
Does DPP-IV Inhibition Offer New Avenues for Therapeutic Intervention in Malignant Disease?


Abstract
Simple Summary
Abstract
1. Introduction
2. DPP-IV (CD26)—A Multifunctional Molecule with Enzyme-Activity-Dependent and -Independent Functions
3. DPP-IV in Cancer—A Matter of Controversy
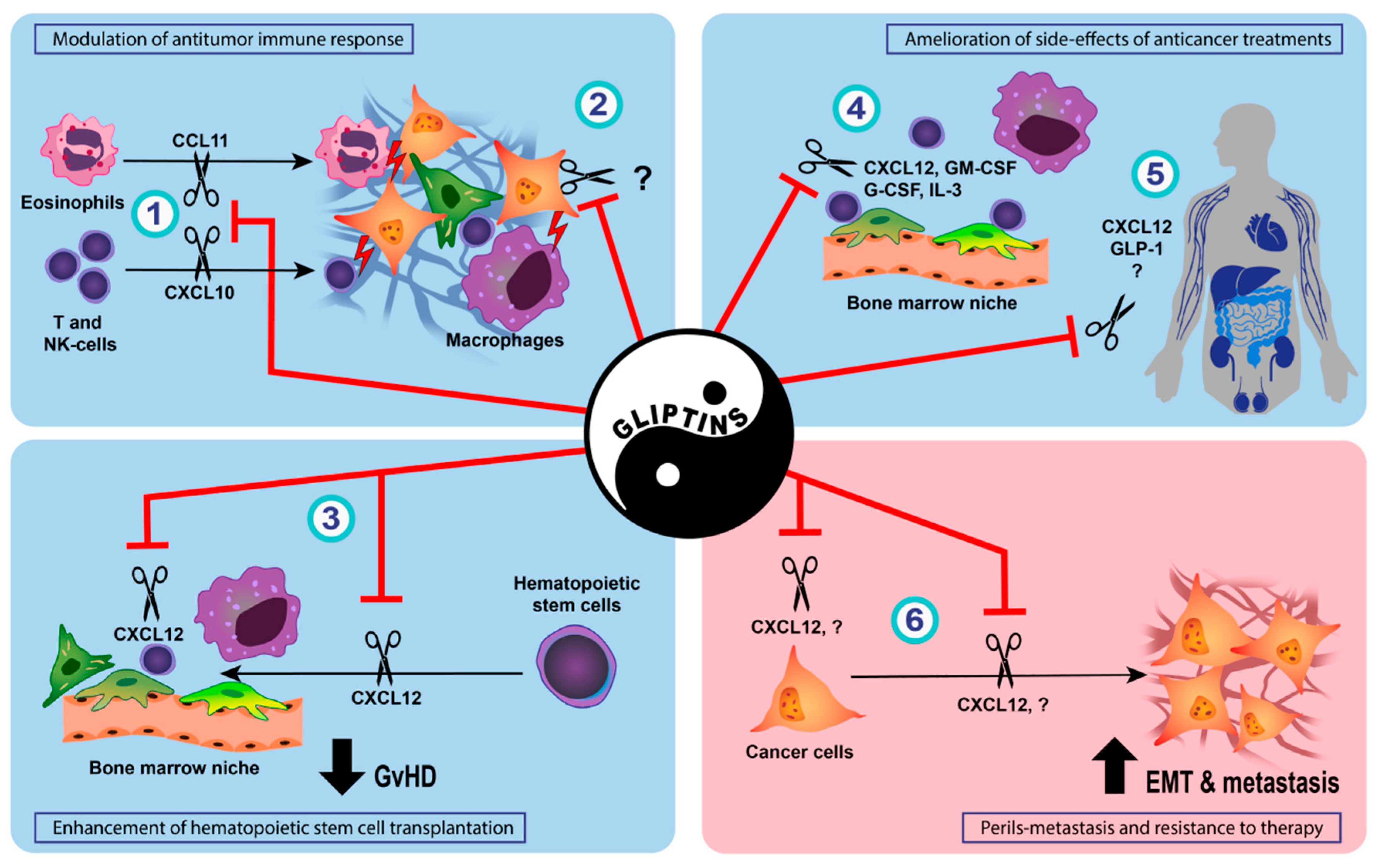
4. DPP-IV Inhibition and Cancer Initiation and Progression

{kind=link}
| Tumor Type | Model | Gliptin | Reference |
|---|---|---|---|
| Hepatocellular carcinoma | Diethylnitrosamine + high-fat diet-induced carcinogenesis in rats | Vildagliptin | [112] |
| Thiacetamide-induced carcinogenesis in rats | Saxagliptin | [117] | |
| Hepatocellular carcinoma associated with nonalcoholic steatohepatitis | Melanocortin 4 receptor (MC4R)-deficient mice fed Western-type diet | Anagliptin | [119] |
| STAM mouse model | Sitagliptin | [120] | |
| Choline deficiency-induced steatohepatitis in rats | Teneligliptin | [121] | |
| Choline deficiency-induced steatohepatitis in rats | Sitagliptin | [122] | |
| Colorectal cancer | 1,2-dimethylhydrazine and high-fat diet in rats | Sitagliptin | [129] |
| Leptin-deficient mice administered 1,2-dimethylhydrazine and dextran sulfate sodium-induced colitis | Sitagliptin | [130] | |
| Mice with heterozygous Apc mutation fed high-fat diet | Sitagliptin | [131] | |
| Renal cell carcinoma | Diethylnitrosamine-induced carcinogenesis in rats | Sitagliptin | [132] |
5. DPP-IV Inhibition in Anticancer Treatment
5.1. Direct Cytotoxic Effects of DPP-IV Inhibition on Cancer Cells
5.2. Effect of DPP-IV Inhibition on Anti-Tumor Immune Response
5.3. Enhancement of Hematopoietic Stem Cell Transplantation and Prophylaxis of Acute Graft-Versus-Host Disease by DPP-IV Inhibition
5.4. Amelioration of Side-Effects of Conventional Anticancer Treatments by DPP-IV Inhibition
6. Conclusions and Unanswered Questions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Murgai, M.; Giles, A.; Kaplan, R. Physiological, Tumor, and Metastatic Niches: Opportunities and Challenges for Targeting the Tumor Microenvironment. Crit. Rev. Oncog. 2015, 20, 301–314. [Google Scholar] [CrossRef]
- Mulvihill, E.E.; Drucker, D.J. Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr. Rev. 2014, 35, 992–1019. [Google Scholar] [CrossRef]
- Klemann, C.; Wagner, L.; Stephan, M.; von Horsten, S. Cut to the chase: A review of CD26/dipeptidyl peptidase-4’s (DPP4) entanglement in the immune system. Clin. Exp. Immunol. 2016, 185, 1–21. [Google Scholar] [CrossRef]
- Busek, P.; Sedo, A. Dipeptidyl Peptidase-IV and Related Proteases in Brain Tumors. In Evolution of the Molecular Biology of Brain Tumors and the Therapeutic Implications; Lichtor, T., Ed.; InTech: London, UK, 2013. [Google Scholar]
- Lambeir, A.M.; Durinx, C.; Scharpe, S.; De Meester, I. Dipeptidyl-peptidase IV from bench to bedside: An update on structural properties, functions, and clinical aspects of the enzyme DPP IV. Crit. Rev. Clin. Lab. Sci. 2003, 40, 209–294. [Google Scholar] [CrossRef]
- Mentlein, R. Dipeptidyl-peptidase IV (CD26)—Role in the inactivation of regulatory peptides. Regul. Pept. 1999, 85, 9–24. [Google Scholar] [CrossRef]
- Wang, H.; Liu, X.; Long, M.; Huang, Y.; Zhang, L.; Zhang, R.; Zheng, Y.; Liao, X.; Wang, Y.; Liao, Q.; et al. NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis. Sci. Transl. Med. 2016, 8, 334–351. [Google Scholar] [CrossRef]
- He, L.; Zhang, T.; Sun, W.; Qin, Y.; Wang, Z.; Dong, W.; Zhang, H. The DPP-IV inhibitor saxagliptin promotes the migration and invasion of papillary thyroid carcinoma cells via the NRF2/HO1 pathway. Med. Oncol. 2020, 37, 97. [Google Scholar] [CrossRef] [PubMed]
- Durinx, C.; Lambeir, A.M.; Bosmans, E.; Falmagne, J.B.; Berghmans, R.; Haemers, A.; Scharpe, S.; De Meester, I. Molecular characterization of dipeptidyl peptidase activity in serum: Soluble CD26/dipeptidyl peptidase IV is responsible for the release of X-Pro dipeptides. Eur. J. Biochem. 2000, 267, 5608–5613. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Barrett, A.J.; Bateman, A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2012, 40, D343–D350. [Google Scholar] [CrossRef]
- Elmansi, A.M.; Awad, M.E.; Eisa, N.H.; Kondrikov, D.; Hussein, K.A.; Aguilar-Perez, A.; Herberg, S.; Periyasamy-Thandavan, S.; Fulzele, S.; Hamrick, M.W.; et al. What doesn’t kill you makes you stranger: Dipeptidyl peptidase-4 (CD26) proteolysis differentially modulates the activity of many peptide hormones and cytokines generating novel cryptic bioactive ligands. Pharmacol. Ther. 2019, 198, 90–108. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E.; Drucker, D.J. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 2013, 17, 819–837. [Google Scholar] [CrossRef]
- American Diabetes Association. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2021. Diabetes Care 2021, 44, S111–S124. [Google Scholar] [CrossRef]
- Deacon, C.F. A review of dipeptidyl peptidase-4 inhibitors. Hot topics from randomized controlled trials. Diabetes Obes. Metab. 2018, 20, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Kaji, K.; Yoshiji, H.; Ikenaka, Y.; Noguchi, R.; Aihara, Y.; Douhara, A.; Moriya, K.; Kawaratani, H.; Shirai, Y.; Yoshii, J.; et al. Dipeptidyl peptidase-4 inhibitor attenuates hepatic fibrosis via suppression of activated hepatic stellate cell in rats. J. Gastroenterol. 2014, 49, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Tomovic, K.; Lazarevic, J.; Kocic, G.; Deljanin-Ilic, M.; Anderluh, M.; Smelcerovic, A. Mechanisms and pathways of anti-inflammatory activity of DPP-4 inhibitors in cardiovascular and renal protection. Med. Res. Rev. 2019, 39, 404–422. [Google Scholar] [CrossRef]
- Makdissi, A.; Ghanim, H.; Vora, M.; Green, K.; Abuaysheh, S.; Chaudhuri, A.; Dhindsa, S.; Dandona, P. Sitagliptin exerts an antinflammatory action. J. Clin. Endocrinol. Metab. 2012, 97, 3333–3341. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S.; Rai, U.; Kosuru, R.; Tiwari, V.; Singh, S. Amelioration of diet-induced metabolic syndrome and fatty liver with sitagliptin via regulation of adipose tissue inflammation and hepatic Adiponectin/AMPK levels in mice. Biochimie 2020, 168, 198–209. [Google Scholar] [CrossRef]
- Lee, T.M.; Chen, W.T.; Yang, C.C.; Lin, S.Z.; Chang, N.C. Sitagliptin attenuates sympathetic innervation via modulating reactive oxygen species and interstitial adenosine in infarcted rat hearts. J. Cell. Mol. Med. 2015, 19, 418–429. [Google Scholar] [CrossRef]
- He, Y.; Yang, G.; Yao, F.; Xian, Y.; Wang, G.; Chen, L.; Lv, X.; Gao, H.; Zheng, Z.; Sun, L.; et al. Sitagliptin inhibits vascular inflammation via the SIRT6-dependent signaling pathway. Int. Immunopharmacol. 2019, 75, 105805. [Google Scholar] [CrossRef]
- Kim, S.H.; Yoo, J.H.; Lee, W.J.; Park, C.Y. Gemigliptin: An Update of Its Clinical Use in the Management of Type 2 Diabetes Mellitus. Diabetes Metab. J. 2016, 40, 339–353. [Google Scholar] [CrossRef]
- Weber, A.E.; Thornberry, N.A. Dipeptidyl Peptidase 4 Inhibitors. In Burger’s Medicinal Chemistry and Drug Discovery; 2021; pp. 1–48. Available online: https://onlinelibrary.wiley.com/doi/book/10.1002/0471266949 (accessed on 20 March 2022).
