google Scholar] [CrossRef] [PubMed]
- Revenco, T.; Nicodeme, A.; Pastushenko, I.; Sznurkowska, M.K.; Latil, M.; Sotiropoulou, P.A.; Dubois, C.; Moers, V.; Lemaire, S.; de Maertelaer, V.; et al. Context dependency of epithelial-to-mesenchymal transition for metastasis. Cell Rep. 2019, 29, 1458–1468.e1453. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Jung, A.; Reu, S.; Porzner, M.; Hlubek, F.; Kunz-Schughart, L.A.; Knuechel, R.; Kirchner, T. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc. Natl. Acad. Sci. USA 2001, 98, 10356–10361. [Google Scholar] [CrossRef]
- Qin, Q.; Li, X.; Liang, X.; Zeng, L.; Wang, J.; Sun, L.; Zhong, D. Targeting the emt transcription factor snail overcomes resistance to osimertinib in egfr-mutant non-small cell lung cancer. Thorac. Cancer 2021, 12, 1708–1715. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. Emt, cscs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Nagaraj, N.S.; Datta, P.K. Targeting the transforming growth factor-beta signaling pathway in human cancer. Expert Opin. Investig. Drugs 2010, 19, 77–91. [Google Scholar] [CrossRef]
- Kelley, R.K.; Gane, E.; Assenat, E.; Siebler, J.; Galle, P.R.; Merle, P.; Hourmand, I.O.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; et al. A phase 2 study of galunisertib (tgf-beta1 receptor type i inhibitor) and sorafenib in patients with advanced hepatocellular carcinoma. Clin. Transl. Gastroenterol. 2019, 10, e00056. [Google Scholar] [CrossRef]
- Wilson, M.M.; Weinberg, R.A.; Lees, J.A.; Guen, V.J. Emerging mechanisms by which emt programs control stemness. Trends Cancer 2020, 6, 775–780. [Google Scholar] [CrossRef]
- Morath, I.; Hartmann, T.N.; Orian-Rousseau, V. Cd44: More than a mere stem cell marker. Int. J. Biochem. Cell Biol. 2016, 81, 166–173. [Google Scholar] [CrossRef]
- Ko, Y.H.; Won, H.S.; Jeon, E.K.; Hong, S.H.; Roh, S.Y.; Hong, Y.S.; Byun, J.H.; Jung, C.K.; Kang, J.H. Prognostic significance of cd44s expression in resected non-small cell lung cancer. BMC Cancer 2011, 11, 340. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Kulkarni, P.; Weninger, K.; Orban, J.; Levine, H. Phenotypic plasticity, bet-hedging, and androgen independence in prostate cancer: Role of non-genetic heterogeneity. Front. Oncol. 2018, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://celllines.tron-mainz.de/ (accessed on 1 December 2021).
- Scholtalbers, J.; Boegel, S.; Bukur, T.; Byl, M.; Goerges, S.; Sorn, P.; Loewer, M.; Sahin, U.; Castle, J.C. Tclp: An online cancer cell line catalogue integrating hla type, predicted neo-epitopes, virus and gene expression. Genome Med. 2015, 7, 118. [Google Scholar] [PubMed]
- Adachi, Y.; Kimura, R.; Hirade, K.; Ebi, H. Escaping kras: Gaining autonomy and resistance to kras inhibition in kras mutant cancers. Cancers 2021, 13, 5081. [Google Scholar] [CrossRef]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A gene expression signature associated with “k-ras addiction” reveals regulators of emt and tumor cell survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef]
- Jiao, D.; Yang, S. Overcoming resistance to drugs targeting kras(g12c) mutation. Innovation 2020, 1, 100035. [Google Scholar]
- Ryan, M.B.; Fece de la Cruz, F.; Phat, S.; Myers, D.T.; Wong, E.; Shahzade, H.A.; Hong, C.B.; Corcoran, R.B. Vertical pathway inhibition overcomes adaptive feedback resistance to kras(g12c) inhibition. Clin. Cancer Res. 2020, 26, 1633–1643. [Google Scholar] [CrossRef]
- Du, R.; Huang, C.; Liu, K.; Li, X.; Dong, Z. Targeting aurka in cancer: Molecular mechanisms and opportunities for cancer therapy. Mol. Cancer 2021, 20, 15. [Google Scholar] [CrossRef]
