
Arginine Methyltransferase PRMT7 Deregulates Expression of RUNX1 Target Genes in T-Cell Acute Lymphoblastic Leukemia

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Gene Expression Analysis
2.2. Protein Expression and Immunohistochemistry
2.3. Cell Culture and Knockout Lines
2.4. Functional Studies
2.5. Mass Spectrometry-Based Arginine Monomethylation Analysis
2.6. LC-MS/MS Analysis
2.7. Survival Analysis
2.8. RNA Sequencing
2.9. Enrichment Analysis
2.10. Statistical Analyses
3. Results
3.1. PRMT7 Is Strongly Expressed in Mature T-ALL
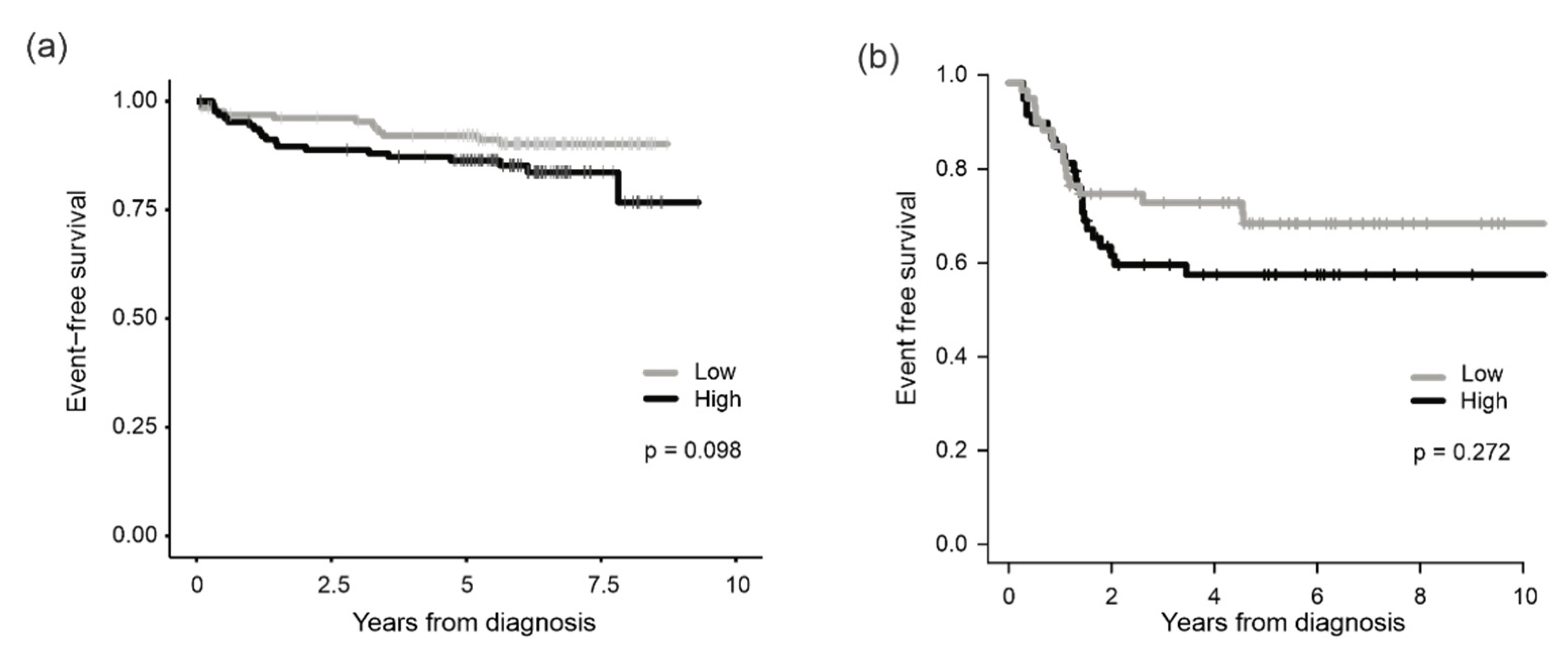
3.2. Association of PRMT7 Expression with Survival
3.3. Reduced Colony Formation and Cell Viability after Genetic Deletion of PRMT7
3.4. Arginine Monomethylation Is Disrupted after PRMT7 Knockout in T-ALL
3.5. PRMT7 Deletion Deregulates Expression of RUNX1 Target Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Girardi, T.; Vicente, C.; Cools, J.; De Keersmaecker, K. The Genetics and Molecular Biology of T-ALL. Blood 2017, 129, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Roti, G.; Stegmaier, K. New Approaches to Target T-ALL. Front. Oncol. 2014, 4, 170. [Google Scholar] [CrossRef] [PubMed]
- Quist-Paulsen, P.; Toft, N.; Heyman, M.; Abrahamsson, J.; Griškevičius, L.; Hallböök, H.; Jónsson, Ó.G.; Palk, K.; Vaitkeviciene, G.; Vettenranta, K.; et al. T-Cell Acute Lymphoblastic Leukemia in Patients 1–45 Years Treated with the Pediatric NOPHO ALL2008 Protocol. Leukemia 2020, 34, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Schrappe, M.; Valsecchi, M.G.; Bartram, C.R.; Schrauder, A.; Panzer-Grümayer, R.; Möricke, A.; Parasole, R.; Zimmermann, M.; Dworzak, M.; Buldini, B.; et al. Late MRD Response Determines Relapse Risk Overall and in Subsets of Childhood T-Cell ALL: Results of the AIEOP-BFM-ALL 2000 Study. Blood 2011, 118, 2077–2084. [Google Scholar] [CrossRef]
- Winter, S.S.; Dunsmore, K.P.; Devidas, M.; Wood, B.L.; Esiashvili, N.; Chen, Z.; Eisenberg, N.; Briegel, N.; Hayashi, R.J.; Gastier-Foster, J.M.; et al. Improved Survival for Children and Young Adults with T-Lineage Acute Lymphoblastic Leukemia: Results from the Children’s Oncology Group AALL0434 Methotrexate Randomization. J. Clin. Oncol. 2018, 36, 2926–2934. [Google Scholar] [CrossRef]
- Patrick, K.; Wade, R.; Goulden, N.; Mitchell, C.; Moorman, A.V.; Rowntree, C.; Jenkinson, S.; Hough, R.; Vora, A. Outcome for Children and Young People with Early T-Cell Precursor Acute Lymphoblastic Leukaemia Treated on a Contemporary Protocol, UKALL 2003. Br. J. Haematol. 2014, 166, 421–424. [Google Scholar] [CrossRef]
- Miranda, T.B.; Miranda, M.; Frankel, A.; Clarke, S. PRMT7 Is a Member of the Protein Arginine Methyltransferase Family with a Distinct Substrate Specificity. J. Biol. Chem. 2004, 279, 22902–22907. [Google Scholar] [CrossRef]
