
Oncogenic Kras-Mediated Cytokine CCL15 Regulates Pancreatic Cancer Cell Migration and Invasion through ROS

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines, Antibodies, and Reagents
2.2. Gene Knockdown
2.3. Cell Lysates Collection and Immunoblotting
2.4. RNA Isolation and Real-Time Quantitative PCR
2.5. Immunohistochemistry
2.6. Measurement of Cellular ROS Levels
2.7. Wound Healing Assay/Cell Migration Assay
2.8. Cell Growth Assay
2.9. Cell Invasion Assay
2.10. Statistical Analysis
3. Results
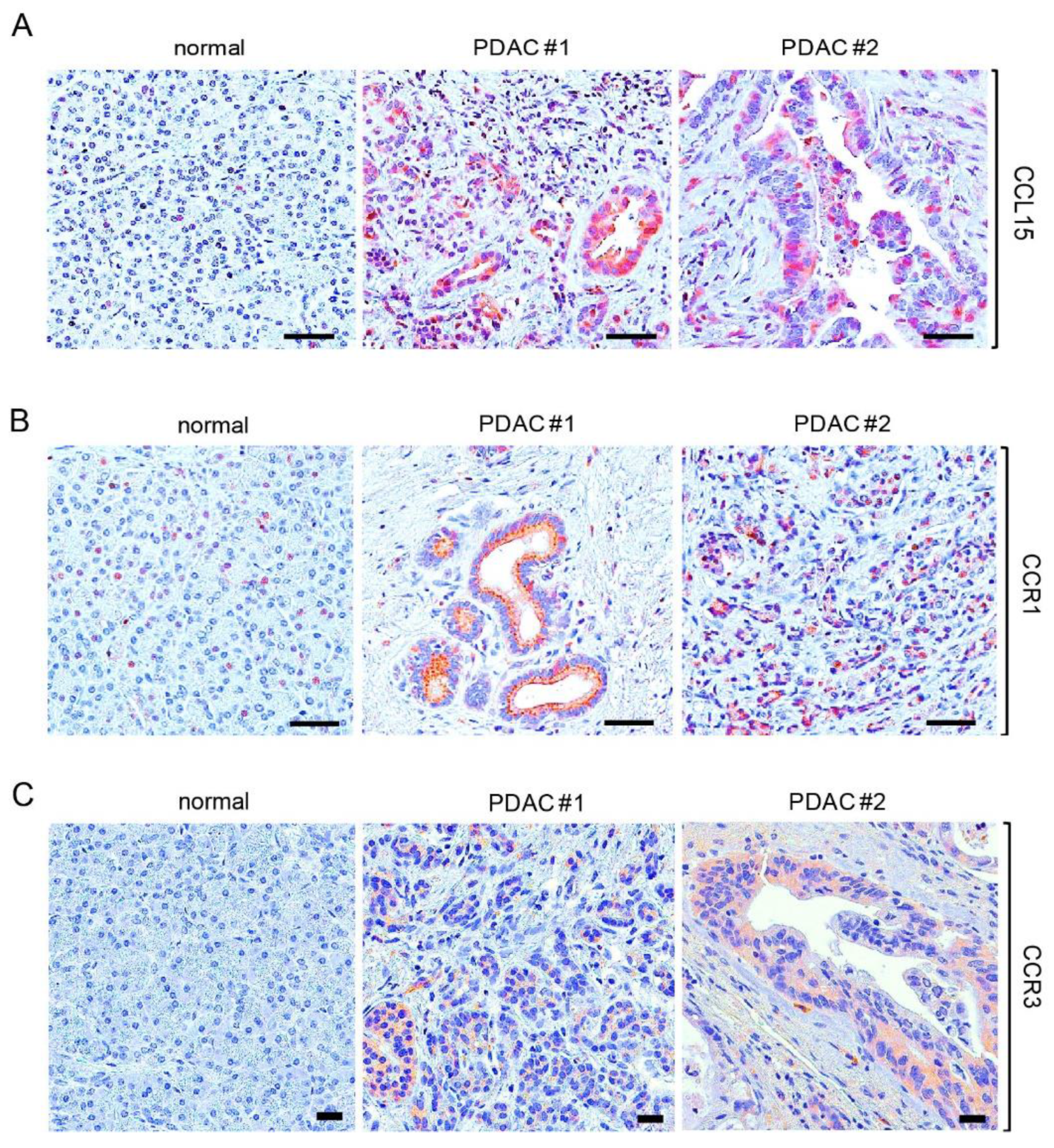
3.1. Elevated Expression of CCL15 and Its Receptors in Human PDAC
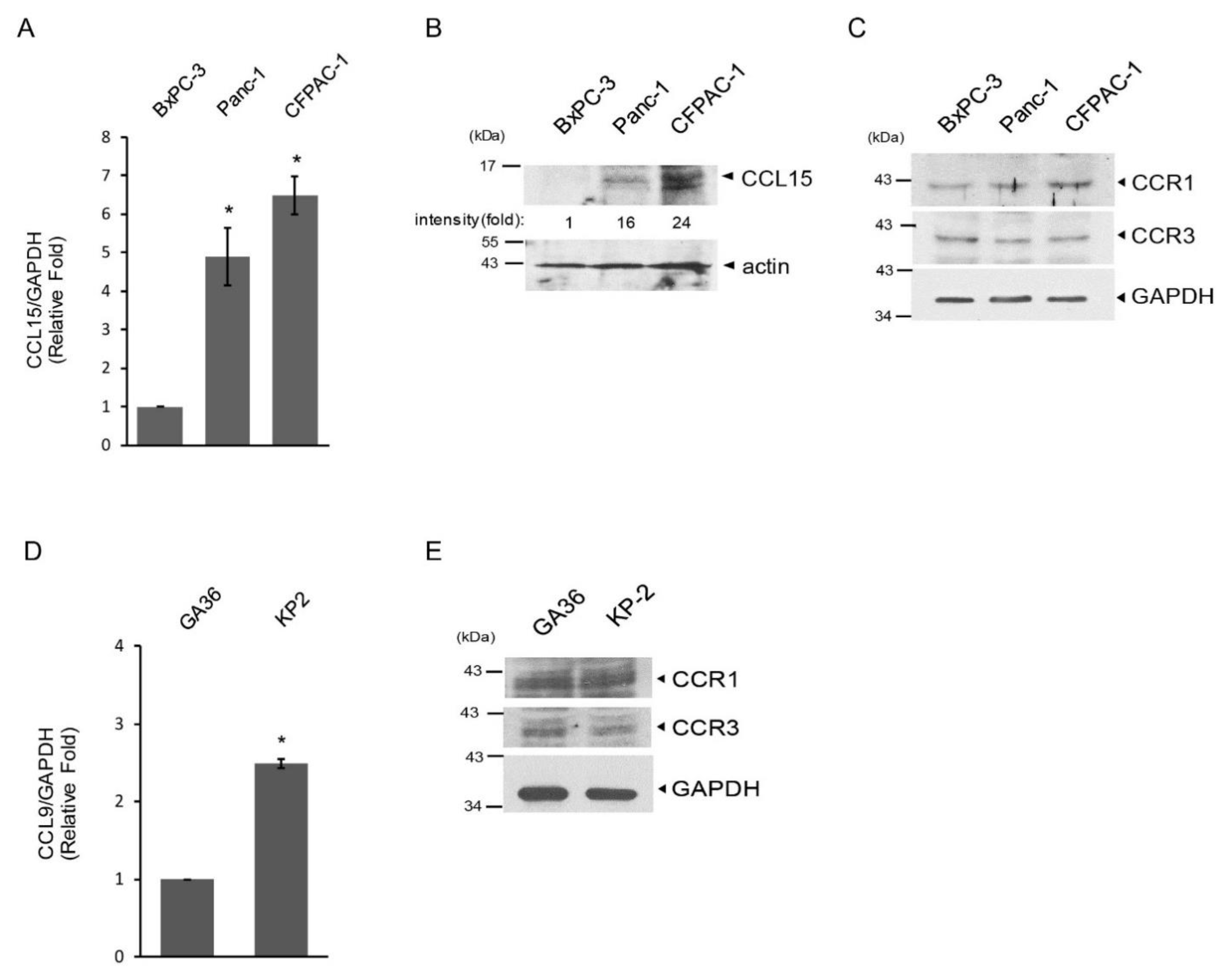
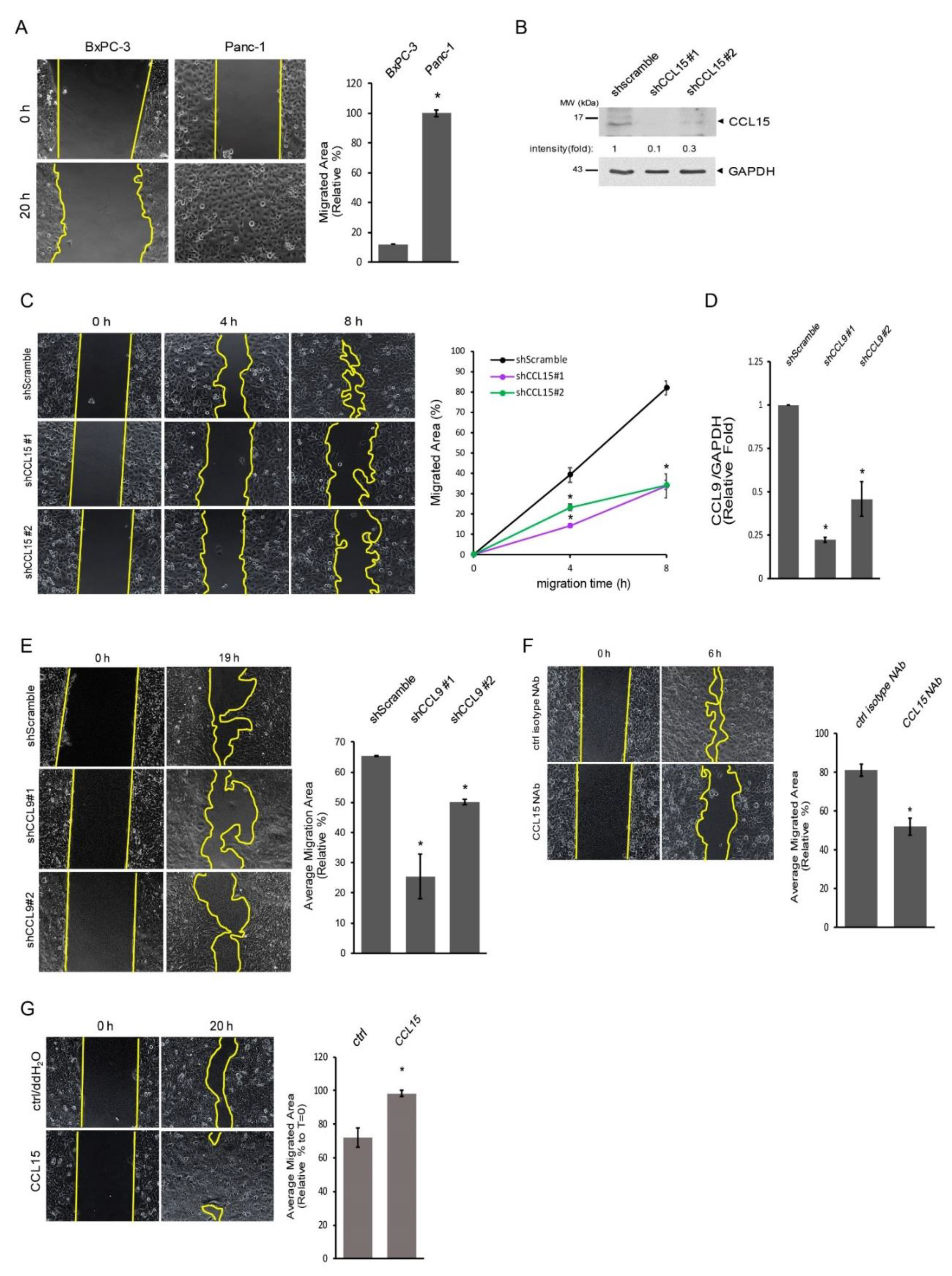
3.2. Upregulation of CCL15 in Metastatic PDAC Cells versus Non-Metastatic PDAC Cells
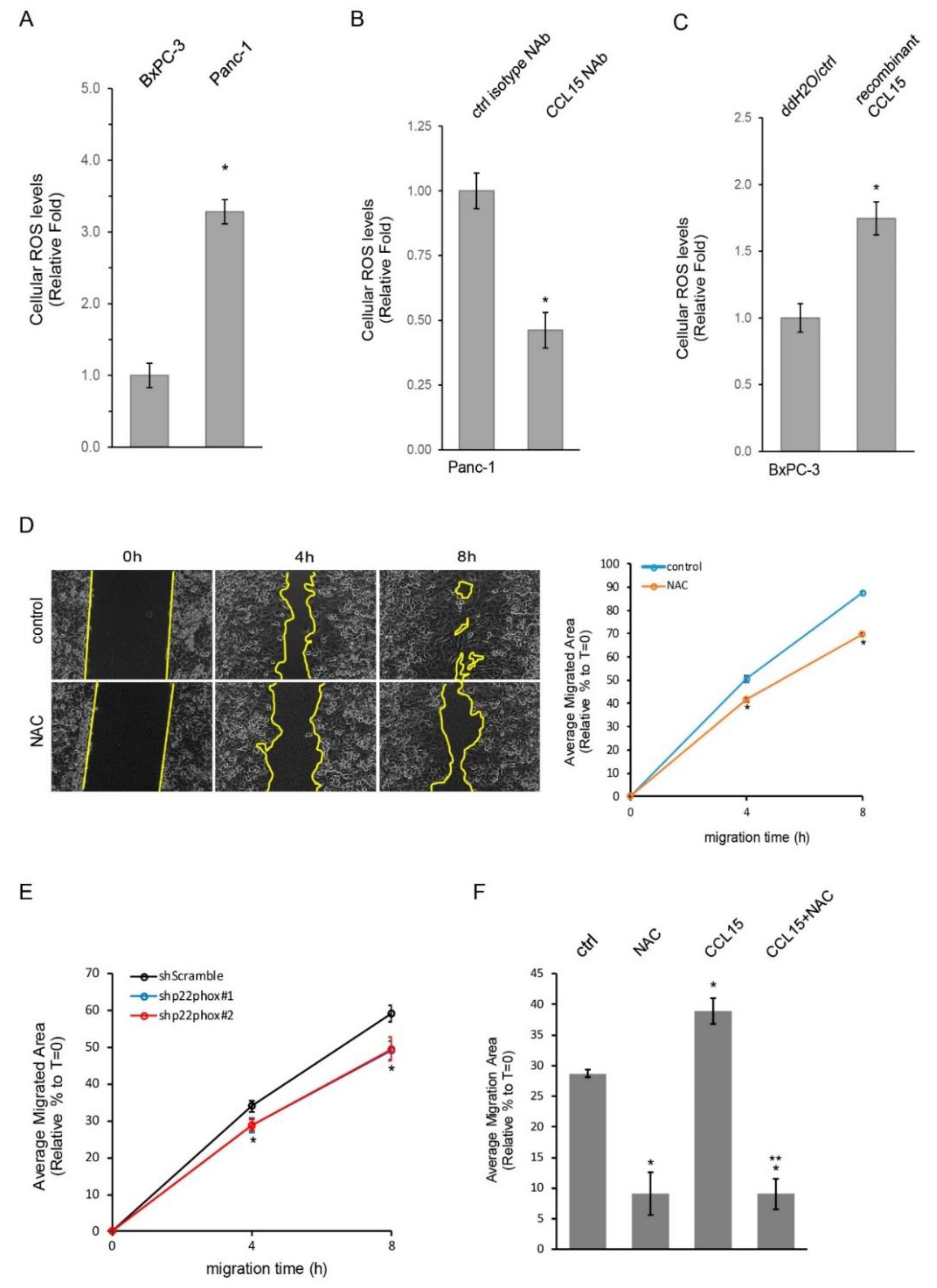
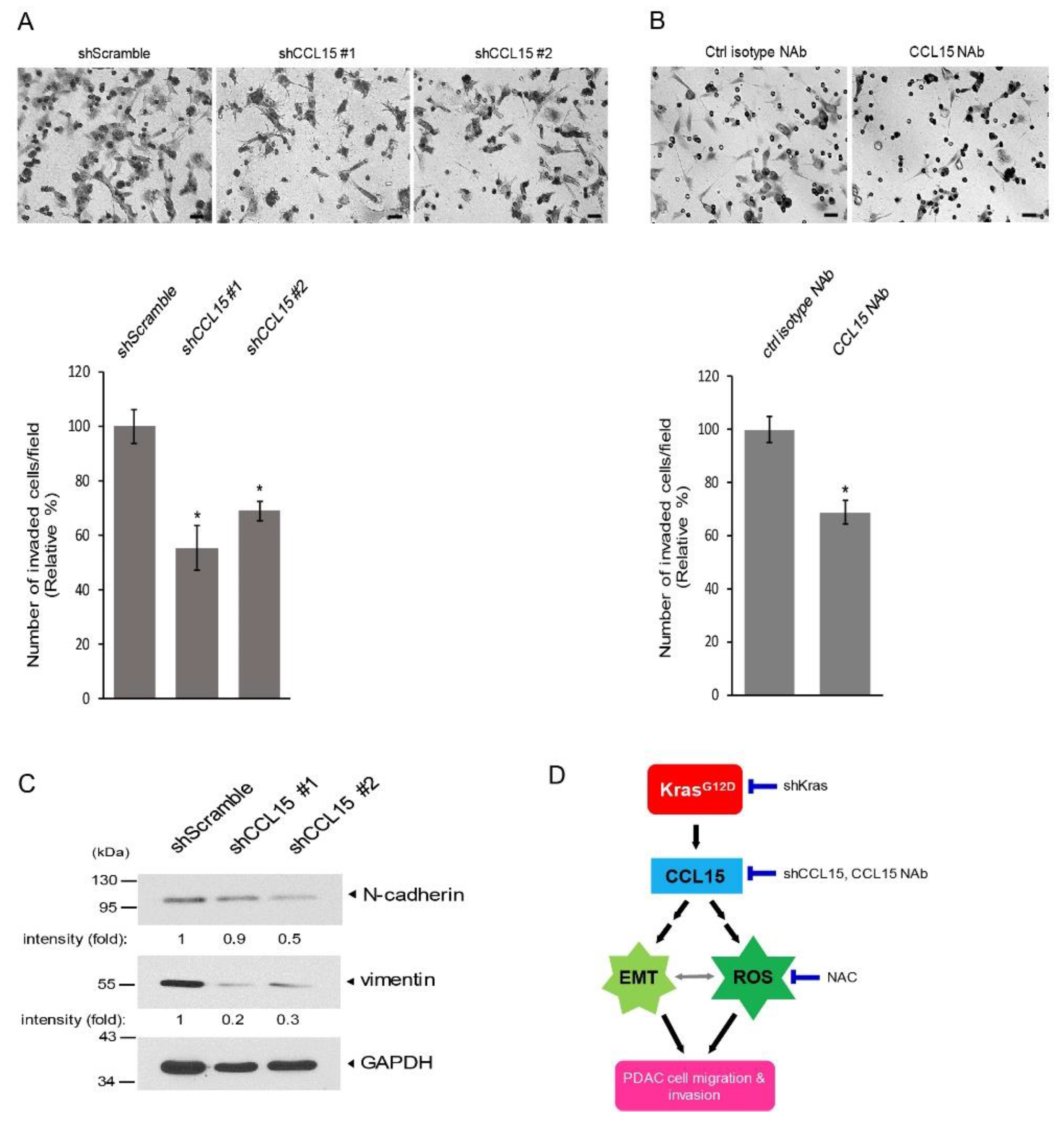
3.3. CCL15-Modulated PDAC Cell Migration through Reactive Oxygen Species (ROS)
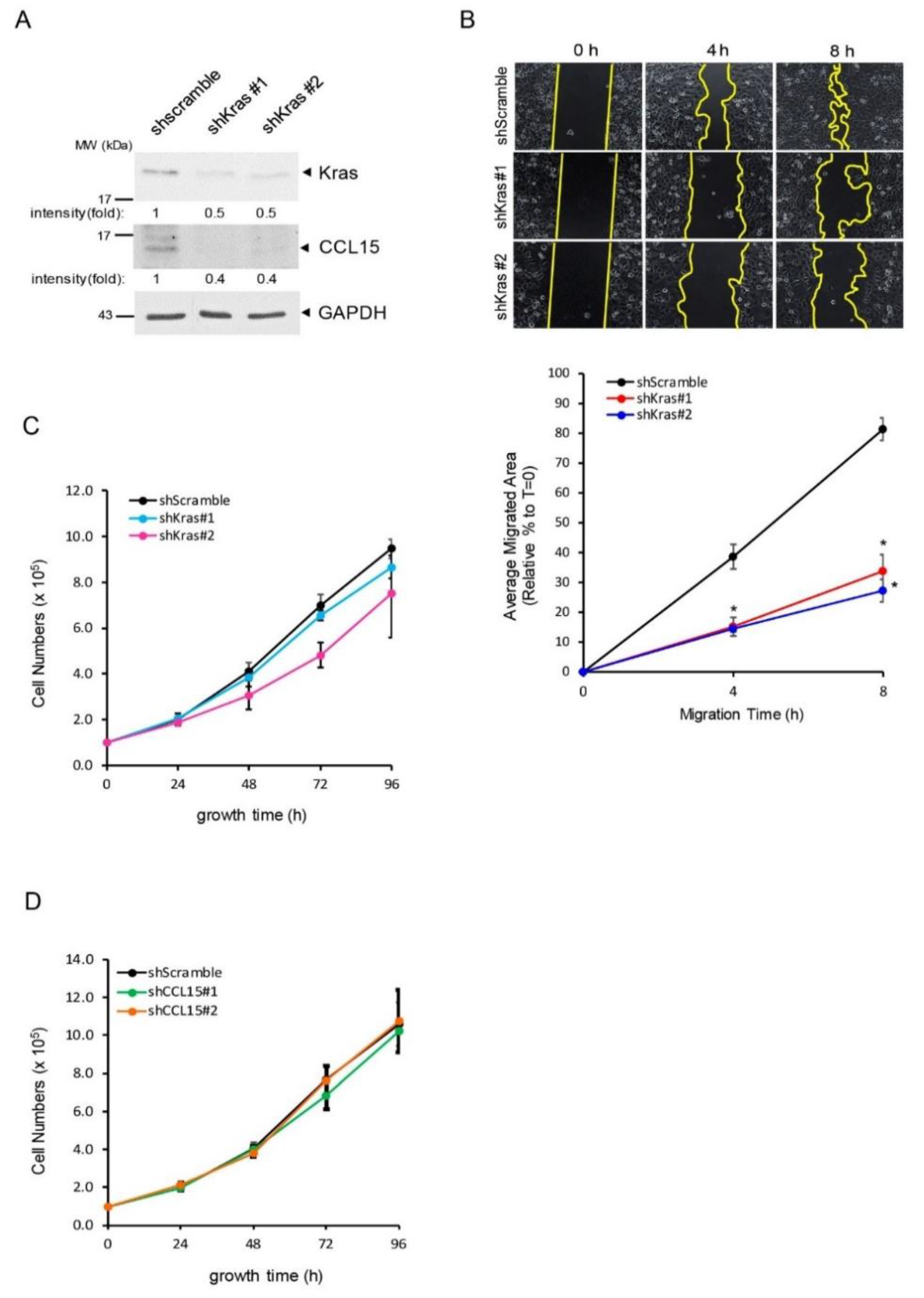
3.4. CCL15 Promoted PDAC Cell Migration and Invasion and Was Mediated through Oncogenic Kras
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Collins, M.A.; Bednar, F.; Zhang, Y.; Brisset, J.-C.; Galbán, S.; Galbán, C.J.; Rakshit, S.; Flannagan, K.S.; Adsay, N.V.; di Magliano, M.P. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Investig. 2012, 122, 639–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephen, A.G.; Esposito, D.; Bagni, R.K.; McCormick, F. Dragging ras back in the ring. Cancer Cell 2014, 25, 272–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef] [PubMed]
- Coradini, D.; Casarsa, C.; Oriana, S. Oriana, Epithelial cell polarity and tumorigenesis: New perspectives for cancer detection and treatment. Acta Pharmacol. Sin. 2011, 32, 552–564. [Google Scholar] [CrossRef] [Green Version]