- Naso, F.D.; Boi, D.; Ascanelli, C.; Pamfil, G.; Lindon, C.; Paiardini, A.; Guarguaglini, G. Nuclear localisation of aurora-a: Its regulation and significance for aurora-a functions in cancer. Oncogene 2021, 40, 3917–3928. [Google Scholar]
- Zheng, X.; Chi, J.; Zhi, J.; Zhang, H.; Yue, D.; Zhao, J.; Li, D.; Li, Y.; Gao, M.; Guo, J. Aurora-a-mediated phosphorylation of lkb1 compromises lkb1/ampk signaling axis to facilitate nsclc growth and migration. Oncogene 2018, 37, 502–511. [Google Scholar] [CrossRef]
- Dauch, D.; Rudalska, R.; Cossa, G.; Nault, J.C.; Kang, T.W.; Wuestefeld, T.; Hohmeyer, A.; Imbeaud, S.; Yevsa, T.; Hoenicke, L.; et al. A myc-aurora kinase a protein complex represents an actionable drug target in p53-altered liver cancer. Nat. Med. 2016, 22, 744–753. [Google Scholar] [CrossRef]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting kras mutant cancers with a covalent g12c-specific inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Kim, S.; Yang, C.; Burtness, B. Abstract p078: Aurora a kinase inhibition with vic-1911 overcomes intrinsic and acquired resistance to krasg12c inhibition in kras(g12c)-mutated lung cancer. Mol. Cancer Ther. 2021, 20, P078. [Google Scholar]
- Breitenbach, T.; Liang, C.; Beyersdorf, N.; Dandekar, T. Analyzing pharmacological intervention points: A method to calculate external stimuli to switch between steady states in regulatory networks. PLoS Comput. Biol. 2019, 15, e1007075. [Google Scholar] [CrossRef]
- Sodir, N.M.; Kortlever, R.M.; Barthet, V.J.A.; Campos, T.; Pellegrinet, L.; Kupczak, S.; Anastasiou, P.; Swigart, L.B.; Soucek, L.; Arends, M.J.; et al. Myc instructs and maintains pancreatic adenocarcinoma phenotype. Cancer Discov. 2020, 10, 588–607. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peindl, M.; Göttlich, C.; Crouch, S.; Hoff, N.; Lüttgens, T.; Schmitt, F.; Pereira, J.G.N.; May, C.; Schliermann, A.; Kronenthaler, C.; et al. EMT, Stemness, and Drug Resistance in Biological Context: A 3D Tumor Tissue/In Silico Platform for Analysis of Combinatorial Treatment in NSCLC with Aggressive KRAS-Biomarker Signatures. Cancers 2022, 14, 2176. https://doi.org/10.3390/cancers14092176
Peindl M, Göttlich C, Crouch S, Hoff N, Lüttgens T, Schmitt F, Pereira JGN, May C, Schliermann A, Kronenthaler C, et al. EMT, Stemness, and Drug Resistance in Biological Context: A 3D Tumor Tissue/In Silico Platform for Analysis of Combinatorial Treatment in NSCLC with Aggressive KRAS-Biomarker Signatures. Cancers. 2022; 14(9):2176. https://doi.org/10.3390/cancers14092176
Chicago/Turabian StylePeindl, Matthias, Claudia Göttlich, Samantha Crouch, Niklas Hoff, Tamara Lüttgens, Franziska Schmitt, Jesús Guillermo Nieves Pereira, Celina May, Anna Schliermann, Corinna Kronenthaler, and et al. 2022. "EMT, Stemness, and Drug Resistance in Biological Context: A 3D Tumor Tissue/In Silico Platform for Analysis of Combinatorial Treatment in NSCLC with Aggressive KRAS-Biomarker Signatures" Cancers 14, no. 9: 2176. https://doi.org/10.3390/cancers14092176
APA StylePeindl, M., Göttlich, C., Crouch, S., Hoff, N., Lüttgens, T., Schmitt, F., Pereira, J. G. N., May, C., Schliermann, A., Kronenthaler, C., Cheufou, D., Reu-Hofer, S., Rosenwald, A., Weigl, E., Walles, T., Schüler, J., Dandekar, T., Nietzer, S., & Dandekar, G. (2022). EMT, Stemness, and Drug Resistance in Biological Context: A 3D Tumor Tissue/In Silico Platform for Analysis of Combinatorial Treatment in NSCLC with Aggressive KRAS-Biomarker Signatures. Cancers, 14(9), 2176. https://doi.org/10.3390/cancers14092176