
Human Papillomavirus 16 E6 Suppresses Transporter Associated with Antigen-Processing Complex in Human Tongue Keratinocyte Cells by Activating Lymphotoxin Pathway

 ,
,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Clinical Specimen of OSCCs
2.3. Cell Lines and Cultures
2.4. Measurement of Gene Expression by Quantitative RT-PCR (qRT-PCR)
2.5. Determination of Protein Concentrations Using Western Blotting
2.6. Gene Silencing by Retroviruses Expressing Short Hairpin RNAs (shRNA)
2.7. siRNA and Cell Transfection
2.8. Analysis of RNA Sequence Data from TCGA
2.9. Tissue Microarray (TMA) Construction
2.10. Detection of HPV DNA by Polymerase Chain Reaction (PCR)
2.11. Detection of High-Risk HPV in OSCC Tissues by In-Situ Hybridization (ISH)
2.12. Immunohistochemistry (IHC) Assay
2.13. Systematic Review and Meta-Analysis
2.14. Statistical Analysis
3. Results
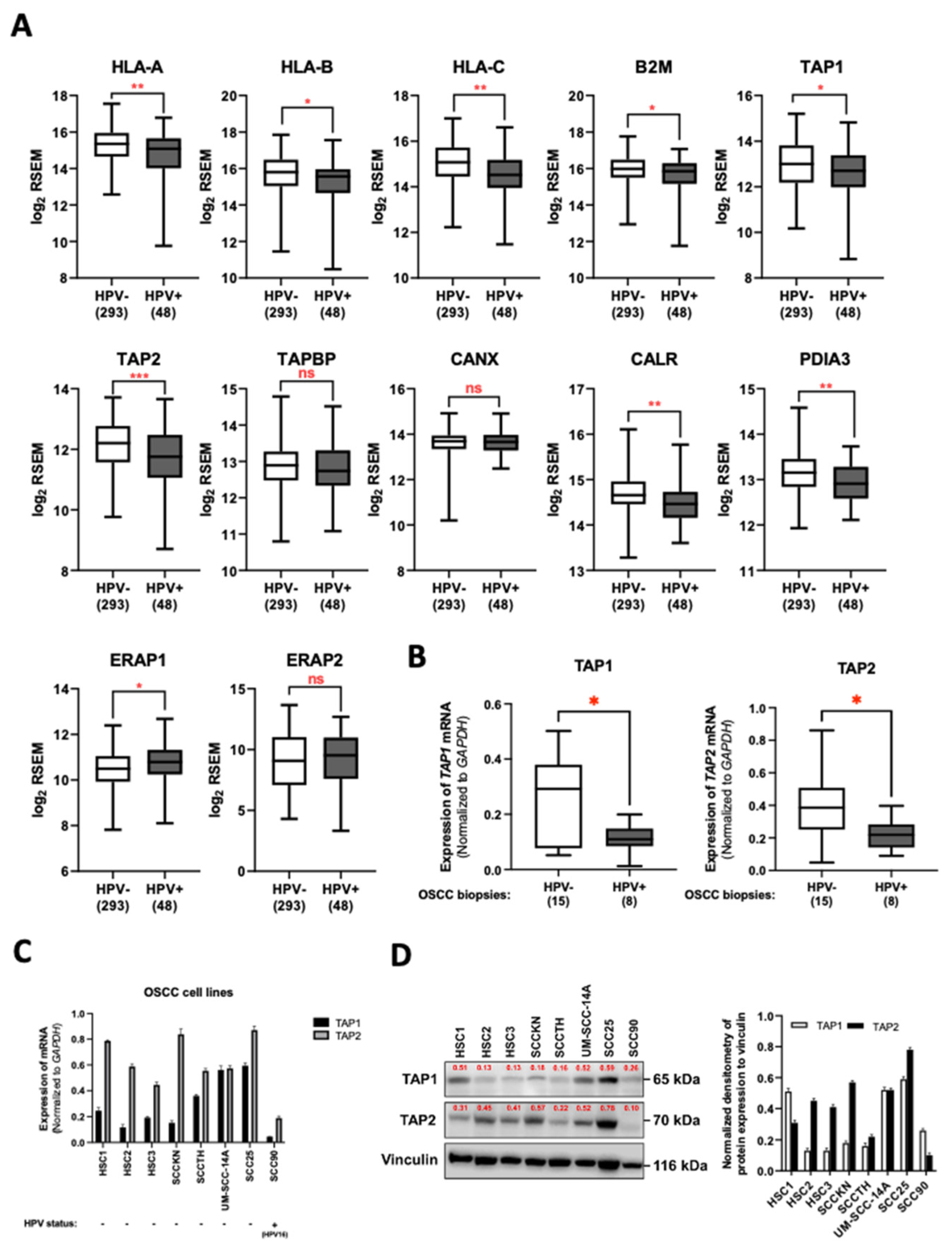
3.1. HPV Infection Downregulated TAP1 and TAP2 Expression in OSCC Tissues
3.2. HPV16 Infection Is Associated with Increased Risk of OSCC
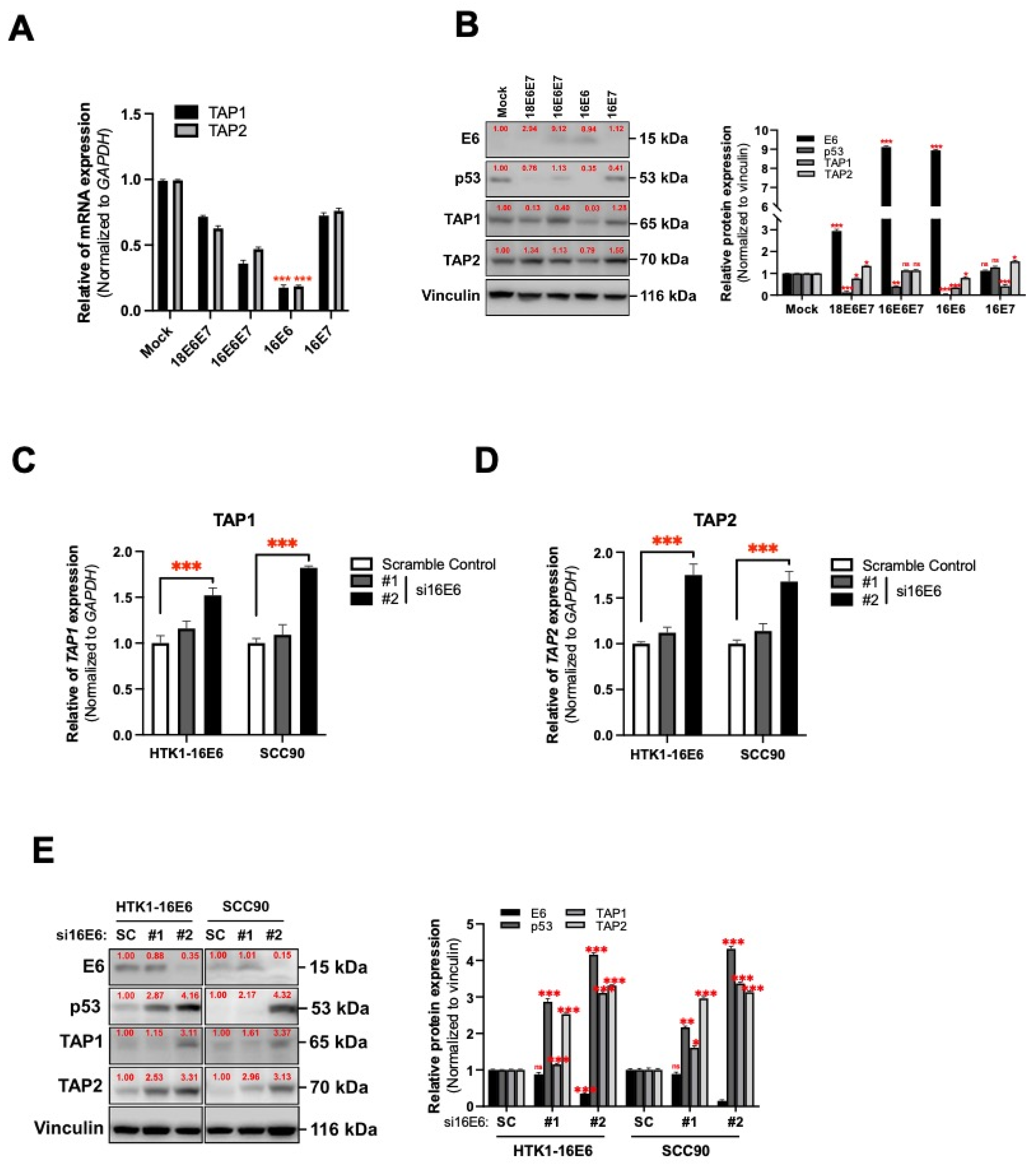
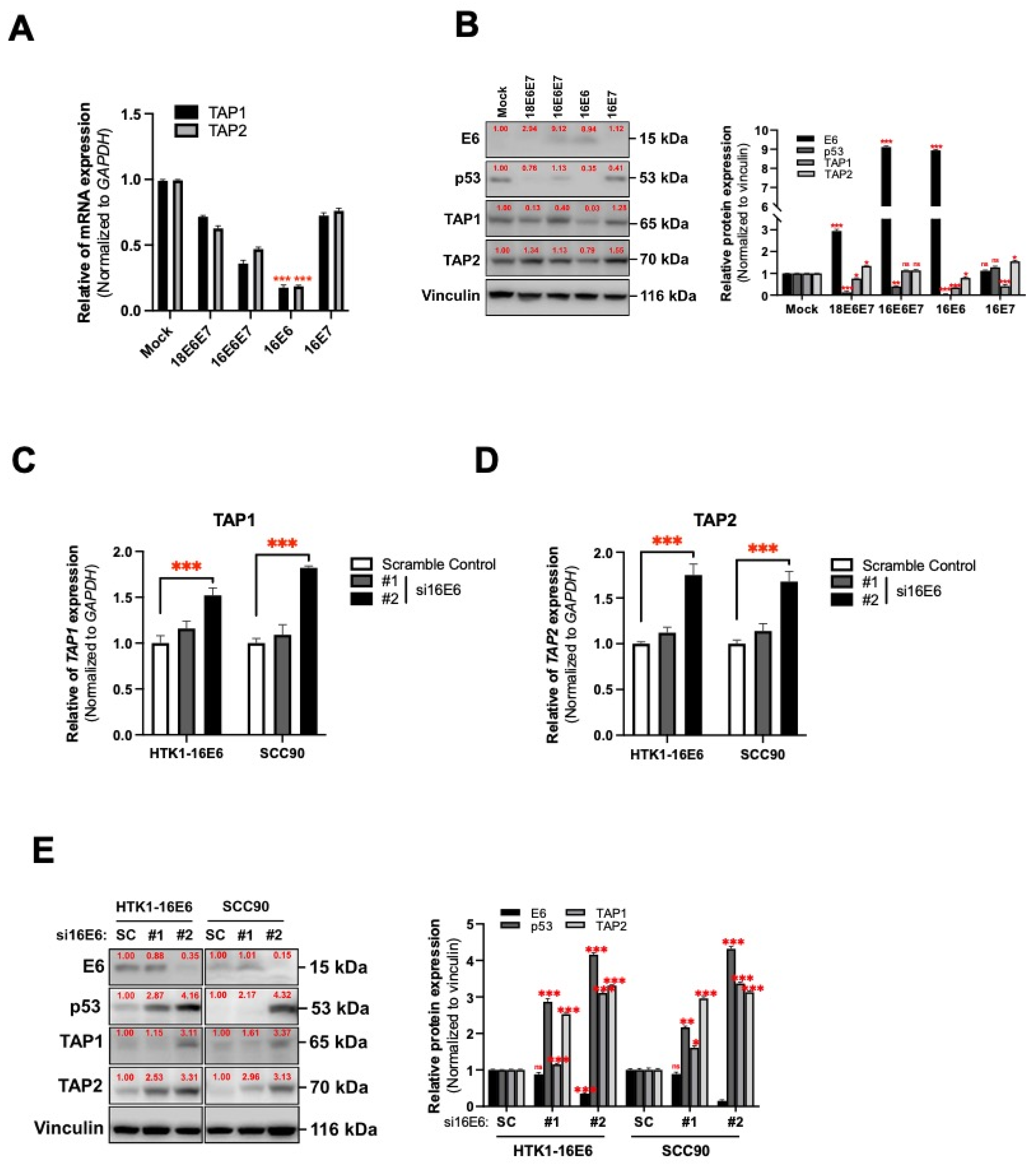
3.3. HPV16 Oncogene-Mediated Suppression of TAP1 and TAP2 in Human Tongue Keratinocytes
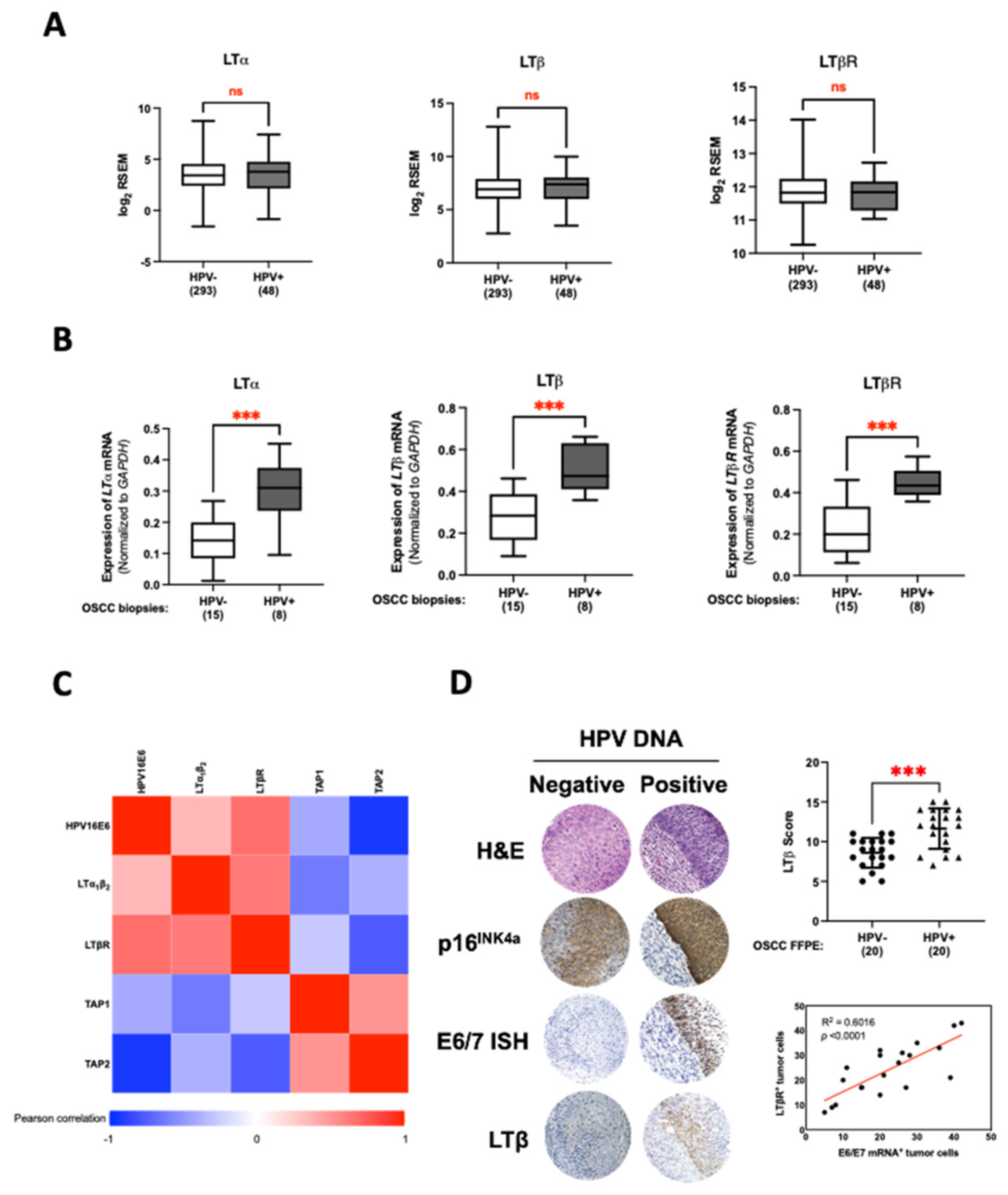
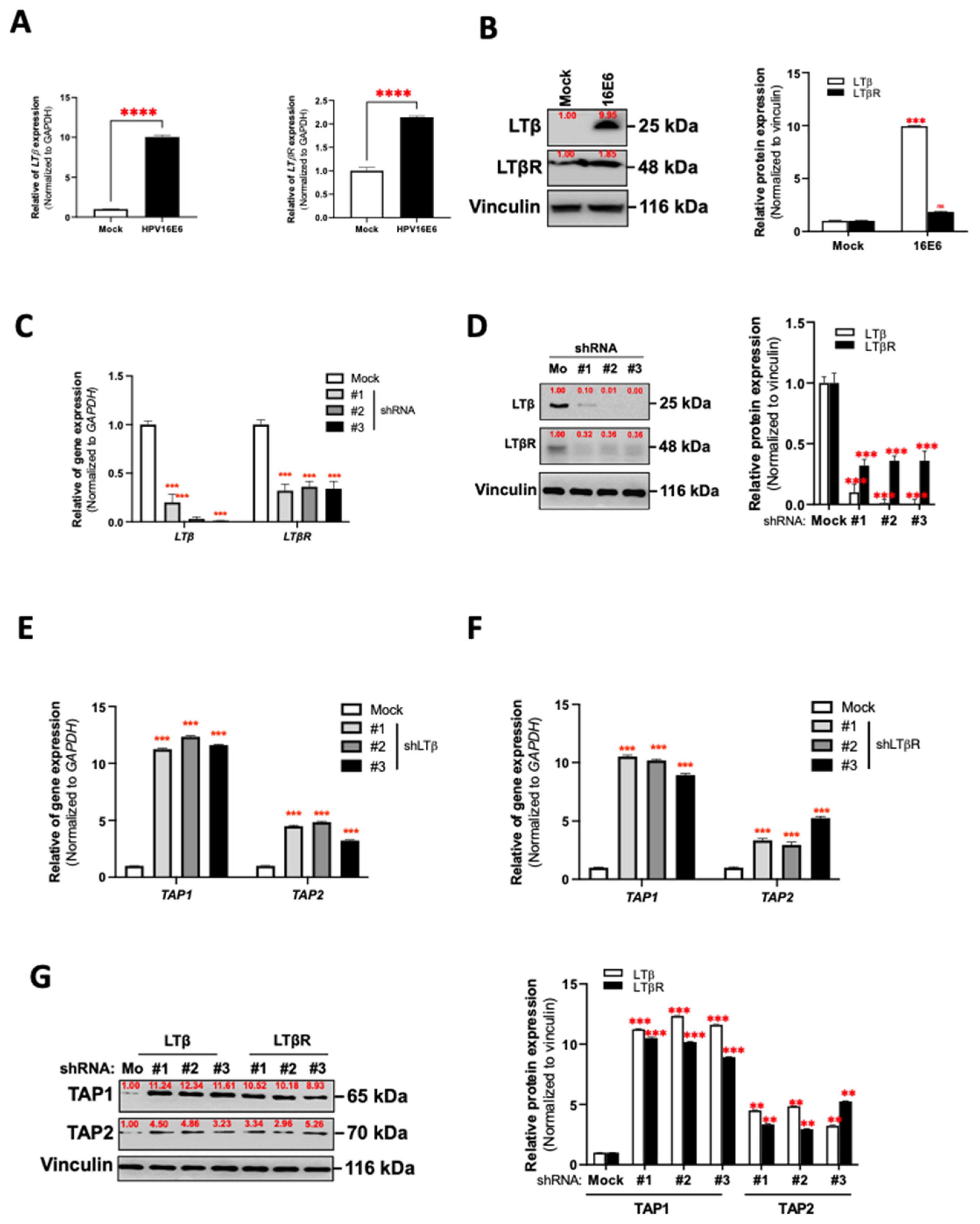
3.4. LT Network Linked to TAP1 and TAP2 Suppression in HPV16 E6-Expressing Oral Epithelial Cells
3.5. HPV16 E6 Regulates TAP1 and TAP2 Expression via LTα1β2-LTβR Axis in Oral Epithelial Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Bagan, J.; Sarrion, G.; Jimenez, Y. Oral cancer: Clinical features. Oral Oncol. 2010, 46, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tan, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Souza, G.; Kreimer, A.R.; Viscidi, R.; Pawlita, M.; Fakhry, C.; Koch, W.M.; Westra, W.H.; Gillison, M.L. Case-control study of human papillomavirus and oropharyngeal cancer. N. Engl. J. Med. 2007, 356, 1944–1956. [Google Scholar] [CrossRef] [Green Version]