- Kim, S.H.; Jung, E.; Yoon, M.K.; Kwon, O.H.; Hwang, D.M.; Kim, D.W.; Kim, J.; Lee, S.M.; Yim, H.J. Pharmacological profiles of gemigliptin (LC15-0444), a novel dipeptidyl peptidase-4 inhibitor, in vitro and in vivo. Eur. J. Pharmacol. 2016, 788, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.; Eckhardt, M.; Langkopf, E.; Tadayyon, M.; Himmelsbach, F.; Mark, M. (R)-8-(3-amino-piperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylm ethyl)-3,7-dihydro-purine-2,6-dione (BI 1356), a novel xanthine-based dipeptidyl peptidase 4 inhibitor, has a superior potency and longer duration of action compared with other dipeptidyl peptidase-4 inhibitors. J. Pharmacol. Exp. Ther. 2008, 325, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.S.; Yasuda, Y.; Kojima, Y.; Okada, S.; Motoyama, T.; Takahashi, R.; Oka, M. Anagliptin, a potent dipeptidyl peptidase IV inhibitor: Its single-crystal structure and enzyme interactions. J. Enzym. Inhib. Med. Chem. 2015, 30, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Proost, P.; Struyf, S.; Schols, D.; Durinx, C.; Wuyts, A.; Lenaerts, J.P.; De Clercq, E.; De Meester, I.; Van Damme, J. Processing by CD26/dipeptidyl-peptidase IV reduces the chemotactic and anti-HIV-1 activity of stromal-cell-derived factor-1alpha. FEBS Lett. 1998, 432, 73–76. [Google Scholar] [CrossRef]
- Christopherson, K.W.; Cooper, S.; Hangoc, G.; Broxmeyer, H.E. CD26 is essential for normal G-CSF-induced progenitor cell mobilization as determined by CD26(−/−) mice. Exp. Hematol. 2003, 31, 1126–1134. [Google Scholar] [CrossRef]
- Christopherson, K.W., 2nd; Hangoc, G.; Mantel, C.R.; Broxmeyer, H.E. Modulation of hematopoietic stem cell homing and engraftment by CD26. Science 2004, 305, 1000–1003. [Google Scholar] [CrossRef]
- Hansen, H.H.; Gronlund, R.V.; Baader-Pagler, T.; Haebel, P.; Tammen, H.; Larsen, L.K.; Jelsing, J.; Vrang, N.; Klein, T. Characterization of combined linagliptin and Y2R agonist treatment in diet-induced obese mice. Sci. Rep. 2021, 11, 8060. [Google Scholar] [CrossRef]
- Gutheil, W.G.; Subramanyam, M.; Flentke, G.R.; Sanford, D.G.; Munoz, E.; Huber, B.T.; Bachovchin, W.W. Human immunodeficiency virus 1 Tat binds to dipeptidyl aminopeptidase IV (CD26): A possible mechanism for Tat’s immunosuppressive activity. Proc. Natl. Acad. Sci. USA 1994, 91, 6594–6598. [Google Scholar] [CrossRef]
- Blanco, J.; Valenzuela, A.; Herrera, C.; Lluis, C.; Hovanessian, A.G.; Franco, R. The HIV-1 gp120 inhibits the binding of adenosine deaminase to CD26 by a mechanism modulated by CD4 and CXCR4 expression. FEBS Lett. 2000, 477, 123–128. [Google Scholar] [CrossRef]
- Lu, G.; Hu, Y.; Wang, Q.; Qi, J.; Gao, F.; Li, Y.; Zhang, Y.; Zhang, W.; Yuan, Y.; Bao, J.; et al. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature 2013, 500, 227–231. [Google Scholar] [CrossRef]
- Fox, D.A.; Hussey, R.E.; Fitzgerald, K.A.; Acuto, O.; Poole, C.; Palley, L.; Daley, J.F.; Schlossman, S.F.; Reinherz, E.L. Ta1, a novel 105 KD human T cell activation antigen defined by a monoclonal antibody. J. Immunol. 1984, 133, 1250–1256. [Google Scholar]
- Lepore, M.; Lewinsohn, D.A.; Lewinsohn, D.M. T cell receptor diversity, specificity and promiscuity of functionally heterogeneous human MR1-restricted T cells. Mol. Immunol. 2021, 130, 64–68. [Google Scholar] [CrossRef]
- Waumans, Y.; Baerts, L.; Kehoe, K.; Lambeir, A.M.; De Meester, I. The Dipeptidyl Peptidase Family, Prolyl Oligopeptidase, and Prolyl Carboxypeptidase in the Immune System and Inflammatory Disease, Including Atherosclerosis. Front. Immunol. 2015, 6, 387. [Google Scholar] [CrossRef]
- Bengsch, B.; Seigel, B.; Flecken, T.; Wolanski, J.; Blum, H.E.; Thimme, R. Human Th17 cells express high levels of enzymatically active dipeptidylpeptidase IV (CD26). J. Immunol. 2012, 188, 5438–5447. [Google Scholar] [CrossRef]
- Ohnuma, K.; Yamochi, T.; Uchiyama, M.; Nishibashi, K.; Yoshikawa, N.; Shimizu, N.; Iwata, S.; Tanaka, H.; Dang, N.H.; Morimoto, C. CD26 up-regulates expression of CD86 on antigen-presenting cells by means of caveolin-1. Proc. Natl. Acad. Sci. USA 2004, 101, 14186–14191. [Google Scholar] [CrossRef]
- Ohnuma, K.; Uchiyama, M.; Yamochi, T.; Nishibashi, K.; Hosono, O.; Takahashi, N.; Kina, S.; Tanaka, H.; Lin, X.; Dang, N.H.; et al. Caveolin-1 triggers T-cell activation via CD26 in association with CARMA1. J. Biol. Chem. 2007, 282, 10117–10131. [Google Scholar] [CrossRef]
- Hatano, R.; Ohnuma, K.; Yamamoto, J.; Dang, N.H.; Morimoto, C. CD26-mediated co-stimulation in human CD8(+) T cells provokes effector function via pro-inflammatory cytokine production. Immunology 2013, 138, 165–172. [Google Scholar] [CrossRef]
- Huang, S.; Apasov, S.; Koshiba, M.; Sitkovsky, M. Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood 1997, 90, 1600–1610. [Google Scholar] [CrossRef]
- Kameoka, J.; Tanaka, T.; Nojima, Y.; Schlossman, S.F.; Morimoto, C. Direct association of adenosine deaminase with a T cell activation antigen, CD26. Science 1993, 261, 466–469. [Google Scholar] [CrossRef]
- Gines, S.; Marino, M.; Mallol, J.; Canela, E.I.; Morimoto, C.; Callebaut, C.; Hovanessian, A.; Casado, V.; Lluis, C.; Franco, R. Regulation of epithelial and lymphocyte cell adhesion by adenosine deaminase-CD26 interaction. Biochem. J. 2002, 361, 203–209. [Google Scholar] [CrossRef]
- Yu, D.M.; Slaitini, L.; Gysbers, V.; Riekhoff, A.G.; Kahne, T.; Knott, H.M.; De Meester, I.; Abbott, C.A.; McCaughan, G.W.; Gorrell, M.D. Soluble CD26/dipeptidyl peptidase IV enhances human lymphocyte proliferation in vitro independent of dipeptidyl peptidase enzyme activity and adenosine deaminase binding. Scand. J. Immunol. 2011, 73, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Huhn, J.; Ehrlich, S.; Fleischer, B.; von Bonin, A. Molecular analysis of CD26-mediated signal transduction in T cells. Immunol. Lett. 2000, 72, 127–132. [Google Scholar] [CrossRef]
- Metzemaekers, M.; Van Damme, J.; Mortier, A.; Proost, P. Regulation of Chemokine Activity—A Focus on the Role of Dipeptidyl Peptidase IV/CD26. Front. Immunol. 2016, 7, 483. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, K.; Daniel, P.; Wisbrun, N.; Fuchs, H.; Fan, H. Delayed allogeneic skin graft rejection in CD26-deficient mice. Cell. Mol. Immunol. 2019, 16, 557–567. [Google Scholar] [CrossRef]
- Klemann, C.; Schade, J.; Pabst, R.; Leitner, S.; Stiller, J.; von Horsten, S.; Stephan, M. CD26/dipeptidyl peptidase 4-deficiency alters thymic emigration patterns and leukcocyte subsets in F344-rats age-dependently. Clin. Exp. Immunol. 2009, 155, 357–365. [Google Scholar] [CrossRef]
- Vora, K.A.; Porter, G.; Peng, R.; Cui, Y.; Pryor, K.; Eiermann, G.; Zaller, D.M. Genetic ablation or pharmacological blockade of dipeptidyl peptidase IV does not impact T cell-dependent immune responses. BMC Immunol. 2009, 10, 19. [Google Scholar] [CrossRef]
- Reinhold, D.; Goihl, A.; Wrenger, S.; Reinhold, A.; Kuhlmann, U.C.; Faust, J.; Neubert, K.; Thielitz, A.; Brocke, S.; Tager, M.; et al. Role of dipeptidyl peptidase IV (DP IV)-like enzymes in T lymphocyte activation: Investigations in DP IV/CD26-knockout mice. Clin. Chem. Lab. Med. 2009, 47, 268–274. [Google Scholar] [CrossRef]
- The Immunological Genome Project. ImmGen at 15. Nat. Immunol. 2020, 21, 700–703. [Google Scholar] [CrossRef]
- Cheng, H.C.; Abdel-Ghany, M.; Pauli, B.U. A novel consensus motif in fibronectin mediates dipeptidyl peptidase IV adhesion and metastasis. J. Biol. Chem. 2003, 278, 24600–24607. [Google Scholar] [CrossRef]
- Sedo, A.; Stremenova, J.; Busek, P.; Duke-Cohan, J.S. Dipeptidyl peptidase-IV and related molecules: Markers of malignancy? Expert Opin. Med. Diagn. 2008, 2, 677–689. [Google Scholar] [CrossRef]
- Enz, N.; Vliegen, G.; De Meester, I.; Jungraithmayr, W. CD26/DPP4—A potential biomarker and target for cancer therapy. Pharmacol. Ther. 2019, 198, 135–159. [Google Scholar] [CrossRef]
- Beckenkamp, A.; Davies, S.; Willig, J.B.; Buffon, A. DPPIV/CD26: A tumor suppressor or a marker of malignancy? Tumour Biol. 2016, 37, 7059–7073. [Google Scholar] [CrossRef] [PubMed]