- Klymkowsky, M.W.; Savagner, P. Epithelial-mesenchymal transition: A cancer researcher’s conceptual friend and foe. Am. J. Pathol. 2009, 174, 1588–1593. [Google Scholar] [CrossRef] [Green Version]
- Le Bras, G.F.; Taubenslag, K.J.; Andl, C.D. The regulation of cell-cell adhesion during epithelial-mesenchymal transition, motility and tumor progression. Cell Adhes. Migr. 2012, 6, 365–373. [Google Scholar] [CrossRef] [Green Version]
- Leggett, S.E.; Hruska, A.M.; Guo, M.; Wong, I.Y. The epithelial-mesenchymal transition and the cytoskeleton in bioengineered systems. Cell Commun. Signal. 2021, 19, 32. [Google Scholar] [CrossRef]
- Liu, M.; Hancock, S.E.; Sultani, G.; Wilkins, B.; Ding, E.; Osborne, B.; Quek, L.-E.; Turner, N. Snail-Overexpression Induces Epithelial-mesenchymal Transition and Metabolic Reprogramming in Human Pancreatic Ductal Adenocarcinoma and Non-tumorigenic Ductal Cells. J. Clin. Med. 2019, 8, 822. [Google Scholar] [CrossRef] [Green Version]
- Morvaridi, S.; Dhall, D.; Greene, M.I.; Pandol, S.J.; Wang, Q. Role of YAP and TAZ in pancreatic ductal adenocarcinoma and in stellate cells associated with cancer and chronic pancreatitis. Sci. Rep. 2015, 5, 16759. [Google Scholar] [CrossRef]
- Wang, S.; Huang, S.; Sun, Y.L. Epithelial-Mesenchymal Transition in Pancreatic Cancer: A Review. BioMed Res. Int. 2017, 2017, 2646148. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, Y.; Dobashi, K. CC-chemokine CCL15 expression and possible implications for the pathogenesis of IgE-related severe asthma. Mediat. Inflamm. 2012, 2012, 475253. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Dobashi, K.; Fueki, N.; Fueki, M.; Okada, T.; Tomioka, S.; Makino, S.; Mori, M. Changes of immunomodulatory cytokines associated with omalizumab therapy for severe persistent asthma. J. Biol. Regul. Homeost. Agents 2011, 25, 177–186. [Google Scholar]