- Dyson, N.; Guida, P.; Munger, K.; Harlow, E. Homologous sequences in adenovirus E1A and human papillomavirus E7 proteins mediate interaction with the same set of cellular proteins. J. Virol. 1992, 66, 6893–6902. [Google Scholar] [CrossRef] [Green Version]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996, 56, 4620–4624. [Google Scholar]
- Brehm, A.; Miska, E.A.; McCance, D.J.; Reid, J.L.; Bannister, A.J.; Kouzarides, T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 1998, 391, 597–601. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Campisi, G.; Panzarella, V.; Giuliani, M.; Lajolo, C.; Di Fede, O.; Falaschini, S.; Di Liberto, C.; Scully, C.; Lo Muzio, L. Human papillomavirus: Its identity and controversial role in oral oncogenesis, premalignant and malignant lesions (review). Int. J. Oncol. 2007, 30, 813–823. [Google Scholar] [CrossRef] [Green Version]
- Westrich, J.A.; Warren, C.J.; Pyeon, D. Evasion of host immune defenses by human papillomavirus. Virus Res. 2017, 231, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.H.; Haghshenas, M.; Marchetti, B.; Campo, M.S. E5 protein of human papillomavirus 16 downregulates HLA class I and interacts with the heavy chain via its first hydrophobic domain. Int. J. Cancer 2006, 119, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kim, E.M.; Lee, E.H.; Ji, K.Y.; Yi, J.; Park, M.; Kim, K.D.; Cho, Y.Y.; Kang, H.S. Human papillomavirus 16E6 suppresses major histocompatibility complex class I by upregulating lymphotoxin expression in human cervical cancer cells. Biochem. Biophys. Res. Commun. 2011, 409, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Bottley, G.; Watherston, O.G.; Hiew, Y.L.; Norrild, B.; Cook, G.P.; Blair, G.E. High-risk human papillomavirus E7 expression reduces cell-surface MHC class I molecules and increases susceptibility to natural killer cells. Oncogene 2008, 27, 1794–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgopoulos, N.T.; Proffitt, J.L.; Blair, G.E. Transcriptional regulation of the major histocompatibility complex (MHC) class I heavy chain, TAP1 and LMP2 genes by the human papillomavirus (HPV) type 6b, 16 and 18 E7 oncoproteins. Oncogene 2000, 19, 4930–4935. [Google Scholar] [CrossRef] [Green Version]