- Wesley, U.V.; Tiwari, S.; Houghton, A.N. Role for dipeptidyl peptidase IV in tumor suppression of human non small cell lung carcinoma cells. Int. J. Cancer 2004, 109, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Arscott, W.T.; LaBauve, A.E.; May, V.; Wesley, U.V. Suppression of neuroblastoma growth by dipeptidyl peptidase IV: Relevance of chemokine regulation and caspase activation. Oncogene 2009, 28, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Wesley, U.V.; Albino, A.P.; Tiwari, S.; Houghton, A.N. A role for dipeptidyl peptidase IV in suppressing the malignant phenotype of melanocytic cells. J. Exp. Med. 1999, 190, 311–322. [Google Scholar] [CrossRef]
- Narducci, M.G.; Scala, E.; Bresin, A.; Caprini, E.; Picchio, M.C.; Remotti, D.; Ragone, G.; Nasorri, F.; Frontani, M.; Arcelli, D.; et al. Skin homing of Sezary cells involves SDF-1-CXCR4 signaling and down-regulation of CD26/dipeptidylpeptidase IV. Blood 2006, 107, 1108–1115. [Google Scholar] [CrossRef]
- Angevin, E.; Isambert, N.; Trillet-Lenoir, V.; You, B.; Alexandre, J.; Zalcman, G.; Vielh, P.; Farace, F.; Valleix, F.; Podoll, T.; et al. First-in-human phase 1 of YS110, a monoclonal antibody directed against CD26 in advanced CD26-expressing cancers. Br. J. Cancer 2017, 116, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Wang, T.Y.; Liu, C.L.; Chien, M.N.; Chen, M.J.; Hsu, Y.C.; Leung, C.H.; Cheng, S.P. Dipeptidyl Peptidase IV as a Prognostic Marker and Therapeutic Target in Papillary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2017, 102, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.X.; Yang, S.W.; Li, P.A.; Luo, X.; Li, Z.Y.; Hao, Y.X.; Yu, P.W. The promotion of the transformation of quiescent gastric cancer stem cells by IL-17 and the underlying mechanisms. Oncogene 2017, 36, 1256–1264. [Google Scholar] [CrossRef]
- Larrinaga, G.; Perez, I.; Sanz, B.; Beitia, M.; Errarte, P.; Fernandez, A.; Blanco, L.; Etxezarraga, M.C.; Gil, J.; Lopez, J.I. Dipeptidyl-peptidase IV activity is correlated with colorectal cancer prognosis. PLoS ONE 2015, 10, e0119436. [Google Scholar] [CrossRef]
- Pang, R.; Law, W.L.; Chu, A.C.; Poon, J.T.; Lam, C.S.; Chow, A.K.; Ng, L.; Cheung, L.W.; Lan, X.R.; Lan, H.Y.; et al. A subpopulation of CD26+ cancer stem cells with metastatic capacity in human colorectal cancer. Cell Stem Cell 2010, 6, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; Baerts, L.; Waumans, Y.; De Meester, I.; Yamada, Y.; Limani, P.; Gil-Bazo, I.; Weder, W.; Jungraithmayr, W. Suppression of lung metastases by the CD26/DPP4 inhibitor Vildagliptin in mice. Clin. Exp. Metastasis 2015, 32, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Goscinski, M.A.; Suo, Z.H.; Nesland, J.M.; Florenes, V.A.; Giercksky, K.E. Dipeptidyl peptidase IV expression in cancer and stromal cells of human esophageal squamous cell carcinomas, adenocarcinomas and squamous cell carcinoma cell lines. APMIS 2008, 116, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, P.; Wormser, L.; Fritz, V.; Seitz, T.; De Maria, M.; Schambony, A.; Kremer, A.E.; Gunther, C.; Itzel, T.; Thasler, W.E.; et al. Molecular crosstalk between Y5 receptor and neuropeptide Y drives liver cancer. J. Clin. Investig. 2020, 130, 2509–2526. [Google Scholar] [CrossRef] [PubMed]
- Hollande, C.; Boussier, J.; Ziai, J.; Nozawa, T.; Bondet, V.; Phung, W.; Lu, B.; Duffy, D.; Paradis, V.; Mallet, V.; et al. Inhibition of the dipeptidyl peptidase DPP4 (CD26) reveals IL-33-dependent eosinophil-mediated control of tumor growth. Nat. Immunol. 2019, 20, 257–264. [Google Scholar] [CrossRef]
- Nishina, S.; Yamauchi, A.; Kawaguchi, T.; Kaku, K.; Goto, M.; Sasaki, K.; Hara, Y.; Tomiyama, Y.; Kuribayashi, F.; Torimura, T.; et al. Dipeptidyl Peptidase 4 Inhibitors Reduce Hepatocellular Carcinoma by Activating Lymphocyte Chemotaxis in Mice. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 115–134. [Google Scholar] [CrossRef]
- Henderson, J.M.; Xiang, M.S.W.; Huang, J.C.; Wetzel, S.; Jiang, L.; Lai, J.H.; Wu, W.; Kench, J.G.; Bachovchin, W.W.; Roediger, B.; et al. Dipeptidyl Peptidase Inhibition Enhances CD8 T Cell Recruitment and Activates Intrahepatic Inflammasome in a Murine Model of Hepatocellular Carcinoma. Cancers 2021, 13, 5495. [Google Scholar] [CrossRef]
- Wilson, M.J.; Ruhland, A.R.; Quast, B.J.; Reddy, P.K.; Ewing, S.L.; Sinha, A.A. Dipeptidylpeptidase IV activities are elevated in prostate cancers and adjacent benign hyperplastic glands. J. Androl. 2000, 21, 220–226. [Google Scholar]
- Lu, Z.; Qi, L.; Bo, X.J.; Liu, G.D.; Wang, J.M.; Li, G. Expression of CD26 and CXCR4 in prostate carcinoma and its relationship with clinical parameters. J. Res. Med. Sci. 2013, 18, 647–652. [Google Scholar]
- Wesley, U.V.; McGroarty, M.; Homoyouni, A. Dipeptidyl peptidase inhibits malignant phenotype of prostate cancer cells by blocking basic fibroblast growth factor signaling pathway. Cancer Res. 2005, 65, 1325–1334. [Google Scholar] [CrossRef]
- Sun, Y.X.; Pedersen, E.A.; Shiozawa, Y.; Havens, A.M.; Jung, Y.; Wang, J.; Pienta, K.J.; Taichman, R.S. CD26/dipeptidyl peptidase IV regulates prostate cancer metastasis by degrading SDF-1/CXCL12. Clin. Exp. Metastasis 2008, 25, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.W.; Gao, C.; Bhasin, S.S.; Voznesensky, O.S.; Calagua, C.; Arai, S.; Nelson, P.S.; Montgomery, B.; Mostaghel, E.A.; Corey, E.; et al. Downregulation of Dipeptidyl Peptidase 4 Accelerates Progression to Castration-Resistant Prostate Cancer. Cancer Res. 2018, 78, 6354–6362. [Google Scholar] [CrossRef] [PubMed]
- Stremenova, J.; Krepela, E.; Mares, V.; Trim, J.; Dbaly, V.; Marek, J.; Vanickova, Z.; Lisa, V.; Yea, C.; Sedo, A. Expression and enzymatic activity of dipeptidyl peptidase-IV in human astrocytic tumours are associated with tumour grade. Int. J. Oncol. 2007, 31, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Busek, P.; Stremenova, J.; Krepela, E.; Sedo, A. Modulation of substance P signaling by dipeptidyl peptidase-IV enzymatic activity in human glioma cell lines. Physiol. Res. 2008, 57, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Busek, P.; Stremenova, J.; Sromova, L.; Hilser, M.; Balaziova, E.; Kosek, D.; Trylcova, J.; Strnad, H.; Krepela, E.; Sedo, A. Dipeptidyl peptidase-IV inhibits glioma cell growth independent of its enzymatic activity. Int. J. Biochem. Cell Biol. 2012, 44, 738–747. [Google Scholar] [CrossRef]
- Choi, H.J.; Kim, J.Y.; Lim, S.C.; Kim, G.; Yun, H.J.; Choi, H.S. Dipeptidyl peptidase 4 promotes epithelial cell transformation and breast tumourigenesis via induction of PIN1 gene expression. Br. J. Pharmacol. 2015, 172, 5096–5109. [Google Scholar] [CrossRef]
- Leccia, F.; Nardone, A.; Corvigno, S.; Vecchio, L.D.; De Placido, S.; Salvatore, F.; Veneziani, B.M. Cytometric and biochemical characterization of human breast cancer cells reveals heterogeneous myoepithelial phenotypes. Cytom. A 2012, 81, 960–972. [Google Scholar] [CrossRef]
- Yang, F.; Takagaki, Y.; Yoshitomi, Y.; Ikeda, T.; Li, J.; Kitada, M.; Kumagai, A.; Kawakita, E.; Shi, S.; Kanasaki, K.; et al. Inhibition of Dipeptidyl Peptidase-4 Accelerates Epithelial-Mesenchymal Transition and Breast Cancer Metastasis via the CXCL12/CXCR4/mTOR Axis. Cancer Res. 2019, 79, 735–746. [Google Scholar] [CrossRef]
- Li, S.; Fan, Y.; Kumagai, A.; Kawakita, E.; Kitada, M.; Kanasaki, K.; Koya, D. Deficiency in Dipeptidyl Peptidase-4 Promotes Chemoresistance through the CXCL12/CXCR4/mTOR/TGFbeta Signaling Pathway in Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 805. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, X.; Wu, R.; Huang, Q.; Jiang, Y.; Qin, J.; Yao, F.; Jin, G.; Zhang, Y. DPPIV promotes endometrial carcinoma cell proliferation, invasion and tumorigenesis. Oncotarget 2017, 8, 8679–8692. [Google Scholar] [CrossRef]
- Khin, E.E.; Kikkawa, F.; Ino, K.; Kajiyama, H.; Suzuki, T.; Shibata, K.; Tamakoshi, K.; Nagasaka, T.; Mizutani, S. Dipeptidyl peptidase IV expression in endometrial endometrioid adenocarcinoma and its inverse correlation with tumor grade. Am. J. Obstet. Gynecol. 2003, 188, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Kikkawa, F.; Kajiyama, H.; Shibata, K.; Ino, K.; Nomura, S.; Mizutani, S. Dipeptidyl peptidase IV in tumor progression. Biochim. Biophys. Acta 2005, 1751, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Kajiyama, H.; Kikkawa, F.; Khin, E.; Shibata, K.; Ino, K.; Mizutani, S. Dipeptidyl peptidase IV overexpression induces up-regulation of E-cadherin and tissue inhibitors of matrix metalloproteinases, resulting in decreased invasive potential in ovarian carcinoma cells. Cancer Res. 2003, 63, 2278–2283. [Google Scholar] [PubMed]