- Feng, Y.; Maity, R.; Whitelegge, J.P.; Hadjikyriacou, A.; Li, Z.; Zurita-Lopez, C.; Al-Hadid, Q.; Clark, A.T.; Bedford, M.T.; Masson, J.Y.; et al. Mammalian Protein Arginine Methyltransferase 7 (PRMT7) Specifically Targets RXR Sites in Lysine- and Arginine-Rich Regions. J. Biol. Chem. 2013, 288, 37010–37025. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, M.; Liu, Z.; Mu, Y.; Li, K. PRMT7: A Pivotal Arginine Methyltransferase in Stem Cells and Development. Stem Cells Int. 2021, 2021, 6241600. [Google Scholar] [CrossRef]
- Bedford, M.T.; Frankel, A.; Yaffe, M.B.; Clarke, S.; Leder, P.; Richard, S. Arginine Methylation Inhibits the Binding of Proline-Rich Ligands to Src Homology 3, but Not WW, Domains. J. Biol. Chem. 2000, 275, 16030–16036. [Google Scholar] [CrossRef]
- Côté, J.; Richard, S. Tudor Domains Bind Symmetrical Dimethylated Arginines. J. Biol. Chem. 2005, 280, 28476–28483. [Google Scholar] [CrossRef]
- Liu, H.; Wang, J.Y.; Huang, Y.; Li, Z.; Gong, W.; Lehmann, R.; Xu, R.M. Structural Basis for Methylarginine-Dependent Recognition of Aubergine by Tudor. Genes Dev. 2010, 24, 1876–1881. [Google Scholar] [CrossRef]
- Liu, R.; Gao, J.; Yang, Y.; Qiu, R.; Zheng, Y.; Huang, W.; Zeng, Y.; Hou, Y.; Wang, S.; Leng, S.; et al. PHD Finger Protein 1 (PHF1) Is a Novel Reader for Histone H4R3 Symmetric Dimethylation and Coordinates with PRMT5-WDR77/CRL4B Complex to Promote Tumorigenesis. Nucleic Acids Res. 2018, 46, 6608–6626. [Google Scholar] [CrossRef] [PubMed]
- Li, W.J.; He, Y.H.; Yang, J.J.; Hu, G.S.; Lin, Y.A.; Ran, T.; Peng, B.L.; Xie, B.L.; Huang, M.F.; Gao, X.; et al. Profiling PRMT Methylome Reveals Roles of HnRNPA1 Arginine Methylation in RNA Splicing and Cell Growth. Nat. Commun. 2021, 12, 1946. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, R.M.; Morettin, A.; Côté, J. Role of PRMTs in Cancer: Could Minor Isoforms Be Leaving a Mark? World J. Biol. Chem. 2014, 5, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.; Jiang, H.; Ma, Y.; Wang, L.; Wang, L.; Du, J.; Hou, P.; Gao, Y.; Zhao, L.; Wang, G.; et al. PRMT7 Induces Epithelial-to-Mesenchymal Transition and Promotes Metastasis in Breast Cancer. Cancer Res. 2014, 74, 5656–5667. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wan, L.; Zou, H.; Pan, Z.; Zhou, W.; Lu, X. PRMT7 Promotes the Growth of Renal Cell Carcinoma through Modulating the β-Catenin/C-MYC Axis. Int. J. Biochem. Cell Biol. 2020, 120, 105686. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; He, Z.; Zheng, L.; Xie, D.; Dong, S.; Zhang, P. PRMT7 Contributes to the Metastasis Phenotype in Human Non-Small-Cell Lung Cancer Cells Possibly through the Interaction with HSPA5 and EEF2. Onco. Targets. Ther. 2018, 11, 4869–4876. [Google Scholar] [CrossRef]
- Szewczyk, M.M.; Ishikawa, Y.; Organ, S.; Sakai, N.; Li, F.; Halabelian, L.; Ackloo, S.; Couzens, A.L.; Eram, M.; Dilworth, D.; et al. Pharmacological Inhibition of PRMT7 Links Arginine Monomethylation to the Cellular Stress Response. Nat. Commun. 2020, 11, 2396. [Google Scholar] [CrossRef]