- Heller, C.; Weisser, T.; Mueller-Schickert, A.; Rufer, E.; Hoh, A.; Leonhardt, R.M.; Knittler, M.R. Identification of key amino acid residues that determine the ability of high risk HPV16-E7 to dysregulate major histocompatibility complex class I expression. J. Biol. Chem. 2011, 286, 10983–10997. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Ou, X.; Xiong, J.; Wang, T. HPV16E7 mediates HADC chromatin repression and downregulation of MHC class I genes in HPV16 tumorigenic cells through interaction with an MHC class I promoter. Biochem. Biophys. Res. Commun. 2006, 349, 1315–1321. [Google Scholar] [CrossRef]
- Cresswell, P.; Bangia, N.; Dick, T.; Diedrich, G. The nature of the MHC class I peptide loading complex. Immunol. Rev. 1999, 172, 21–28. [Google Scholar] [CrossRef]
- Parcej, D.; Tampe, R. ABC proteins in antigen translocation and viral inhibition. Nat. Chem. Biol. 2010, 6, 572–580. [Google Scholar] [CrossRef]
- Abele, R.; Tampe, R. Modulation of the antigen transport machinery TAP by friends and enemies. FEBS Lett. 2006, 580, 1156–1163. [Google Scholar] [CrossRef] [Green Version]
- Seliger, B.; Hohne, A.; Knuth, A.; Bernhard, H.; Ehring, B.; Tampe, R.; Huber, C. Reduced membrane major histocompatibility complex class I density and stability in a subset of human renal cell carcinomas with low TAP and LMP expression. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1996, 2, 1427–1433. [Google Scholar]
- Zhou, F.; Chen, J.; Zhao, K.N. Human papillomavirus 16-encoded E7 protein inhibits IFN-gamma-mediated MHC class I antigen presentation and CTL-induced lysis by blocking IRF-1 expression in mouse keratinocytes. J. Gen. Virol. 