- Youn, B.S.; Zhang, S.M.; Lee, E.K.; Park, D.H.; Broxmeyer, H.E.; Murphy, P.M.; Locati, M.; Pease, J.; Kim, K.K.; Antol, K.; et al. Molecular cloning of leukotactin-1: A novel human beta-chemokine, a chemoattractant for neutrophils, monocytes, and lymphocytes, and a potent agonist at CC chemokine receptors 1 and 3. J. Immunol. 1997, 159, 5201–5205. [Google Scholar] [PubMed]
- Gao, Y.; Zhou, Z.; Lu, S.; Huang, X.; Zhang, C.; Jiang, R.; Yao, A.; Sun, B.; Wang, X. Chemokine CCL15 Mediates Migration of Human Bone Marrow-Derived Mesenchymal Stem Cells Toward Hepatocellular Carcinoma. Stem Cells 2016, 34, 1112–1122. [Google Scholar] [PubMed] [Green Version]
- Inamoto, S.; Itatani, Y.; Yamamoto, T.; Minamiguchi, S.; Hirai, H.; Iwamoto, M.; Hasegawa, S.; Taketo, M.M.; Sakai, Y.; Kawada, K. Loss of SMAD4 Promotes Colorectal Cancer Progression by Accumulation of Myeloid-Derived Suppressor Cells through the CCL15-CCR1 Chemokine Axis. Clin. Cancer Res. 2016, 22, 492–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Yu, H.P.; Zhang, P. CCL15 overexpression predicts poor prognosis for hepatocellular carcinoma. Hepatol. Int. 2016, 10, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Milz, F.; Harder, A.; Neuhaus, P.; Breitkreuz-Korff, O.; Walhorn, V.; Lübke, T.; Anselmetti, D.; Dierks, T. Cooperation of binding sites at the hydrophilic domain of cell-surface sulfatase Sulf1 allows for dynamic interaction of the enzyme with its substrate heparan sulfate. Biochim. Biophys. Acta 2013, 1830, 5287–5298. [Google Scholar] [CrossRef]
- Agbunag, C.; Lee, K.E.; Buontempo, S.; Bar-Sagi, D. Pancreatic duct epithelial cell isolation and cultivation in two-dimensional and three-dimensional culture systems. Meth. Enzymol. 2006, 407, 703–710. [Google Scholar]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338.e6. [Google Scholar] [CrossRef]