- Kajiyama, H.; Shibata, K.; Ino, K.; Mizutani, S.; Nawa, A.; Kikkawa, F. The expression of dipeptidyl peptidase IV (DPPIV/CD26) is associated with enhanced chemosensitivity to paclitaxel in epithelial ovarian carcinoma cells. Cancer Sci. 2010, 101, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xu, L.; Wang, X.; Sun, B.; Ding, J. Expression levels of seprase/FAPalpha and DPPIV/CD26 in epithelial ovarian carcinoma. Oncol. Lett. 2015, 10, 34–42. [Google Scholar] [CrossRef]
- Busek, P.; Vanickova, Z.; Hrabal, P.; Brabec, M.; Fric, P.; Zavoral, M.; Skrha, J.; Kmochova, K.; Laclav, M.; Bunganic, B.; et al. Increased tissue and circulating levels of dipeptidyl peptidase-IV enzymatic activity in patients with pancreatic ductal adenocarcinoma. Pancreatology 2016, 16, 829–838. [Google Scholar] [CrossRef]
- Matveyenko, A.V.; Dry, S.; Cox, H.I.; Moshtaghian, A.; Gurlo, T.; Galasso, R.; Butler, A.E.; Butler, P.C. Beneficial endocrine but adverse exocrine effects of sitagliptin in the human islet amyloid polypeptide transgenic rat model of type 2 diabetes: Interactions with metformin. Diabetes 2009, 58, 1604–1615. [Google Scholar] [CrossRef]
- Butler, A.E.; Campbell-Thompson, M.; Gurlo, T.; Dawson, D.W.; Atkinson, M.; Butler, P.C. Marked expansion of exocrine and endocrine pancreas with incretin therapy in humans with increased exocrine pancreas dysplasia and the potential for glucagon-producing neuroendocrine tumors. Diabetes 2013, 62, 2595–2604. [Google Scholar] [CrossRef]
- Ueberberg, S.; Jutte, H.; Uhl, W.; Schmidt, W.; Nauck, M.; Montanya, E.; Tannapfel, A.; Meier, J. Histological changes in endocrine and exocrine pancreatic tissue from patients exposed to incretin-based therapies. Diabetes Obes. Metab. 2016, 18, 1253–1262. [Google Scholar] [CrossRef]
- Aston-Mourney, K.; Subramanian, S.L.; Zraika, S.; Samarasekera, T.; Meier, D.T.; Goldstein, L.C.; Hull, R.L. One year of sitagliptin treatment protects against islet amyloid-associated beta-cell loss and does not induce pancreatitis or pancreatic neoplasia in mice. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E475–E484. [Google Scholar] [CrossRef]
- Cox, A.R.; Lam, C.J.; Rankin, M.M.; Rios, J.S.; Chavez, J.; Bonnyman, C.W.; King, K.B.; Wells, R.A.; Anthony, D.; Tu, J.X.; et al. Incretin Therapies Do Not Expand beta-Cell Mass or Alter Pancreatic Histology in Young Male Mice. Endocrinology 2017, 158, 1701–1714. [Google Scholar] [CrossRef] [PubMed]
- Busch, S.J.; Hoffmann, P.; Sahota, P.; Johnson, R.; Kothny, W.; Meyer, F.; Foley, J.E. Studies in rodents with the dipeptidyl peptidase-4 inhibitor vildagliptin to evaluate possible drug-induced pancreatic histological changes that are predictive of pancreatitis and cancer development in man. Diabetes Obes. Metab. 2013, 15, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Pinto, L.C.; Rados, D.V.; Barkan, S.S.; Leitao, C.B.; Gross, J.L. Dipeptidyl peptidase-4 inhibitors, pancreatic cancer and acute pancreatitis: A meta-analysis with trial sequential analysis. Sci. Rep. 2018, 8, 782. [Google Scholar] [CrossRef] [PubMed]
- Abd El Aziz, M.; Cahyadi, O.; Meier, J.J.; Schmidt, W.E.; Nauck, M.A. Incretin-based glucose-lowering medications and the risk of acute pancreatitis and malignancies: A meta-analysis based on cardiovascular outcomes trials. Diabetes Obes. Metab. 2020, 22, 699–704. [Google Scholar] [CrossRef]
- Dicembrini, I.; Montereggi, C.; Nreu, B.; Mannucci, E.; Monami, M. Pancreatitis and pancreatic cancer in patientes treated with Dipeptidyl Peptidase-4 inhibitors: An extensive and updated meta-analysis of randomized controlled trials. Diabetes Res. Clin. Pract. 2020, 159, 107981. [Google Scholar] [CrossRef] [PubMed]
- Dicembrini, I.; Nreu, B.; Montereggi, C.; Mannucci, E.; Monami, M. Risk of cancer in patients treated with dipeptidyl peptidase-4 inhibitors: An extensive meta-analysis of randomized controlled trials. Acta Diabetol. 2020, 57, 689–696. [Google Scholar] [CrossRef]
- Lee, D.Y.; Yu, J.H.; Park, S.; Han, K.; Kim, N.H.; Yoo, H.J.; Choi, K.M.; Baik, S.H.; Kim, N.H.; Seo, J.A. The influence of diabetes and antidiabetic medications on the risk of pancreatic cancer: A nationwide population-based study in Korea. Sci. Rep. 2018, 8, 9719. [Google Scholar] [CrossRef]
- Boniol, M.; Franchi, M.; Bota, M.; Leclercq, A.; Guillaume, J.; van Damme, N.; Corrao, G.; Autier, P.; Boyle, P. Incretin-Based Therapies and the Short-term Risk of Pancreatic Cancer: Results From Two Retrospective Cohort Studies. Diabetes Care 2018, 41, 286–292. [Google Scholar] [CrossRef]
- Lee, M.; Sun, J.; Han, M.; Cho, Y.; Lee, J.Y.; Nam, C.M.; Kang, E.S. Nationwide Trends in Pancreatitis and Pancreatic Cancer Risk Among Patients With Newly Diagnosed Type 2 Diabetes Receiving Dipeptidyl Peptidase 4 Inhibitors. Diabetes Care 2019, 42, 2057–2064. [Google Scholar] [CrossRef]
- Abrahami, D.; Douros, A.; Yin, H.; Yu, O.H.; Faillie, J.L.; Montastruc, F.; Platt, R.W.; Bouganim, N.; Azoulay, L. Incretin based drugs and risk of cholangiocarcinoma among patients with type 2 diabetes: Population based cohort study. BMJ 2018, 363, k4880. [Google Scholar] [CrossRef]
- Pech, V.; Abusaada, K.; Alemany, C. Dipeptidyl Peptidase-4 Inhibition May Stimulate Progression of Carcinoid Tumor. Case Rep. Endocrinol. 2015, 2015, 952019. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.R.; Rhee, S.D.; Kim, H.Y.; Jung, W.H.; Yang, S.D.; Kim, S.S.; Ahn, J.H.; Cheon, H.G. KR-62436, 6-{2-[2-(5-cyano-4,5-dihydropyrazol-1-yl)-2-oxoethylamino]ethylamino}nicotinonitr ile, is a novel dipeptidyl peptidase-IV (DPP-IV) inhibitor with anti-hyperglycemic activity. Eur. J. Pharmacol. 2005, 518, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Rathmann, W.; Kostev, K. Association of dipeptidyl peptidase 4 inhibitors with risk of metastases in patients with type 2 diabetes and breast, prostate or digestive system cancer. J. Diabetes Complicat. 2017, 31, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Noh, Y.; Jeon, S.M.; Shin, S. Association between glucose-lowering treatment and cancer metastasis among patients with preexisting type 2 diabetes and incident malignancy. Int. J. Cancer 2019, 144, 1530–1539. [Google Scholar] [CrossRef]
- Tseng, C.H. Sitagliptin May Reduce Breast Cancer Risk in Women With Type 2 Diabetes. Clin. Breast Cancer 2017, 17, 211–218. [Google Scholar] [CrossRef]
- Tseng, C.H. Sitagliptin may reduce prostate cancer risk in male patients with type 2 diabetes. Oncotarget 2017, 8, 19057–19064. [Google Scholar] [CrossRef]
- Hsu, W.H.; Sue, S.P.; Liang, H.L.; Tseng, C.W.; Lin, H.C.; Wen, W.L.; Lee, M.Y. Dipeptidyl Peptidase 4 Inhibitors Decrease the Risk of Hepatocellular Carcinoma in Patients With Chronic Hepatitis C Infection and Type 2 Diabetes Mellitus: A Nationwide Study in Taiwan. Front. Public Health 2021, 9, 711723. [Google Scholar] [CrossRef]
- Kamada, S.; Namekawa, T.; Ikeda, K.; Suzuki, T.; Kagawa, M.; Takeshita, H.; Yano, A.; Okamoto, K.; Ichikawa, T.; Horie-Inoue, K.; et al. Functional inhibition of cancer stemness-related protein DPP4 rescues tyrosine kinase inhibitor resistance in renal cell carcinoma. Oncogene 2021, 40, 3899–3913. [Google Scholar] [CrossRef]
- Stecca, B.A.; Nardo, B.; Chieco, P.; Mazziotti, A.; Bolondi, L.; Cavallari, A. Aberrant dipeptidyl peptidase IV (DPP IV/CD26) expression in human hepatocellular carcinoma. J. Hepatol. 1997, 27, 337–345. [Google Scholar] [CrossRef]
- Qin, C.J.; Zhao, L.H.; Zhou, X.; Zhang, H.L.; Wen, W.; Tang, L.; Zeng, M.; Wang, M.D.; Fu, G.B.; Huang, S.; et al. Inhibition of dipeptidyl peptidase IV prevents high fat diet-induced liver cancer angiogenesis by downregulating chemokine ligand 2. Cancer Lett. 2018, 420, 26–37. [Google Scholar] [CrossRef]
- Khalil, R.; Shata, A.; Abd El-Kader, E.M.; Sharaf, H.; Abdo, W.S.; Amin, N.A.; Saber, S. Vildagliptin, a DPP-4 inhibitor, attenuates carbon tetrachloride-induced liver fibrosis by targeting ERK1/2, p38alpha, and NF-kappaB signaling. Toxicol. Appl. Pharmacol. 2020, 407, 115246. [Google Scholar] [CrossRef] [PubMed]