2013, 94, 2504–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attaran, N.; Gu, X.; Coates, P.J.; Fahraeus, R.; Boldrup, L.; Wilms, T.; Wang, L.; Sgaramella, N.; Zborayova, K.; Nylander, K. Downregulation of TAP1 in Tumor-Free Tongue Contralateral to Squamous Cell Carcinoma of the Oral Tongue, an Indicator of Better Survival. Int. J. Mol. Sci. 2020, 21, 6220. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Pan, H.; Jiang, L.; Wei, K.; Zou, D.; Zhang, Z. A novel approach to rescue immune escape in oral squamous cell carcinoma: Combined use of interferon-gamma and LY294002. Oncol. Rep. 2011, 25, 181–187. [Google Scholar]
- Paul, N.L.; Ruddle, N.H. Lymphotoxin. Annu. Rev. Immunol. 1988, 6, 407–438. [Google Scholar] [CrossRef]
- Spies, T.; Morton, C.C.; Nedospasov, S.A.; Fiers, W.; Pious, D.; Strominger, J.L. Genes for the tumor necrosis factors alpha and beta are linked to the human major histocompatibility complex. Proc. Natl. Acad. Sci. USA 1986, 83, 8699–8702. [Google Scholar] [CrossRef] [Green Version]
- Moseman, E.A.; Iannacone, M.; Bosurgi, L.; Tonti, E.; Chevrier, N.; Tumanov, A.; Fu, Y.X.; Hacohen, N.; von Andrian, U.H. B cell maintenance of subcapsular sinus macrophages protects against a fatal viral infection independent of adaptive immunity. Immunity 2012, 36, 415–426. [Google Scholar] [CrossRef] [Green Version]
- Schneider, K.; Loewendorf, A.; De Trez, C.; Fulton, J.; Rhode, A.; Shumway, H.; Ha, S.; Patterson, G.; Pfeffer, K.; Nedospasov, S.A.; et al. Lymphotoxin-mediated crosstalk between B cells and splenic stroma promotes the initial type I interferon response to cytomegalovirus. Cell Host Microbe 2008, 3, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.C. Non-canonical NF-kappaB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [Green Version]
- de Camargo Cancela, M.; de Souza, D.L.; Curado, M.P. International incidence of oropharyngeal cancer: A population-based study. Oral Oncol. 2012, 48, 484–490. [Google Scholar] [CrossRef]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nature reviews. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Zushi, Y.; Narisawa-Saito, M.; Noguchi, K.; Yoshimatsu, Y.; Yugawa, T.; Egawa, N.; Fujita, M.; Urade, M.; Kiyono, T. An in vitro multistep carcinogenesis model for both HPV-positive and -negative human oral squamous cell carcinomas. Am. J. Cancer Res. 2011, 1, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Ghani, F.I.; Dendo, K.; Watanabe, R.; Yamada, K.; Yoshimatsu, Y.; Yugawa, T.; Nakahara, T.; Tanaka, K.; Yoshida, H.; Yoshida, M.; et al. An Ex-Vivo Culture System of Ovarian Cancer Faithfully Recapitulating the Pathological Features of Primary Tumors. Cells 2019, 8, 644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yugawa, T.; Nishino, K.; Ohno, S.; Nakahara, T.; Fujita, M.; Goshima, N.; Umezawa, A.; Kiyono, T. Noncanonical NOTCH signaling limits self-renewal of human epithelial and induced pluripotent stem cells through ROCK activation. Mol. Cell Biol. 2013, 33, 4434–4447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gewin, L.; Myers, H.; Kiyono, T.; Galloway, D.A. Identification of a novel telomerase repressor that interacts with the human papillomavirus type-16 E6/E6-AP complex. Genes Dev. 2004, 18, 2269–2282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yugawa, T.; Handa, K.; Narisawa-Saito, M.; Ohno, S.; Fujita, M.; Kiyono, T. Regulation of Notch1 gene expression by p53 in epithelial cells. Mol. Cell Biol. 2007, 27, 3732–3742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gameiro, S.F.; Zhang, A.; Ghasemi, F.; Barrett, J.W.; Nichols, A.C.; Mymryk, J.S. Analysis of Class I Major Histocompatibility Complex Gene Transcription in Human Tumors Caused by Human Papillomavirus Infection. Viruses 2017, 9, 252. [Google Scholar] [CrossRef]
- Phusingha, P.; Ekalaksananan, T.; Vatanasapt, P.; Loyha, K.; Promthet, S.; Kongyingyoes, B.; Patarapadungkit, N.; Chuerduangphui, J.; Pientong, C. Human papillomavirus (HPV) infection in a case-control study of oral squamous cell carcinoma and its increasing trend in northeastern Thailand. J. Med. Virol. 2017, 89, 1096–1101. [Google Scholar] [CrossRef]