- Coulin, F.; Power, C.A.; Alouani, S.; Peitsch, M.C.; Schroeder, J.M.; Moshizuki, M.; Clark-Lewis, I.; Wells, T.N. Characterisation of macrophage inflammatory protein-5/human CC cytokine-2, a member of the macrophage-inflammatory-protein family of chemokines. Eur. J. Biochem. 1997, 248, 507–515. [Google Scholar] [CrossRef]
- Pardigol, A.; Forssmann, U.; Zucht, H.D.; Loetscher, P.; Schulz-Knappe, P.; Baggiolini, M.; Forssmann, W.G.; Magert, H.J. HCC-2, a human chemokine: Gene structure, expression pattern, and biological activity. Proc. Natl. Acad. Sci. USA 1998, 95, 6308–6313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhagwandin, V.J.; Bishop, J.M.; Wright, W.E.; Shay, J.W. The Metastatic Potential and Chemoresistance of Human Pancreatic Cancer Stem Cells. PLoS ONE 2016, 11, e0148807. [Google Scholar] [CrossRef] [PubMed]
- Deer, E.L.; González-Hernández, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Ou, Y.; Wu, K.; Chen, Y.; Sun, W. miR-143 inhibits the metastasis of pancreatic cancer and an associated signaling pathway. Tumor Biol. 2012, 33, 1863–1870. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free. Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomou, E.; Gatzoulis, K.; Skiadas, I.; Doundoulakis, I.; Arsenos, P.; Dilaveris, P.; Sideris, S.; Tousoulis, D. Upgrade to cardiac resynchronization therapy difibrillator device of a pacemaker-dependent patient with end-stage hypertrophic cardiomyopathy. Hell. J. Cardiol. 2020, 61, 65–67. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef]
- Cerutti, P.A. Prooxidant states and tumor promotion. Science 1985, 227, 375–381. [Google Scholar] [CrossRef]
- Weinberg, F.; Chandel, N.S. Reactive oxygen species-dependent signaling regulates cancer. Cell. Mol. Life Sci. 2009, 66, 3663–3673. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.