- Abd Elmaaboud, M.; Khattab, H.; Shalaby, S. Hepatoprotective effect of linagliptin against liver fibrosis induced by carbon tetrachloride in mice. Can. J. Physiol. Pharmacol. 2021, 99, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Sokar, S.S.; El-Sayad, M.E.; Ghoneim, M.E.; Shebl, A.M. Combination of Sitagliptin and Silymarin ameliorates liver fibrosis induced by carbon tetrachloride in rats. Biomed. Pharmacother. 2017, 89, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Holz, L.E.; Chowdhury, S.; Cordoba, S.P.; Evans, K.A.; Gall, M.G.; de Ribeiro, A.J.V.; Zheng, Y.Z.; Levy, M.T.; Yu, D.M.; et al. The pro-fibrotic role of dipeptidyl peptidase 4 in carbon tetrachloride-induced experimental liver injury. Immunol. Cell Biol. 2017, 95, 443–453. [Google Scholar] [CrossRef]
- Abd Elhameed, A.G.; Helal, M.G.; Said, E.; Salem, H.A. Saxagliptin defers thioacetamide-induced hepatocarcinogenesis in rats: A novel suppressive impact on Wnt/Hedgehog/Notch1 signaling. Environ. Toxicol. Pharmacol. 2021, 86, 103668. [Google Scholar] [CrossRef]
- Baumeier, C.; Schluter, L.; Saussenthaler, S.; Laeger, T.; Rodiger, M.; Alaze, S.A.; Fritsche, L.; Haring, H.U.; Stefan, N.; Fritsche, A.; et al. Elevated hepatic DPP4 activity promotes insulin resistance and non-alcoholic fatty liver disease. Mol. Metab. 2017, 6, 1254–1263. [Google Scholar] [CrossRef]
- Kawakubo, M.; Tanaka, M.; Ochi, K.; Watanabe, A.; Saka-Tanaka, M.; Kanamori, Y.; Yoshioka, N.; Yamashita, S.; Goto, M.; Itoh, M.; et al. Dipeptidyl peptidase-4 inhibition prevents nonalcoholic steatohepatitis-associated liver fibrosis and tumor development in mice independently of its anti-diabetic effects. Sci. Rep. 2020, 10, 983. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Nakano, D.; Koga, H.; Torimura, T. Effects of a DPP4 Inhibitor on Progression of NASH-related HCC and the p62/Keap1/Nrf2-Pentose Phosphate Pathway in a Mouse Model. Liver Cancer 2019, 8, 359–372. [Google Scholar] [CrossRef]
- Ozutsumi, T.; Namisaki, T.; Shimozato, N.; Kaji, K.; Tsuji, Y.; Kaya, D.; Fujinaga, Y.; Furukawa, M.; Nakanishi, K.; Sato, S.; et al. Combined Treatment with Sodium-Glucose Cotransporter-2 Inhibitor (Canagliflozin) and Dipeptidyl Peptidase-4 Inhibitor (Teneligliptin) Alleviates NASH Progression in A Non-Diabetic Rat Model of Steatohepatitis. Int. J. Mol. Sci. 2020, 21, 2164. [Google Scholar] [CrossRef]
- Okura, Y.; Namisaki, T.; Moriya, K.; Kitade, M.; Takeda, K.; Kaji, K.; Noguchi, R.; Nishimura, N.; Seki, K.; Kawaratani, H.; et al. Combined treatment with dipeptidyl peptidase-4 inhibitor (sitagliptin) and angiotensin-II type 1 receptor blocker (losartan) suppresses progression in a non-diabetic rat model of steatohepatitis. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2017, 47, 1317–1328. [Google Scholar] [CrossRef]
- Billeschou, A.; Hunt, J.E.; Ghimire, A.; Holst, J.J.; Kissow, H. Intestinal Adaptation upon Chemotherapy-Induced Intestinal Injury in Mice Depends on GLP-2 Receptor Activation. Biomedicines 2021, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Masur, K.; Schwartz, F.; Entschladen, F.; Niggemann, B.; Zaenker, K.S. DPPIV inhibitors extend GLP-2 mediated tumour promoting effects on intestinal cancer cells. Regul. Pept. 2006, 137, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Abrahami, D.; Yin, H.; Yu, O.H.Y.; Pollak, M.N.; Azoulay, L. Incretin-based Drugs and the Incidence of Colorectal Cancer in Patients with Type 2 Diabetes. Epidemiology 2018, 29, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.; Foo, D.C.; Wong, C.K.; Man, A.T.; Lo, O.S.; Law, W.L. Repurposing DPP-4 Inhibitors for Colorectal Cancer: A Retrospective and Single Center Study. Cancers 2021, 13, 3588. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Fuentes, A.; Skelton, W.I.; Wang, Y.; McGorray, S.; Shah, C.; Bishnoi, R.; Dang, L.H.; Dang, N.H. A multi-center retrospective analysis of the effect of DPP4 inhibitors on progression-free survival in advanced airway and colorectal cancers. Mol. Clin. Oncol. 2019, 10, 118–124. [Google Scholar] [CrossRef]
- Bishnoi, R.; Hong, Y.R.; Shah, C.; Ali, A.; Skelton, W.P.t.; Huo, J.; Dang, N.H.; Dang, L.H. Dipeptidyl peptidase 4 inhibitors as novel agents in improving survival in diabetic patients with colorectal cancer and lung cancer: A Surveillance Epidemiology and Endpoint Research Medicare study. Cancer Med. 2019, 8, 3918–3927. [Google Scholar] [CrossRef]
- Femia, A.P.; Raimondi, L.; Maglieri, G.; Lodovici, M.; Mannucci, E.; Caderni, G. Long-term treatment with Sitagliptin, a dipeptidyl peptidase-4 inhibitor, reduces colon carcinogenesis and reactive oxygen species in 1,2-dimethylhydrazine-induced rats. Int. J. Cancer 2013, 133, 2498–2503. [Google Scholar] [CrossRef]
- Yorifuji, N.; Inoue, T.; Iguchi, M.; Fujiwara, K.; Kakimoto, K.; Nouda, S.; Okada, T.; Kawakami, K.; Abe, Y.; Takeuchi, T.; et al. The dipeptidyl peptidase-4 inhibitor sitagliptin suppresses mouse colon tumorigenesis in type 2 diabetic mice. Oncol. Rep. 2016, 35, 676–682. [Google Scholar] [CrossRef]
- Fujiwara, K.; Inoue, T.; Henmi, Y.; Hirata, Y.; Naka, Y.; Hara, A.; Kakimoto, K.; Nouda, S.; Okada, T.; Kawakami, K.; et al. Sitagliptin, a dipeptidyl peptidase-4 inhibitor, suppresses CXCL5 and SDF-1 and does not accelerate intestinal neoplasia formation in Apc(Min/+) mice fed a high-fat diet. Oncol. Lett. 2017, 14, 4355–4360. [Google Scholar] [CrossRef]
- Kabel, A.M.; Atef, A.; Estfanous, R.S. Ameliorative potential of sitagliptin and/or resveratrol on experimentally-induced clear cell renal cell carcinoma. Biomed. Pharmacother. 2018, 97, 667–674. [Google Scholar] [CrossRef]
- Arwert, E.N.; Mentink, R.A.; Driskell, R.R.; Hoste, E.; Goldie, S.J.; Quist, S.; Watt, F.M. Upregulation of CD26 expression in epithelial cells and stromal cells during wound-induced skin tumour formation. Oncogene 2012, 31, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Chen, J.; Yuan, Y.; Zou, Z.; Lai, X.; Rahmani, D.M.; Wang, F.; Xi, Y.; Huang, Q.; Bu, S. Dipeptidyl peptidase-4 inhibitors and cancer risk in patients with type 2 diabetes: A meta-analysis of randomized clinical trials. Sci. Rep. 2017, 7, 8273. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, J.; Terauchi, Y. Potential linkage between dipeptidyl peptidase-4 inhibitor use and the risk of pancreatitis/pancreatic cancer. J. Diabetes Investig. 2020, 11, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, J.A.; Bakker, M.; van der Heijden, A.; van Herk-Sukel, M.P.P.; Herings, R.M.C.; Nijpels, G. Risk of dipeptidyl peptidase-4 (DPP-4) inhibitors on site-specific cancer: A systematic review and meta-analysis. Diabetes Metab. Res. Rev. 2018, 34, e3004. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Kim, D.J.; Shin, S. Incident cancer risk in dipeptidyl peptidase-4 inhibitor-treated patients with type 2 diabetes mellitus. Cancer Manag. Res. 2019, 11, 7427–7438. [Google Scholar] [CrossRef]
- Kim, S.H.; Kang, J.G.; Kim, C.S.; Ihm, S.H.; Choi, M.G.; Yoo, H.J.; Lee, S.J. Synergistic cytotoxicity of the dipeptidyl peptidase-IV inhibitor gemigliptin with metformin in thyroid carcinoma cells. Endocrine 2018, 59, 383–394. [Google Scholar] [CrossRef]
- Kim, S.H.; Kang, J.G.; Kim, C.S.; Ihm, S.H.; Choi, M.G.; Yoo, H.J.; Lee, S.J. Gemigliptin, a novel dipeptidyl peptidase-IV inhibitor, exerts a synergistic cytotoxicity with the histone deacetylase inhibitor PXD101 in thyroid carcinoma cells. J. Endocrinol. Investig. 2018, 41, 677–689. [Google Scholar] [CrossRef]
- Kim, S.H.; Kang, J.G.; Kim, C.S.; Ihm, S.H.; Choi, M.G.; Yoo, H.J.; Lee, S.J. The dipeptidyl peptidase-IV inhibitor gemigliptin alone or in combination with NVP-AUY922 has a cytotoxic activity in thyroid carcinoma cells. Tumour Biol. 2017, 39, 1010428317722068. [Google Scholar] [CrossRef]
- Wang, Q.; Lu, P.; Wang, T.; Zheng, Q.; Li, Y.; Leng, S.X.; Meng, X.; Wang, B.; Xie, J.; Zhang, H. Sitagliptin affects gastric cancer cells proliferation by suppressing Melanoma-associated antigen-A3 expression through Yes-associated protein inactivation. Cancer Med. 2020, 9, 3816–3828. [Google Scholar] [CrossRef]