- Xiang, T.; Lin, Y.X.; Ma, W.; Zhang, H.J.; Chen, K.M.; He, G.P.; Zhang, X.; Xu, M.; Feng, Q.S.; Chen, M.Y.; et al. Vasculogenic mimicry formation in EBV-associated epithelial malignancies. Nat. Commun. 2018, 9, 5009. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Rapado-Gonzalez, O.; Martinez-Reglero, C.; Salgado-Barreira, A.; Rodriguez-Fernandez, A.; Aguin-Losada, S.; Leon-Mateos, L.; Muinelo-Romay, L.; Lopez-Lopez, R.; Suarez-Cunqueiro, M.M. Association of Salivary Human Papillomavirus Infection and Oral and Oropharyngeal Cancer: A Meta-Analysis. J. Clin. Med. 2020, 9, 1305. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, M.; Bhawal, U.K.; Dohmen, T.; Ono, S.; Miyauchi, M.; Ishikawa, T. Detection of human papillomavirus-16 and HPV-18 DNA in normal, dysplastic, and malignant oral epithelium. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2003, 95, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Dong, Y. Human papillomavirus and oral squamous cell carcinoma: A review of HPV-positive oral squamous cell carcinoma and possible strategies for future. Curr. Probl. Cancer 2017, 41, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.J.; Seleznik, G.M.; Zeller, N.; Heikenwalder, M. The unexpected role of lymphotoxin beta receptor signaling in carcinogenesis: From lymphoid tissue formation to liver and prostate cancer development. Oncogene 2010, 29, 5006–5018. [Google Scholar] [CrossRef] [Green Version]
- Hansen, T.H.; Bouvier, M. MHC class I antigen presentation: Learning from viral evasion strategies. Nat. Rev. Immunol. 2009, 9, 503–513. [Google Scholar] [CrossRef]
- Breitburd, F.; Ramoz, N.; Salmon, J.; Orth, G. HLA control in the progression of human papillomavirus infections. Semin. Cancer Biol. 1996, 7, 359–371. [Google Scholar] [CrossRef]
- Krul, E.J.; Schipper, R.F.; Schreuder, G.M.; Fleuren, G.J.; Kenter, G.G.; Melief, C.J. HLA and susceptibility to cervical neoplasia. Hum. Immunol. 1999, 60, 337–342. [Google Scholar] [CrossRef]
- Castellsague, X.; Bosch, F.X.; Munoz, N. Environmental co-factors in HPV carcinogenesis. Virus Res. 2002, 89, 191–199. [Google Scholar] [CrossRef]