- Smil, D.; Eram, M.S.; Li, F.; Kennedy, S.; Szewczyk, M.M.; Brown, P.J.; Barsyte-Lovejoy, D.; Arrowsmith, C.H.; Vedadi, M.; Schapira, M. Discovery of a Dual PRMT5-PRMT7 Inhibitor. ACS Med. Chem. Lett. 2015, 6, 408–412. [Google Scholar] [CrossRef]
- Mehtonen, J.; Pölönen, P.; Häyrynen, S.; Dufva, O.; Lin, J.; Liuksiala, T.; Granberg, K.; Lohi, O.; Hautamäki, V.; Nykter, M.; et al. Data-Driven Characterization of Molecular Phenotypes across Heterogeneous Sample Collections. Nucleic Acids Res. 2019, 47, e76. [Google Scholar] [CrossRef] [PubMed]
- Pölönen, P.; Mehtonen, J.; Lin, J.; Liuksiala, T.; Häyrynen, S.; Teppo, S.; Mäkinen, A.; Kumar, A.; Malani, D.; Pohjolainen, V.; et al. Hemap: An Interactive Online Resource for Characterizing Molecular Phenotypes across Hematologic Malignancies. Cancer Res. 2019, 79, 2466–2479. [Google Scholar] [CrossRef] [PubMed]
- Laukkanen, S.; Grönroos, T.; Pölönen, P.; Kuusanmäki, H.; Mehtonen, J.; Cloos, J.; Ossenkoppele, G.; Gjertsen, B.; Øystein, B.; Heckman, C.; et al. In Silico and Preclinical Drug Screening Identifies Dasatinib as a Targeted Therapy for T-ALL. Blood Cancer J. 2017, 7, e604. [Google Scholar] [CrossRef] [PubMed]
- Laukkanen, S.; Oksa, L.; Nikkilä, A.; Lahnalampi, M.; Parikka, M.; Seki, M.; Takita, J.; Degerman, S.; de Bock, C.E.; Heinäniemi, M.; et al. SIX6 Is a TAL1-Regulated Transcription Factor in T-ALL and Associated with Inferior Outcome. Leuk. Lymphoma 2020, 61, 3089–3100. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Botting, R.A.; Domínguez Conde, C.; Popescu, D.M.; Lavaert, M.; Kunz, D.J.; Goh, I.; Stephenson, E.; Ragazzini, R.; Tuck, E.; et al. A Cell Atlas of Human Thymic Development Defines T Cell Repertoire Formation. Science 2020, 367, eaay3224. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Mäkinen, A.; Nikkilä, A.; Haapaniemi, T.; Oksa, L.; Mehtonen, J.; Vänskä, M.; Heinäniemi, M.; Paavonen, T.; Lohi, O. IGF2BP3 Associates with Proliferative Phenotype and Prognostic Features in B-Cell Acute Lymphoblastic Leukemia. Cancers 2021, 13, 1505. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.; et al. QuPath: Open Source Software for Digital Pathology Image Analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bursteinas, B.; et al. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Sood, R.; Kamikubo, Y.; Liu, P. Role of RUNX1 in Hematological Malignancies. Blood 2017, 129, 2070–2082. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Kimura, S.; Isobe, T.; Yoshida, K.; Ueno, H.; Nakajima-Takagi, Y.; Wang, C.; Lin, L.; Kon, A.; Suzuki, H.; et al. Recurrent SPI1 (PU.1) Fusions in High-Risk Pediatric T Cell Acute Lymphoblastic Leukemia. Nat. Genet. 2017, 49, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable Kinase-Activating Lesions in Ph-like Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2; Springer: New York, NY, USA, 2016. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and Collaborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Tenenbaum, D.; Maintainer, B. KEGGREST: Client-Side REST Access to the Kyoto Encyclopedia of Genes and Genomes (KEGG); R Package Version 1.35.0; version: Release (3.14); Bioconductor: Dortmund, Germany, 2021; Available online: https://bioconductor.org/packages/release/bioc/html/KEGGREST.html (accessed on 1 March 2019). [CrossRef]