- Varela-Calvino, R.; Rodriguez-Quiroga, M.; Carvalho, P.D.; Martins, F.; Serra-Roma, A.; Vazquez-Iglesias, L.; de la Cadena, M.P.; Velho, S.; Cordero, O.J. The mechanism of sitagliptin inhibition of colorectal cancer cell lines’ metastatic functionalities. IUBMB Life 2021, 73, 761–773. [Google Scholar] [CrossRef]
- Sliwinska, A.; Rogalska, A.; Marczak, A.; Kasznicki, J.; Drzewoski, J. Metformin, but not sitagliptin, enhances WP 631-induced apoptotic HepG2 cell death. Toxicol. Vitr. 2015, 29, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Beckenkamp, A.; Willig, J.B.; Santana, D.B.; Nascimento, J.; Paccez, J.D.; Zerbini, L.F.; Bruno, A.N.; Pilger, D.A.; Wink, M.R.; Buffon, A. Differential Expression and Enzymatic Activity of DPPIV/CD26 Affects Migration Ability of Cervical Carcinoma Cells. PLoS ONE 2015, 10, e0134305. [Google Scholar] [CrossRef] [PubMed]
- Spagnuolo, P.A.; Hurren, R.; Gronda, M.; MacLean, N.; Datti, A.; Basheer, A.; Lin, F.H.; Wang, X.; Wrana, J.; Schimmer, A.D. Inhibition of intracellular dipeptidyl peptidases 8 and 9 enhances parthenolide’s anti-leukemic activity. Leukemia 2013, 27, 1236–1244. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Tatekoshi, A.; Takada, K.; Iyama, S.; Kamihara, Y.; Jawaid, P.; Rehman, M.U.; Noguchi, K.; Kondo, T.; Kajikawa, S.; et al. DPP8 is a novel therapeutic target for multiple myeloma. Sci. Rep. 2019, 9, 18094. [Google Scholar] [CrossRef]
- Herrmann, H.; Sadovnik, I.; Cerny-Reiterer, S.; Rulicke, T.; Stefanzl, G.; Willmann, M.; Hoermann, G.; Bilban, M.; Blatt, K.; Herndlhofer, S.; et al. Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood 2014, 123, 3951–3962. [Google Scholar] [CrossRef]
- Willmann, M.; Sadovnik, I.; Eisenwort, G.; Entner, M.; Bernthaler, T.; Stefanzl, G.; Hadzijusufovic, E.; Berger, D.; Herrmann, H.; Hoermann, G.; et al. Evaluation of cooperative antileukemic effects of nilotinib and vildagliptin in Ph(+) chronic myeloid leukemia. Exp. Hematol. 2018, 57, 50–59.e56. [Google Scholar] [CrossRef]
- Bergman, A.J.; Stevens, C.; Zhou, Y.; Yi, B.; Laethem, M.; De Smet, M.; Snyder, K.; Hilliard, D.; Tanaka, W.; Zeng, W.; et al. Pharmacokinetic and pharmacodynamic properties of multiple oral doses of sitagliptin, a dipeptidyl peptidase-IV inhibitor: A double-blind, randomized, placebo-controlled study in healthy male volunteers. Clin. Ther. 2006, 28, 55–72. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Kodama, T.; Hikita, H.; Makino, Y.; Saito, Y.; Tanaka, S.; Shimizu, S.; Sakamori, R.; Miyagi, T.; Wada, H.; et al. Synthetic lethal interaction of combined CD26 and Bcl-xL inhibition is a powerful anticancer therapy against hepatocellular carcinoma. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2015, 45, 1023–1033. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Watanabe, A.; Tanaka, M.; Shiota, M.; Osada-Oka, M.; Sano, S.; Yoshiyama, M.; Miura, K.; Kitajima, S.; Matsunaga, S.; et al. A dipeptidyl peptidase-4 (DPP-4) inhibitor, linagliptin, attenuates cardiac dysfunction after myocardial infarction independently of DPP-4. J. Pharmacol. Sci. 2019, 139, 112–119. [Google Scholar] [CrossRef]
- Batchu, S.N.; Yerra, V.G.; Liu, Y.; Advani, S.L.; Klein, T.; Advani, A. The Dipeptidyl Peptidase-4 Inhibitor Linagliptin Directly Enhances the Contractile Recovery of Mouse Hearts at a Concentration Equivalent to that Achieved with Standard Dosing in Humans. Int. J. Mol. Sci. 2020, 21, 5756. [Google Scholar] [CrossRef]
- Heo, R.; Seo, M.S.; An, J.R.; Kang, M.; Park, H.; Han, E.T.; Han, J.H.; Chun, W.; Park, W.S. The anti-diabetic drug trelagliptin induces vasodilation via activation of Kv channels and SERCA pumps. Life Sci. 2021, 283, 119868. [Google Scholar] [CrossRef]
- Bailey, S.R.; Nelson, M.H.; Majchrzak, K.; Bowers, J.S.; Wyatt, M.M.; Smith, A.S.; Neal, L.R.; Shirai, K.; Carpenito, C.; June, C.H.; et al. Human CD26(high) T cells elicit tumor immunity against multiple malignancies via enhanced migration and persistence. Nat. Commun. 2017, 8, 1961. [Google Scholar] [CrossRef]
- Nelson, M.H.; Knochelmann, H.M.; Bailey, S.R.; Huff, L.W.; Bowers, J.S.; Majchrzak-Kuligowska, K.; Wyatt, M.M.; Rubinstein, M.P.; Mehrotra, S.; Nishimura, M.I.; et al. Identification of human CD4(+) T cell populations with distinct antitumor activity. Sci. Adv. 2020, 6, eaba7443. [Google Scholar] [CrossRef] [PubMed]
- Casrouge, A.; Decalf, J.; Ahloulay, M.; Lababidi, C.; Mansour, H.; Vallet-Pichard, A.; Mallet, V.; Mottez, E.; Mapes, J.; Fontanet, A.; et al. Evidence for an antagonist form of the chemokine CXCL10 in patients chronically infected with HCV. J. Clin. Investig. 2011, 121, 308–317. [Google Scholar] [CrossRef]
- da Silva, R.B.; Laird, M.E.; Yatim, N.; Fiette, L.; Ingersoll, M.A.; Albert, M.L. Dipeptidylpeptidase 4 inhibition enhances lymphocyte trafficking, improving both naturally occurring tumor immunity and immunotherapy. Nat. Immunol. 2015, 16, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.J.; Lee, Q.; Wong, A.C.; Spann, B.C.; Vincent, J.N.; Wong, J.J.; Schlitzer, A.; Gorrell, M.D.; Weninger, W.; Roediger, B. Differential chemokine receptor expression and usage by pre-cDC1 and pre-cDC2. Immunol. Cell Biol. 2018, 96, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.L.; Moffitt, L.R.; Wilson, K.L.; Bilandzic, M.; Wright, M.D.; Gorrell, M.D.; Oehler, M.K.; Plebanski, M.; Stephens, A.N. DPP4 Inhibitor Sitagliptin Enhances Lymphocyte Recruitment and Prolongs Survival in a Syngeneic Ovarian Cancer Mouse Model. Cancers 2021, 13, 487. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; Janker, F.; De Meester, I.; Arni, S.; Borgeaud, N.; Yamada, Y.; Gil Bazo, I.; Weder, W.; Jungraithmayr, W. The CD26/DPP4-inhibitor vildagliptin suppresses lung cancer growth via macrophage-mediated NK cell activity. Carcinogenesis 2019, 40, 324–334. [Google Scholar] [CrossRef]
- Farag, S.S.; Srivastava, S.; Messina-Graham, S.; Schwartz, J.; Robertson, M.J.; Abonour, R.; Cornetta, K.; Wood, L.; Secrest, A.; Strother, R.M.; et al. In vivo DPP-4 inhibition to enhance engraftment of single-unit cord blood transplants in adults with hematological malignancies. Stem Cells Dev. 2013, 22, 1007–1015. [Google Scholar] [CrossRef]
- Farag, S.S.; Nelson, R.; Cairo, M.S.; O’Leary, H.A.; Zhang, S.; Huntley, C.; Delgado, D.; Schwartz, J.; Zaid, M.A.; Abonour, R.; et al. High-dose sitagliptin for systemic inhibition of dipeptidylpeptidase-4 to enhance engraftment of single cord umbilical cord blood transplantation. Oncotarget 2017, 8, 110350–110357. [Google Scholar] [CrossRef]
- Farag, S.S.; Abu Zaid, M.; Schwartz, J.E.; Thakrar, T.C.; Blakley, A.J.; Abonour, R.; Robertson, M.J.; Broxmeyer, H.E.; Zhang, S. Dipeptidyl Peptidase 4 Inhibition for Prophylaxis of Acute Graft-versus-Host Disease. N. Engl. J. Med. 2021, 384, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Broxmeyer, H.E.; Hoggatt, J.; O’Leary, H.A.; Mantel, C.; Chitteti, B.R.; Cooper, S.; Messina-Graham, S.; Hangoc, G.; Farag, S.; Rohrabaugh, S.L.; et al. Dipeptidylpeptidase 4 negatively regulates colony-stimulating factor activity and stress hematopoiesis. Nat. Med. 2012, 18, 1786–1796. [Google Scholar] [CrossRef] [PubMed]
- Iwakura, T.; Zhao, Z.; Marschner, J.A.; Devarapu, S.K.; Yasuda, H.; Anders, H.J. Dipeptidyl peptidase-4 inhibitor teneligliptin accelerates recovery from cisplatin-induced acute kidney injury by attenuating inflammation and promoting tubular regeneration. Nephrol. Dial. Transplant. 2019, 34, 1669–1680. [Google Scholar] [CrossRef] [PubMed]
- Salama, R.M.; Nasr, M.M.; Abdelhakeem, J.I.; Roshdy, O.K.; ElGamal, M.A. Alogliptin attenuates cyclophosphamide-induced nephrotoxicity: A novel therapeutic approach through modulating MAP3K/JNK/SMAD3 signaling cascade. Drug Chem. Toxicol. 2020, 45, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- Jo, C.H.; Kim, S.; Park, J.S.; Kim, G.H. Anti-Inflammatory Action of Sitagliptin and Linagliptin in Doxorubicin Nephropathy. Kidney Blood Press. Res. 2018, 43, 987–999. [Google Scholar] [CrossRef]
- Mostafa, R.E.; Morsi, A.H.; Asaad, G.F. Anti-inflammatory effects of saxagliptin and vildagliptin against doxorubicin-induced nephrotoxicity in rats: Attenuation of NLRP3 inflammasome up-regulation and tubulo-interstitial injury. Res. Pharm. Sci. 2021, 16, 547–558. [Google Scholar] [CrossRef]