-Y.; Döppler, H.; DelGiorno, K.E.; Zhang, L.; Leitges, M.; Crawford, H.C.; Murphy, M.P.; Storz, P. Mutant KRas-Induced Mitochondrial Oxidative Stress in Acinar Cells Upregulates EGFR Signaling to Drive Formation of Pancreatic Precancerous Lesions. Cell Rep. 2016, 14, 2325–2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Bae, I.; Fan, S.; Meng, Q.; Rih, J.K.; Kim, H.J.; Kang, H.J.; Xu, J.; Goldberg, I.D.; Jaiswal, A.K.; Rosen, E.M. BRCA1 induces antioxidant gene expression and resistance to oxidative stress. Cancer Res. 2004, 64, 7893–7909. [Google Scholar] [CrossRef] [Green Version]
- Bhat, P.; Thorn, P. Myosin 2 maintains an open exocytic fusion pore in secretory epithelial cells. Mol. Biol. Cell 2009, 20, 1795–1803. [Google Scholar] [CrossRef] [Green Version]
- Delbridge, A.R.D.; Valente, L.J.; Strasser, A. The role of the apoptotic machinery in tumor suppression. Cold Spring Harb. Perspect. Biol. 2012, 4, a008789. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, J.C.; Wallace, D.; O’Brien, C.J.; Cotter, T.G. A novel antioxidant function for the tumor-suppressor gene p53 in the retinal ganglion cell. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4237–4244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.-K.; Choi, Y.W.; Lim, I.K.; Park, T.J. BTG2 suppresses cancer cell migration through inhibition of Src-FAK signaling by downregulation of reactive oxygen species generation in mitochondria. Clin. Exp. Metastasis 2012, 29, 901–913. [Google Scholar] [CrossRef]
- Hurd, T.R.; DeGennaro, M.; Lehmann, R. Redox Regulation of Cell Migration and Adhesion. Trends Cell Biol. 2012, 22, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Burtenshaw, D.; Hakimjavadi, R.; Redmond, E.M.; Cahill, P.A. Nox, Reactive oxygen species and regulation of vascular cell fate. Antioxidants 2017, 6, 90. [Google Scholar] [CrossRef] [Green Version]