- Giurgiu, M.; Reinhard, J.; Brauner, B.; Dunger-Kaltenbach, I.; Fobo, G.; Frishman, G.; Montrone, C.; Ruepp, A. CORUM: The Comprehensive Resource of Mammalian Protein Complexes—2019. Nucleic Acids Res. 2019, 47, D559. [Google Scholar] [CrossRef]
- Smits, A.H.; Ziebell, F.; Joberty, G.; Zinn, N.; Mueller, W.F.; Clauder-Münster, S.; Eberhard, D.; Fälth Savitski, M.; Grandi, P.; Jakob, P.; et al. Biological Plasticity Rescues Target Activity in CRISPR Knock Outs. Nat. Methods 2019, 16, 1087–1093. [Google Scholar] [CrossRef]
- Meador, J.A.; Zhao, M.; Su, Y.; Narayan, G.; Geard, C.R.; Balajee, A.S. Histone H2AX Is a Critical Factor for Cellular Protection against DNA Alkylating Agents. Oncogene 2008, 27, 5662–5671. [Google Scholar] [CrossRef]
- Manzl, C.; Peintner, L.; Krumschnabel, G.; Bock, F.; Labi, V.; Drach, M.; Newbold, A.; Johnstone, R.; Villunger, A. PIDDosome-Independent Tumor Suppression by Caspase-2. Cell Death Differ. 2012, 19, 1722. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Weintrau, A.S.; Day, D.S.; Valton, A.L.; Bak, R.O.; Li, C.H.; Goldmann, J.; Lajoie, B.R.; Fan, Z.P.; Sigova, A.A.; et al. Activation of Proto-Oncogenes by Disruption of Chromosome Neighborhoods. Science 2016, 351, 1454–1458. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, S.; Yoshida, T.; Zhao, X.; Nimer, S.D.; Taniwaki, M.; Okuda, T. Loss of RUNX1/AML1 Arginine-Methylation Impairs Peripheral T Cell Homeostasis. Br. J. Haematol. 2015, 170, 859–873. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Jankovic, V.; Gural, A.; Huang, G.; Pardanani, A.; Menendez, S.; Zhang, J.; Dunne, R.; Xiao, A.; Erdjument-Bromage, H.; et al. Methylation of RUNX1 by PRMT1 Abrogates SIN3A Binding and Potentiates Its Transcriptional Activity. Genes Dev. 2008, 22, 640–653. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Xie, X.; Ye, Y.; Wang, L.; Che, F. BCL11A: A Potential Diagnostic Biomarker and Therapeutic Target in Human Diseases. Biosci. Rep. 2019, 39, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wang, J.; Khaled, W.; Burke, S.; Li, P.; Chen, X.; Yang, W.; Jenkins, N.A.; Copeland, N.G.; Zhang, S.; et al. Bcl11a Is Essential for Lymphoid Development and Negatively Regulates P53. J. Exp. Med. 2012, 209, 2467. [Google Scholar] [CrossRef] [PubMed]
- Yin, B.; Delwel, R.; Valk, P.J.; Wallace, M.R.; Loh, M.L.; Shannon, K.M.; Largaespada, D.A. A Retroviral Mutagenesis Screen Reveals Strong Cooperation between Bcl11a Overexpression and Loss of the Nf1 Tumor Suppressor Gene. Blood 2009, 113, 1075–1085. [Google Scholar] [CrossRef]