- Iwakura, T.; Fukasawa, H.; Kitamura, A.; Ishibuchi, K.; Yasuda, H.; Furuya, R. Effect of dipeptidyl peptidase-4 inhibitors on cisplatin-induced acute nephrotoxicity in cancer patients with diabetes mellitus: A retrospective study. PLoS ONE 2020, 15, e0229377. [Google Scholar] [CrossRef]
- El-Agamy, D.S.; Abo-Haded, H.M.; Elkablawy, M.A. Cardioprotective effects of sitagliptin against doxorubicin-induced cardiotoxicity in rats. Exp. Biol. Med. 2016, 241, 1577–1587. [Google Scholar] [CrossRef]
- Aykan, D.A.; Yaman, S.; Eser, N.; Ozcan Metin, T.; Seyithanoglu, M.; Aykan, A.C.; Kurt, A.H.; Ergun, Y. Bisoprolol and linagliptin ameliorated electrical and mechanical isometric myocardial contractions in doxorubicin-induced cardiomyopathy in rats. Pharmacol. Rep. 2020, 72, 867–876. [Google Scholar] [CrossRef]
- Shigematsu, N.; Kawashiri, T.; Kobayashi, D.; Shimizu, S.; Mine, K.; Hiromoto, S.; Uchida, M.; Egashira, N.; Shimazoe, T. Neuroprotective effect of alogliptin on oxaliplatin-induced peripheral neuropathy in vivo and in vitro. Sci. Rep. 2020, 10, 6734. [Google Scholar] [CrossRef]
- Abo-Haded, H.M.; Elkablawy, M.A.; Al-Johani, Z.; Al-Ahmadi, O.; El-Agamy, D.S. Hepatoprotective effect of sitagliptin against methotrexate induced liver toxicity. PLoS ONE 2017, 12, e0174295. [Google Scholar] [CrossRef]
- Elrashidy, R.A.; Hasan, R.A. Stromal cell-derived factor-1alpha predominantly mediates the ameliorative effect of linagliptin against cisplatin-induced testicular injury in adult male rats. Cytokine 2020, 136, 155260. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Yoo, I.K.; Lee, J.M.; Kim, S.H.; Choi, H.S.; Kim, E.S.; Keum, B.; Seo, Y.S.; Jeen, Y.T.; Chun, H.J.; et al. Dipeptidyl-peptidase-4 (DPP-4) inhibitor ameliorates 5-flurouracil induced intestinal mucositis. BMC Cancer 2019, 19, 1016. [Google Scholar] [CrossRef]
- Proost, P.; Schutyser, E.; Menten, P.; Struyf, S.; Wuyts, A.; Opdenakker, G.; Detheux, M.; Parmentier, M.; Durinx, C.; Lambeir, A.M.; et al. Amino-terminal truncation of CXCR3 agonists impairs receptor signaling and lymphocyte chemotaxis, while preserving antiangiogenic properties. Blood 2001, 98, 3554–3561. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.E.; Abe, K.; Zukowska, Z. Stress, NPY and vascular remodeling: Implications for stress-related diseases. Peptides 2007, 28, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Tokuhara, T.; Nishikawa, M.; Nishizawa, S.; Nishioka, T.; Nozawa, A.; Takahashi, A.; Watanabe, Y.; Wada, R.; Wakasa, K.; et al. Spontaneous regression of hepatocellular carcinoma after improving diabetes mellitus: Possibly responsible for immune system. Kanzo 2012, 53, 164–174. [Google Scholar] [CrossRef]
- Duarte, R.F.; Labopin, M.; Bader, P.; Basak, G.W.; Bonini, C.; Chabannon, C.; Corbacioglu, S.; Dreger, P.; Dufour, C.; Gennery, A.R.; et al. Indications for haematopoietic stem cell transplantation for haematological diseases, solid tumours and immune disorders: Current practice in Europe, 2019. Bone Marrow Transplant. 2019, 54, 1525–1552. [Google Scholar] [CrossRef]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef]
- Peled, A.; Petit, I.; Kollet, O.; Magid, M.; Ponomaryov, T.; Byk, T.; Nagler, A.; Ben-Hur, H.; Many, A.; Shultz, L.; et al. Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science 1999, 283, 845–848. [Google Scholar] [CrossRef]
- Schwaiger, E.; Klaus, C.; Matheeussen, V.; Baranyi, U.; Pilat, N.; Ramsey, H.; Korom, S.; De Meester, I.; Wekerle, T. Dipeptidyl peptidase IV (DPPIV/CD26) inhibition does not improve engraftment of unfractionated syngeneic or allogeneic bone marrow after nonmyeloablative conditioning. Exp. Hematol. 2012, 40, 97–106. [Google Scholar] [CrossRef][Green Version]
- Kawai, T.; Choi, U.; Liu, P.C.; Whiting-Theobald, N.L.; Linton, G.F.; Malech, H.L. Diprotin A infusion into nonobese diabetic/severe combined immunodeficiency mice markedly enhances engraftment of human mobilized CD34+ peripheral blood cells. Stem Cells Dev. 2007, 16, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Kissow, H.; Hartmann, B.; Holst, J.J.; Poulsen, S.S. Glucagon-like peptide-1 as a treatment for chemotherapy-induced mucositis. Gut 2013, 62, 1724–1733. [Google Scholar] [CrossRef] [PubMed]
- Muscogiuri, G.; DeFronzo, R.A.; Gastaldelli, A.; Holst, J.J. Glucagon-like Peptide-1 and the Central/Peripheral Nervous System: Crosstalk in Diabetes. Trends Endocrinol. Metab. 2017, 28, 88–103. [Google Scholar] [CrossRef] [PubMed]
- Davidson, E.P.; Coppey, L.J.; Dake, B.; Yorek, M.A. Treatment of streptozotocin-induced diabetic rats with alogliptin: Effect on vascular and neural complications. Exp. Diabetes Res. 2011, 2011, 810469. [Google Scholar] [CrossRef]
- De La Vega, M.R.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Baek, S.H.; Kim, S.H.; Kim, J.W.; Kim, Y.J.; Lee, K.W.; Na, K.Y. Effects of a DPP4 inhibitor on cisplatin-induced acute kidney injury: Study protocol for a randomized controlled trial. Trials 2015, 16, 239. [Google Scholar] [CrossRef]
- Agarwal, P.; Isringhausen, S.; Li, H.; Paterson, A.J.; He, J.; Gomariz, A.; Nagasawa, T.; Nombela-Arrieta, C.; Bhatia, R. Mesenchymal Niche-Specific Expression of Cxcl12 Controls Quiescence of Treatment-Resistant Leukemia Stem Cells. Cell Stem Cell 2019, 24, 769–784.e6. [Google Scholar] [CrossRef]
- Ladikou, E.E.; Chevassut, T.; Pepper, C.J.; Pepper, A.G. Dissecting the role of the CXCL12/CXCR4 axis in acute myeloid leukaemia. Br. J. Haematol. 2020, 189, 815–825. [Google Scholar] [CrossRef]
- Tavor, S.; Petit, I. Can inhibition of the SDF-1/CXCR4 axis eradicate acute leukemia? Semin. Cancer Biol. 2010, 20, 178–185. [Google Scholar] [CrossRef]
- Devignes, C.S.; Aslan, Y.; Brenot, A.; Devillers, A.; Schepers, K.; Fabre, S.; Chou, J.; Casbon, A.J.; Werb, Z.; Provot, S. HIF signaling in osteoblast-lineage cells promotes systemic breast cancer growth and metastasis in mice. Proc. Natl. Acad. Sci. USA 2018, 115, E992–E1001. [Google Scholar] [CrossRef]
- Sleightholm, R.L.; Neilsen, B.K.; Li, J.; Steele, M.M.; Singh, R.K.; Hollingsworth, M.A.; Oupicky, D. Emerging roles of the CXCL12/CXCR4 axis in pancreatic cancer progression and therapy. Pharmacol. Ther. 2017, 179, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Avogaro, A. Dipeptidyl peptidase-4 inhibition and vascular repair by mobilization of endogenous stem cells in diabetes and beyond. Atherosclerosis 2013, 229, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Zaruba, M.M.; Theiss, H.D.; Vallaster, M.; Mehl, U.; Brunner, S.; David, R.; Fischer, R.; Krieg, L.; Hirsch, E.; Huber, B.; et al. Synergy between CD26/DPP-IV inhibition and G-CSF improves cardiac function after acute myocardial infarction. Cell Stem Cell 2009, 4, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Kim, Y.G.; Cho, H.J.; Park, J.; Jeong, H.; Lee, S.E.; Lee, S.P.; Kang, H.J.; Kim, H.S. Dipeptidyl Peptidase-4 Inhibitor Increases Vascular Leakage in Retina through VE-cadherin Phosphorylation. Sci. Rep. 2016, 6, 29393. [Google Scholar] [CrossRef]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef]
- Pan, K.; Ohnuma, K.; Morimoto, C.; Dang, N.H. CD26/Dipeptidyl Peptidase IV and Its Multiple Biological Functions. Cureus 2021, 13, e13495. [Google Scholar] [CrossRef]
- Mingueneau, M.; Kreslavsky, T.; Gray, D.; Heng, T.; Cruse, R.; Ericson, J.; Bendall, S.; Spitzer, M.H.; Nolan, G.P.; Kobayashi, K.; et al. The transcriptional landscape of alphabeta T cell differentiation. Nat. Immunol. 2013, 14, 619–632. [Google Scholar] [CrossRef]
- Najar, M.; Raicevic, G.; Fayyad-Kazan, H.; De Bruyn, C.; Bron, D.; Toungouz, M.; Lagneaux, L. Impact of different mesenchymal stromal cell types on T-cell activation, proliferation and migration. Int. Immunopharmacol. 2013, 15, 693–702. [Google Scholar] [CrossRef]
- Hinks, T.S.C.; Zhang, X.W. MAIT Cell Activation and Functions. Front. Immunol. 2020, 11, 1014. [Google Scholar] [CrossRef]
- Eble, J.A.; Niland, S. The extracellular matrix in tumor progression and metastasis. Clin. Exp. Metastasis 2019, 36, 171–198. [Google Scholar] [CrossRef]