- Hiraga, R.; Kato, M.; Miyagawa, S.; Kamata, T. Nox4-derived ROS signaling contributes to TGF-beta-induced epithelial-mesenchymal transition in pancreatic cancer cells. Anticancer Res. 2013, 33, 4431–4438. [Google Scholar] [PubMed]
- Ju, H.-Q.; Ying, H.; Tian, T.; Ling, J.; Fu, J.; Lu, Y.; Wu, M.; Yang, L.; Achreja, A.; Chen, G.; et al. Mutant Kras- and p16-regulated NOX4 activation overcomes metabolic checkpoints in development of pancreatic ductal adenocarcinoma. Nat. Commun. 2017, 8, 14437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, P.; He, X.; Rong, Y.; Cao, I.; Li, Y.; Hu, Y.; Liu, Y.; Li, D.; Lou, W.; Liu, M. Kras/NF-κB/YY1/miR-489 signaling axis controls pancreatic cancer metastasis. Cancer Res. 2017, 77, 100–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, A.; Tashiro, K.; Dixit, A.; Soni, A.; Vogel, K.; Hall, B.; Shafqat, I.; Slaughter, J.; Param, N.; Le, A.; et al. Loss of HIF1A from pancreatic cancer cells increases expression of PPP1R1B and degradation of p53 to promote invasion and metastasis. Gastroenterology 2020, 159, 1882–1897. [Google Scholar] [CrossRef]
- Kelber, J.A.; Rebo, T.; Kaushal, S.; Metildi, C.; Wright, T.; Stoletov, K.; Weems, J.M.; Park, F.D.; Mose, E.; Wang, Y.; et al. KRas induces a Src/PEAK1/ErbB2 kinase amplification loop that drives metastatic growth and therapy resistance in pancreatic cancer. Cancer Res 2012, 72, 2554–2564. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Li, Y.; Lian, G.; Lin, H.; Shang, C.; Zeng, L.; Chen, S.; Li, I.; Huang, C.; Huang, K.; et al. KRAS promotes tumor metastasis and chemoresistance by repressing RKIP via the MAPK-ERK pathway in pancreatic cancer. Int. J. Cancer 2018, 142, 2323–2334. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.H.; Philip, M.R.; Chen, Y.; Lu, L.; Dai, W. K-Ras Lys-42 is crucial for its signaling, cell migration, and invasion. J. Biol. Chem. 2018, 293, 17574–17581. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.