- Frazer, I.H.; Thomas, R.; Zhou, J.; Leggatt, G.R.; Dunn, L.; McMillan, N.; Tindle, R.W.; Filgueira, L.; Manders, P.; Barnard, P.; et al. Potential strategies utilised by papillomavirus to evade host immunity. Immunol. Rev. 1999, 168, 131–142. [Google Scholar] [CrossRef]
- Tindle, R.W. Immune evasion in human papillomavirus-associated cervical cancer. Nat. Rev. Cancer 2002, 2, 59–65. [Google Scholar] [CrossRef]
- O’Brien, P.M.; Saveria Campo, M. Evasion of host immunity directed by papillomavirus-encoded proteins. Virus Res. 2002, 88, 103–117. [Google Scholar] [CrossRef]
- Chang, C.C.; Campoli, M.; Ferrone, S. HLA class I defects in malignant lesions: What have we learned? Keio J. Med. 2003, 52, 220–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.H.; Chen, H.L.; Xu, B.Q.; Wei, K.; Ying, X.Y. A preliminary study on the immune responses of HPV16-E7 by combined intranasal immunization with lymphotoxin. Ginekol. Pol. 2020, 91, 301–307. [Google Scholar] [CrossRef]
- Crook, T.; Vousden, K.H. Properties of p53 mutations detected in primary and secondary cervical cancers suggest mechanisms of metastasis and involvement of environmental carcinogens. EMBO J. 1992, 11, 3935–3940. [Google Scholar] [CrossRef]
- Pober, J.S.; Lapierre, L.A.; Stolpen, A.H.; Brock, T.A.; Springer, T.A.; Fiers, W.; Bevilacqua, M.P.; Mendrick, D.L.; Gimbrone, M.A., Jr. Activation of cultured human endothelial cells by recombinant lymphotoxin: Comparison with tumor necrosis factor and interleukin 1 species. J. Immunol. 1987, 138, 3319–3324. [Google Scholar] [PubMed]
- Andrews, J.S.; Berger, A.E.; Ware, C.F. Characterization of the receptor for tumor necrosis factor (TNF) and lymphotoxin (LT) on human T lymphocytes. TNF and LT differ in their receptor binding properties and the induction of MHC class I proteins on a human CD4+ T cell hybridoma. J. Immunol. 1990, 144, 2582–2591. [Google Scholar]