- Saadi, W.; Kermezli, Y.; Dao, L.T.M.; Mathieu, E.; Santiago-Algarra, D.; Manosalva, I.; Torres, M.; Belhocine, M.; Pradel, L.; Loriod, B.; et al. A Critical Regulator of Bcl2 Revealed by Systematic Transcript Discovery of LncRNAs Associated with T-Cell Differentiation. Sci. Rep. 2019, 9, 4707. [Google Scholar] [CrossRef]
- Rothenberg, E.V. Transcriptional Control of Early T and B Cell Developmental Choices. Annu. Rev. Immunol. 2014, 32, 283. [Google Scholar] [CrossRef]
- Filippou, P.S.; Karagiannis, G.S.; Constantinidou, A. Midkine (MDK) Growth Factor: A Key Player in Cancer Progression and a Promising Therapeutic Target. Oncogene 2019, 39, 2040–2054. [Google Scholar] [CrossRef]
- Charmsaz, S.; Al-Ejeh, F.; Yeadon, T.M.; Miller, K.J.; Smith, F.M.; Stringer, B.W.; Moore, A.S.; Lee, F.T.; Cooper, L.T.; Stylianou, C.; et al. EphA3 as a Target for Antibody Immunotherapy in Acute Lymphoblastic Leukemia. Leukemia 2016, 31, 1779–1787. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Zhou, H.; Song, X.; Shi, S.; Zhang, J.; Jia, L. Modification of Sialylation Is Associated with Multidrug Resistance in Human Acute Myeloid Leukemia. Oncogene 2015, 34, 726–740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dong, W.; Zhou, H.; Li, H.; Wang, N.; Miao, X.; Jia, L. α-2,8-Sialyltransferase Is Involved in the Development of Multidrug Resistance via PI3K/Akt Pathway in Human Chronic Myeloid Leukemia. IUBMB Life 2015, 67, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Brady, S.W.; Ma, X.; Shen, S.; Zhang, Y.; Li, Y.; Szlachta, K.; Dong, L.; Liu, Y.; Yang, F.; et al. Therapy-Induced Mutations Drive the Genomic Landscape of Relapsed Acute Lymphoblastic Leukemia. Blood 2020, 135, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.K.E.; Baron, J.; Moore, C.R.; Liu, Y.; Perlman, D.H.; Hart, R.P.; Xie, P. Mutated in Colorectal Cancer (MCC) Is a Novel Oncogene in B Lymphocytes. J. Hematol. Oncol. 2014, 7, 56. [Google Scholar] [CrossRef]
- Gabbasov, R.; Xiao, F.; Howe, C.G.; Bickel, L.E.; O’Brien, S.W.; Benrubi, D.; Do, T.V.; Zhou, Y.; Nicolas, E.; Cai, K.Q.; et al. NEDD9 Promotes Oncogenic Signaling, a Stem/Mesenchymal Gene Signature, and Aggressive Ovarian Cancer Growth in Mice. Oncogene 2018, 37, 4854–4870. [Google Scholar] [CrossRef]
- Shagisultanova, E.; Gaponova, A.V.; Gabbasov, R.; Nicolas, E.; Golemis, E.A. Preclinical and Clinical Studies of the NEDD9 Scaffold Protein in Cancer and Other Diseases. Gene 2015, 567, 1. [Google Scholar] [CrossRef]
- Ribeiro, D.; Melão, A.; Barata, J.T. IL-7R-Mediated Signaling in T-Cell Acute Lymphoblastic Leukemia. Adv. Biol. Regul. 2013, 53, 211–222. [Google Scholar] [CrossRef]
- Clappier, E.; Cuccuini, W.; Cayuela, J.M.; Vecchione, D.; Baruchel, A.; Dombret, H.; Sigaux, F.; Soulier, J. Cyclin D2 Dysregulation by Chromosomal Translocations to TCR Loci in T-Cell Acute Lymphoblastic Leukemias. Leukemia 2006, 20, 82–86. [Google Scholar] [CrossRef]