-Y. Inflammatory Cytokine Signaling during Development of Pancreatic and Prostate Cancers. J. Immunol. Res. 2017, 2017, 7979637. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Fan, J.; Chen, H.; Meng, Z.; Chen, Z.; Wang, P.; Liu, L. The IL-8/CXCR1 axis is associated with cancer stem cell-like properties and correlates with clinical prognosis in human pancreatic cancer cases. Sci. Rep. 2014, 4, 5911. [Google Scholar] [CrossRef]
- Koshiba, T.; Hosotani, R.; Miyamoto, Y.; Ida, J.; Tsuji, S.; Nakajima, S.; Kawaguchi, M.; Kobayashi, H.; Doi, R.; Hori, T.; et al. Expression of stromal cell-derived factor 1 and CXCR4 ligand receptor system in pancreatic cancer: A possible role for tumor progression. Clin. Cancer Res. 2000, 6, 3530–3535. [Google Scholar]
- Mandl, K.D.; Kohane, I.S. A 21st-Century Health IT System—Creating a Real-World Information Economy. N. Engl. J. Med. 2017, 376, 1905–1907. [Google Scholar] [CrossRef] [PubMed]
- Monti, P.; Leone, B.E.; Marchesi, F.; Balzano, G.; Zerbi, A.; Scaltrini, F.; Pasquali, C.; Calori, G.; Pessi, F.; Sperti, C.; et al. The CC chemokine MCP-1/CCL2 in pancreatic cancer progression: Regulation of expression and potential mechanisms of antimalignant activity. Cancer Res. 2003, 63, 7451–7461. [Google Scholar] [PubMed]
- Razidlo, G.L.; Burton, K.M.; McNiven, M.A. Interleukin-6 promotes pancreatic cancer cell migration by rapidly activating the small GTPase CDC42. J. Biol. Chem. 2018, 293, 11143–11153. [Google Scholar] [CrossRef] [Green Version]
- Saur, D.; Seidler, B.; Schneider, G.; Algül, H.; Beck, R.; Senekowitsch–Schmidtke, R.; Schwaiger, M.; Schmid, R.M. CXCR4 expression increases liver and lung metastasis in a mouse model of pancreatic cancer. Gastroenterology 2005, 129, 1237–1250. [Google Scholar] [CrossRef] [PubMed]