- Eugster, H.P.; Muller, M.; Karrer, U.; Car, B.D.; Schnyder, B.; Eng, V.M.; Woerly, G.; Le Hir, M.; di Padova, F.; Aguet, M.; et al. Multiple immune abnormalities in tumor necrosis factor and lymphotoxin-alpha double-deficient mice. Int. Immunol. 1996, 8, 23–36. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, S.I.; Shong, M.; Giuliani, C.; Napolitano, G.; Saji, M.; Montani, V.; Suzuki, K.; Singer, D.S.; Kohn, L.D. Iodide suppression of major histocompatibility class I gene expression in thyroid cells involves enhancer A and the transcription factor NF-kappa B. Mol. Endocrinol. 1998, 12, 19–33. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burassakarn, A.; Phusingha, P.; Yugawa, T.; Noguchi, K.; Ekalaksananan, T.; Vatanasapt, P.; Kiyono, T.; Pientong, C. Human Papillomavirus 16 E6 Suppresses Transporter Associated with Antigen-Processing Complex in Human Tongue Keratinocyte Cells by Activating Lymphotoxin Pathway. Cancers 2022, 14, 1944. https://doi.org/10.3390/cancers14081944
Burassakarn A, Phusingha P, Yugawa T, Noguchi K, Ekalaksananan T, Vatanasapt P, Kiyono T, Pientong C. Human Papillomavirus 16 E6 Suppresses Transporter Associated with Antigen-Processing Complex in Human Tongue Keratinocyte Cells by Activating Lymphotoxin Pathway. Cancers. 2022; 14(8):1944. https://doi.org/10.3390/cancers14081944
Chicago/Turabian StyleBurassakarn, Ati, Pensiri Phusingha, Takashi Yugawa, Kazuma Noguchi, Tipaya Ekalaksananan, Patravoot Vatanasapt, Tohru Kiyono, and Chamsai Pientong. 2022. "Human Papillomavirus 16 E6 Suppresses Transporter Associated with Antigen-Processing Complex in Human Tongue Keratinocyte Cells by Activating Lymphotoxin Pathway" Cancers 14, no. 8: 1944. https://doi.org/10.3390/cancers14081944
APA StyleBurassakarn, A., Phusingha, P., Yugawa, T., Noguchi, K., Ekalaksananan, T., Vatanasapt, P., Kiyono, T., & Pientong, C. (2022). Human Papillomavirus 16 E6 Suppresses Transporter Associated with Antigen-Processing Complex in Human Tongue Keratinocyte Cells by Activating Lymphotoxin Pathway. Cancers, 14(8), 1944. https://doi.org/10.3390/cancers14081944