- Nagel, S.; MacLeod, R.A.F.; Pommerenke, C.; Meyer, C.; Kaufmann, M.; Drexler, H.G. NKL Homeobox Gene NKX2-2 Is Aberrantly Expressed in Hodgkin Lymphoma. Oncotarget 2018, 9, 37480. [Google Scholar] [CrossRef][Green Version]
- Nagel, S.; Pommerenke, C.; Scherr, M.; Meyer, C.; Kaufmann, M.; Battmer, K.; MacLeod, R.A.F.; Drexler, H.G. NKL Homeobox Gene Activities in Hematopoietic Stem Cells, T-Cell Development and T-Cell Leukemia. PLoS ONE 2017, 12, e0171164. [Google Scholar] [CrossRef] [PubMed]
- Vu, L.P.; Perna, F.; Wang, L.; Voza, F.; Figueroa, M.E.; Tempst, P.; Erdjument-Bromage, H.; Gao, R.; Chen, S.; Paietta, E.; et al. PRMT4 Blocks Myeloid Differentiation by Assembling a Methyl-RUNX1-Dependent Repressor Complex. Cell Rep. 2013, 5, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lu, Y.; Espejo, A.; Wu, J.; Xu, W.; Liang, S.; Bedford, M.T. TDRD3 Is an Effector Molecule for Arginine-Methylated Histone Marks. Mol. Cell 2010, 40, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Sims, R.J., 3rd; Rojas, L.A.; Beck, D.B.; Bonasio, R.; Schüller, R.; Drury, W.J., 3rd; Eick, D.; Reinberg, D. The C-Terminal Domain of RNA Polymerase II Is Modified by Site-Specific Methylation. Science 2011, 332, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xu, W. Histone H3R17me2a Mark Recruits Human RNA Polymerase-Associated Factor 1 Complex to Activate Transcription. Proc. Natl. Acad. Sci. USA 2012, 109, 5675–5680. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Bedford, M.T.; Stallcup, M.R. Regulated Recruitment of Tumor Suppressor BRCA1 to the P21 Gene by Coactivator Methylation. Genes Dev. 2011, 25, 176–188. [Google Scholar] [CrossRef]
- Durant, S.T.; Cho, E.C.; La Thangue, N.B. P53 Methylation--the Arg-Ument Is Clear. Cell Cycle 2009, 8, 801–802. [Google Scholar] [CrossRef]
- Cheung, N.; Fung, T.K.; Zeisig, B.B.; Holmes, K.; Rane, J.K.; Mowen, K.A.; Finn, M.G.; Lenhard, B.; Chan, L.C.; So, C.W. Targeting Aberrant Epigenetic Networks Mediated by PRMT1 and KDM4C in Acute Myeloid Leukemia. Cancer Cell 2016, 29, 32–48. [Google Scholar] [CrossRef]
- Shah, M.A.; Denton, E.L.; Arrowsmith, C.H.; Lupien, M.; Schapira, M. A Global Assessment of Cancer Genomic Alterations in Epigenetic Mechanisms. Epigenetics Chromatin 2014, 7, 29. [Google Scholar] [CrossRef]
- Wu, Q.; Schapira, M.; Arrowsmith, C.H.; Barsyte-Lovejoy, D. Protein Arginine Methylation: From Enigmatic Functions to Therapeutic Targeting. Nat. Rev. Drug Discov. 2021, 20, 509–530. [Google Scholar] [CrossRef]
- Bedford, M.T.; Richard, S. Arginine Methylation: An Emerging Regulatorof Protein Function. Mol. Cell 2005, 18, 263–272. [Google Scholar] [CrossRef]