- Takamori, H.; Oades, Z.G.; Hoch, R.C.; Burger, M.; Schraufstatter, I.U. Schraufstatter, Autocrine growth effect of IL-8 and GROalpha on a human pancreatic cancer cell line, Capan-1. Pancreas 2000, 21, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Kim, J.; Revelo-Penafiel, M.P.; Angel, R.; Dawson, D.W.; Lowy, A.M. The chemokine receptor CXCR4 is expressed in pancreatic intraepithelial neoplasia. Gut 2008, 57, 1555–1560. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Groth, S.; Sebens, S.; Lehnert, H.; Gieseler, F.; Fandrich, F. Differential roles of Smad2 and Smad3 in the regulation of TGF-beta1-mediated growth inhibition and cell migration in pancreatic ductal adenocarcinoma cells: Control by Rac1. Mol. Cancer 2011, 10, 67. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Messex, J.K.; Adams, K.L.A.; Hawkins, W.G.; DeNardo, D.; Bardeesy, N.; Billadeau, D.D.; Liou, G.-Y. Oncogenic Kras-Mediated Cytokine CCL15 Regulates Pancreatic Cancer Cell Migration and Invasion through ROS. Cancers 2022, 14, 2153. https://doi.org/10.3390/cancers14092153
Messex JK, Adams KLA, Hawkins WG, DeNardo D, Bardeesy N, Billadeau DD, Liou G-Y. Oncogenic Kras-Mediated Cytokine CCL15 Regulates Pancreatic Cancer Cell Migration and Invasion through ROS. Cancers. 2022; 14(9):2153. https://doi.org/10.3390/cancers14092153
Chicago/Turabian StyleMessex, Justin K., Kiyah L. A. Adams, William G. Hawkins, David DeNardo, Nabeel Bardeesy, Daniel D. Billadeau, and Geou-Yarh Liou. 2022. "Oncogenic Kras-Mediated Cytokine CCL15 Regulates Pancreatic Cancer Cell Migration and Invasion through ROS" Cancers 14, no. 9: 2153. https://doi.org/10.3390/cancers14092153
APA StyleMessex, J. K., Adams, K. L. A., Hawkins, W. G., DeNardo, D., Bardeesy, N., Billadeau, D. D., & Liou, G.-Y. (2022). Oncogenic Kras-Mediated Cytokine CCL15 Regulates Pancreatic Cancer Cell Migration and Invasion through ROS. Cancers, 14(9), 2153. https://doi.org/10.3390/cancers14092153