- Sadir, R.; Imberty, A.; Baleux, F.; Lortat-Jacob, H. Heparan sulfate/heparin oligosaccharides protect stromal cell-derived factor-1 (SDF-1)/CXCL12 against proteolysis induced by CD26/dipeptidyl peptidase IV. J. Biol. Chem. 2004, 279, 43854–43860. [Google Scholar] [CrossRef] [PubMed]
- Busek, P.; Mateu, R.; Zubal, M.; Kotackova, L.; Sedo, A. Targeting fibroblast activation protein in cancer—Prospects and caveats. Front. Biosci. 2018, 23, 1933–1968. [Google Scholar] [CrossRef]
- Fitzgerald, A.A.; Wang, S.; Agarwal, V.; Marcisak, E.F.; Zuo, A.; Jablonski, S.A.; Loth, M.; Fertig, E.J.; MacDougall, J.; Zhukovsky, E.; et al. DPP inhibition alters the CXCR3 axis and enhances NK and CD8+ T cell infiltration to improve anti-PD1 efficacy in murine models of pancreatic ductal adenocarcinoma. J. Immunother. Cancer 2021, 9, e002837. [Google Scholar] [CrossRef] [PubMed]

| Chemokines | CXCL12 (SDF-1), CXCL11 (I-TAC), CXCL10 (IP-10), CXCL9 (Mig), CCL11 (eotaxin), CCL5 (RANTES), CCL3L1, CCL22 (MDC) |
| Incretins | GLP-1, GIP |
| Neuropeptides | SP, PYY, NPY, VIP, PACAP |
| Other | GLP-2, BNP, erythropoietin, GHRH, GRP, glucagon, procalcitonin |
| Generic Name | Daily Dose (mg) Used in Diabetic Patients | IC50 DPP-IV (nM) | IC50 DPP8 (nM) | IC50 DPP9 (nM) | IC50 FAP (nM) | Cmax (nM) in Humans |
|---|---|---|---|---|---|---|
| Gemigliptin | 50 | 6.3 | 277,000 | 233,000 | 418,430 | 128 |
| Sitagliptin | 100 | 19 | 48,000 | >100,000 | >500,000 | 959 |
| Vildagliptin | 100 (2 × 50) | 3 | 810 | 95 | 54,600 | 1309 |
| Saxagliptin | 5 | 1.3 | 520 | 98 | >1000 | 76 |
| Linagliptin | 5 | 1 | >100,000 | >100,000 | 89 | 10 |
| Alogliptin | 25 | 6.9 | >100,000 | >100,000 | >500,000 | 324 |
| Teneligliptin | 20 | 0.37 | 260 | 540 | >10,000 | 645 |
| Anagliptin | 200 (2 × 100) | 3.8 | 63,000 | 60,000 | 72,700 | 1242 |
| Tumor Type | Gliptin | Effective Gliptin Concentrations in In Vitro Studies | References, Notes |
|---|---|---|---|
| Colorectal cancer | Vildagliptin | Cytotoxicity: 2–10 mM for single exposure and >0.328 mM for repeated exposure. Reduced expression of EMT markers: 10–20 µM Reduced cyclin-dependent kinase 1 phosphorylation: 0.08–0.16 mM | Suppression of lung metastases also observed in an animal model [64]. |
| Sitagliptin | Cytotoxicity: above 0.5 mM, lower concentrations did not substantially influence cell growth. Inhibition of motility and invasion: 0.5 mM | [142] | |
| Thyroid cancer | Gemigliptin | Cytotoxicity: 0.5–2 mM Effect in other assays (reduced migration, induction of apoptosis etc.): 0.25–1 mM. At this concentration, some cytotoxicity was seen in normal bronchial epithelial cells. | Synergistic cytotoxic effects with a histone deacetylase inhibitor, metformin, and a Hsp90 inhibitor [138,139,140]. |
| Sitagliptin, vildagliptin | Reduced cell growth: 1 mM Decreased migration and invasion: 10–100 nM | Decreased tumor growth was also observed in a xenotransplantation mouse model [60]. | |
| Gastric cancer | Sitagliptin | Inhibition of growth and colony formation: 1–2 mM Inhibition of YAP signaling: 1–2 mM | [141] |
| Breast cancer | Sitagliptin | Decreased cell viability and activation of apoptotic signaling: 0.5–2.5 mM Decreased colony formation: 0.1–1 mM Inhibition of EGF signaling: 0.5–1 mM | Cells pretreated with sitagliptin (10 mM) form smaller tumors in experimental animals [78]. |
| Endometrial cancer | Sitagliptin | Decreased cell growth: 2–8 mM Decreased migration: 1 mM | [82] |
| Hepatocellular carcinoma | Sitagliptin | No effect on cell growth or synergy with doxurubicine derivative WP 631 toxicity: 0.01–200 µM | [143] |
| Cervical carcinoma | Sitagliptin | Compromised cellular integrity (LDH release): >2 mM Decreased viability: >0.2 mM Decreased cell adhesion 1 mM | Effects are independent of DPP-IV expression [144]. |
| Acute myeloid leukemia | Vildagliptin | 10 µM vildagliptin but not 10 µM sitagliptin enhances the cytotoxic effect of parthenolide. | Effects caused by DPP8 and DPP9 inhibition [145]. |
| Multiple myeloma | Vildagliptin, saxagliptin | Cytotoxicity: 0.005–0.1 mM | Effects caused by DPP9 inhibition [146]. |
| Chronic myeloid leukemia | Vildagliptin, sitagliptin, saxagliptin | No effect on cell growth or synergy with tyrosinkinase inhibitors: 10 nM–10 µM Reduced mobilization of leukemic stem cells from a stroma cell layer: 5–10 µM | Gliptins enhance SDF-1 induced migration, but do not affect colony formation. Preincubation with vildagliptin decreased engraftment of leukemic cells in mice. Gliptin treatment led to decreased BCR/ABL1 transcript levels in two patients [147]. Effect on engraftment was not confirmed in a follow-up study [148]. |
| Tumor TYPE | Gliptin | Proposed Mechanism | Notes, Reference |
|---|---|---|---|
| Melanoma, colorectal carcinoma | Sitagliptin | Preserved bioactivity of CXCL10, leading to increase CXCR3-dependent infiltration of CD4+ and CD8+ lymphocytes. | No effect of sitagliptin on tumor growth in DPP-IV KO mice suggests that sitagliptin does not have a direct cytotoxic effect [157]. |
| Hepatic cancer | Anagliptin, vildagliptin, sitagliptin | Preserved bioactivity of CXCL10, leading to increase CXCR3-dependent infiltration of NK cells. | Gliptins do not affect the growth of hepatocellular carcinoma cells in vitro (up to 100 µM) [68]. |
| Hepatic and breast cancer | Sitagliptin | Preserved bioactivity of CCL11, leading to increased infiltration of eosinophils. | No effect of sitagliptin on hepatic carcinoma cell growth (up to 12.3 µM) [67]. |
| Ovarian cancer | Sitagliptin | Infiltration of the tumors by CXCR3+ T lymphocytes | [82] |
| Lung cancer | Vildagliptin | Increased expression of surfactant proteins in cancer cells, resulting in higher amounts and pro-inflammatory activity of macrophages and NK cells. | The antitumor effect of vildagliptin was preserved in CD26−/− animals. No cytotoxicity observed for 0.3–1.3 mM vildagliptin in vitro, increased surfactant protein expression after treatment with 10–20 µM vildagliptin [160]. |
| Observed Effect | Gliptin | Anticancer Drug | Proposed Mechanism | Notes, Reference |
|---|---|---|---|---|
| Reduced myelotoxicity | Sitagliptin | 5-fluorouracil | Decreased cleavage of GM-CSF, G-CSF, and IL3, leading to increases in recovery of hematopoietic progenitor cells and bone marrow cellularity. | Similar effects observed in CD26−/− mice [164] |
| Nephroprotection | Teneligliptin | Cisplatin | Possible anti-inflammatory effects and inhibition of CXCL12 breakdown. | [165] |
| Alogliptin | Cyclophosphamide | Reduced oxidative stress and inflammation. | [166] | |
| Sitagliptin, Linagliptin | Doxorubicin | Decreased expression of NLRP3 inflammasome-associated genes. | [167] | |
| Vildagliptin, Saxagliptin | Doxorubicin | Decreased inflammation. | [168] | |
| Decreased mucositis | Vildagliptin | 5-fluorouracil | Possibly preserved bioactivity of GLP-1 and 2. | [175] |
| Neuroprotection | Alogliptin | Oxaliplatin | Unknown. | Effect seen in oxaliplatin-induced, but not bortezomib- or paclitaxel-induced neuropathy [172] |
| Reduced cardiotoxicity | Sitagliptin | Doxorubicin | Reduced oxidative damage, inflammation, and apoptosis in cardiac tissue. | [170] |
| Linagliptin | Doxorubicin | Decreased oxidative stress. | [171] | |
| Hepatoprotection | Sitagliptin | Methotrexate | Reduced oxidative stress and inflammation. | [173] |
| Reduced reproductive toxicity | Linagliptin | Cisplatin | Increased bioactivity of CXCL12. | [174] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Busek, P.; Duke-Cohan, J.S.; Sedo, A. Does DPP-IV Inhibition Offer New Avenues for Therapeutic Intervention in Malignant Disease? Cancers 2022, 14, 2072. https://doi.org/10.3390/cancers14092072
Busek P, Duke-Cohan JS, Sedo A. Does DPP-IV Inhibition Offer New Avenues for Therapeutic Intervention in Malignant Disease? Cancers. 2022; 14(9):2072. https://doi.org/10.3390/cancers14092072
Chicago/Turabian StyleBusek, Petr, Jonathan S. Duke-Cohan, and Aleksi Sedo. 2022. "Does DPP-IV Inhibition Offer New Avenues for Therapeutic Intervention in Malignant Disease?" Cancers 14, no. 9: 2072. https://doi.org/10.3390/cancers14092072
APA StyleBusek, P., Duke-Cohan, J. S., & Sedo, A. (2022). Does DPP-IV Inhibition Offer New Avenues for Therapeutic Intervention in Malignant Disease? Cancers, 14(9), 2072. https://doi.org/10.3390/cancers14092072