- Blackwell, E.; Ceman, S. Arginine Methylation of RNA-Binding Proteins Regulates Cell Function and Differentiation. Mol. Reprod. Dev. 2012, 79, 163. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Gao, L.; Teng, L.; Ge, J.; Oo, Z.M.; Kumar, A.R.; Gilliland, D.G.; Mason, P.J.; Tan, K.; Speck, N.A. Runx1 Deficiency Decreases Ribosome Biogenesis and Confers Stress Resistance to Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 2015, 17, 165–177. [Google Scholar] [CrossRef]
- Chuang, L.S.H.; Ito, K.; Ito, Y. RUNX Family: Regulation and Diversification of Roles through Interacting Proteins. Int. J. Cancer 2013, 132, 1260–1271. [Google Scholar] [CrossRef] [PubMed]
- Look, A.T. Oncogenic Transcription Factors in the Human Acute Leukemias. Science 1997, 278, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, T.; Nakamura-Ishizu, A.; Muddineni, S.; Tan, D.Q.; Wang, C.Q.; Tokunaga, K.; Tirado-Magallanes, R.; Sian, S.; Benoukraf, T.; Okuda, T.; et al. Hematopoietic Stem Cells Acquire Survival Advantage by Loss of RUNX1 Methylation Identified in Familial Leukemia. Blood 2020, 136, 1919–1932. [Google Scholar] [CrossRef]
- Fortin Ensign, S.P.; Roos, A.; Mathews, I.T.; Dhruv, H.D.; Tuncali, S.; Sarkaria, J.N.; Symons, M.H.; Loftus, J.C.; Berens, M.E.; Tran, N.L. SGEF Is Regulated via TWEAK/Fn14/NF-ΚB Signaling and Promotes Survival by Modulation of the DNA Repair Response to Temozolomide. Mol. Cancer Res. 2016, 14, 302–312. [Google Scholar] [CrossRef]
- Okuda, S.; Watanabe, Y.; Moriya, Y.; Kawano, S.; Yamamoto, T.; Matsumoto, M.; Takami, T.; Kobayashi, D.; Araki, N.; Yoshizawa, A.C.; et al. JPOSTrepo: An International Standard Data Repository for Proteomes. Nucleic Acids Res. 2017, 45, D1107–D1111. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oksa, L.; Mäkinen, A.; Nikkilä, A.; Hyvärinen, N.; Laukkanen, S.; Rokka, A.; Haapaniemi, P.; Seki, M.; Takita, J.; Kauko, O.; et al. Arginine Methyltransferase PRMT7 Deregulates Expression of RUNX1 Target Genes in T-Cell Acute Lymphoblastic Leukemia. Cancers 2022, 14, 2169. https://doi.org/10.3390/cancers14092169
Oksa L, Mäkinen A, Nikkilä A, Hyvärinen N, Laukkanen S, Rokka A, Haapaniemi P, Seki M, Takita J, Kauko O, et al. Arginine Methyltransferase PRMT7 Deregulates Expression of RUNX1 Target Genes in T-Cell Acute Lymphoblastic Leukemia. Cancers. 2022; 14(9):2169. https://doi.org/10.3390/cancers14092169
Chicago/Turabian StyleOksa, Laura, Artturi Mäkinen, Atte Nikkilä, Noora Hyvärinen, Saara Laukkanen, Anne Rokka, Pekka Haapaniemi, Masafumi Seki, Junko Takita, Otto Kauko, and et al. 2022. "Arginine Methyltransferase PRMT7 Deregulates Expression of RUNX1 Target Genes in T-Cell Acute Lymphoblastic Leukemia" Cancers 14, no. 9: 2169. https://doi.org/10.3390/cancers14092169
APA StyleOksa, L., Mäkinen, A., Nikkilä, A., Hyvärinen, N., Laukkanen, S., Rokka, A., Haapaniemi, P., Seki, M., Takita, J., Kauko, O., Heinäniemi, M., & Lohi, O. (2022). Arginine Methyltransferase PRMT7 Deregulates Expression of RUNX1 Target Genes in T-Cell Acute Lymphoblastic Leukemia. Cancers, 14(9), 2169. https://doi.org/10.3390/cancers14092169