
Cannabinol Inhibits Cellular Proliferation, Invasion, and Angiogenesis of Neuroblastoma via Novel miR-34a/tRiMetF31/PFKFB3 Axis

 ,
,  , and
, and 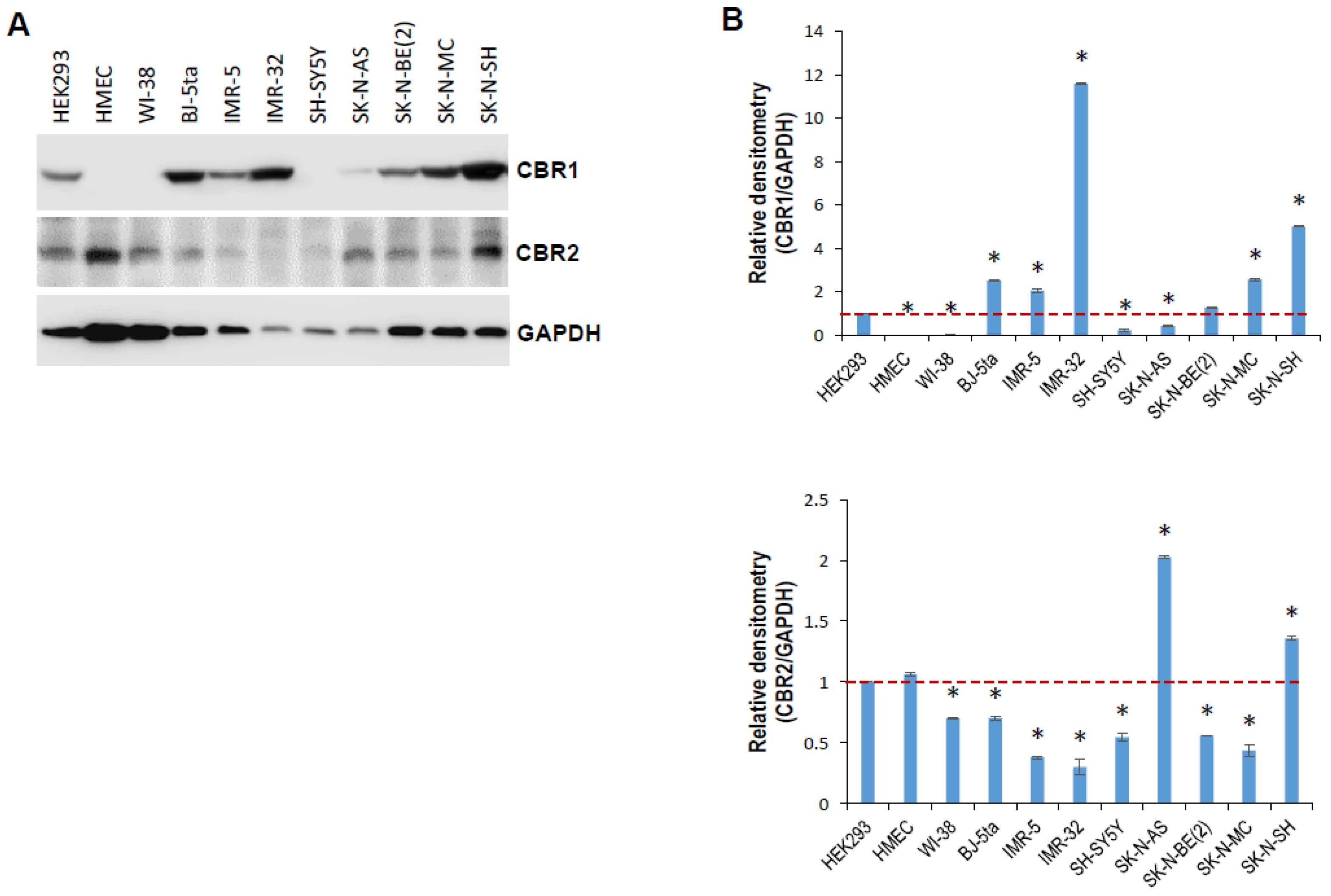{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
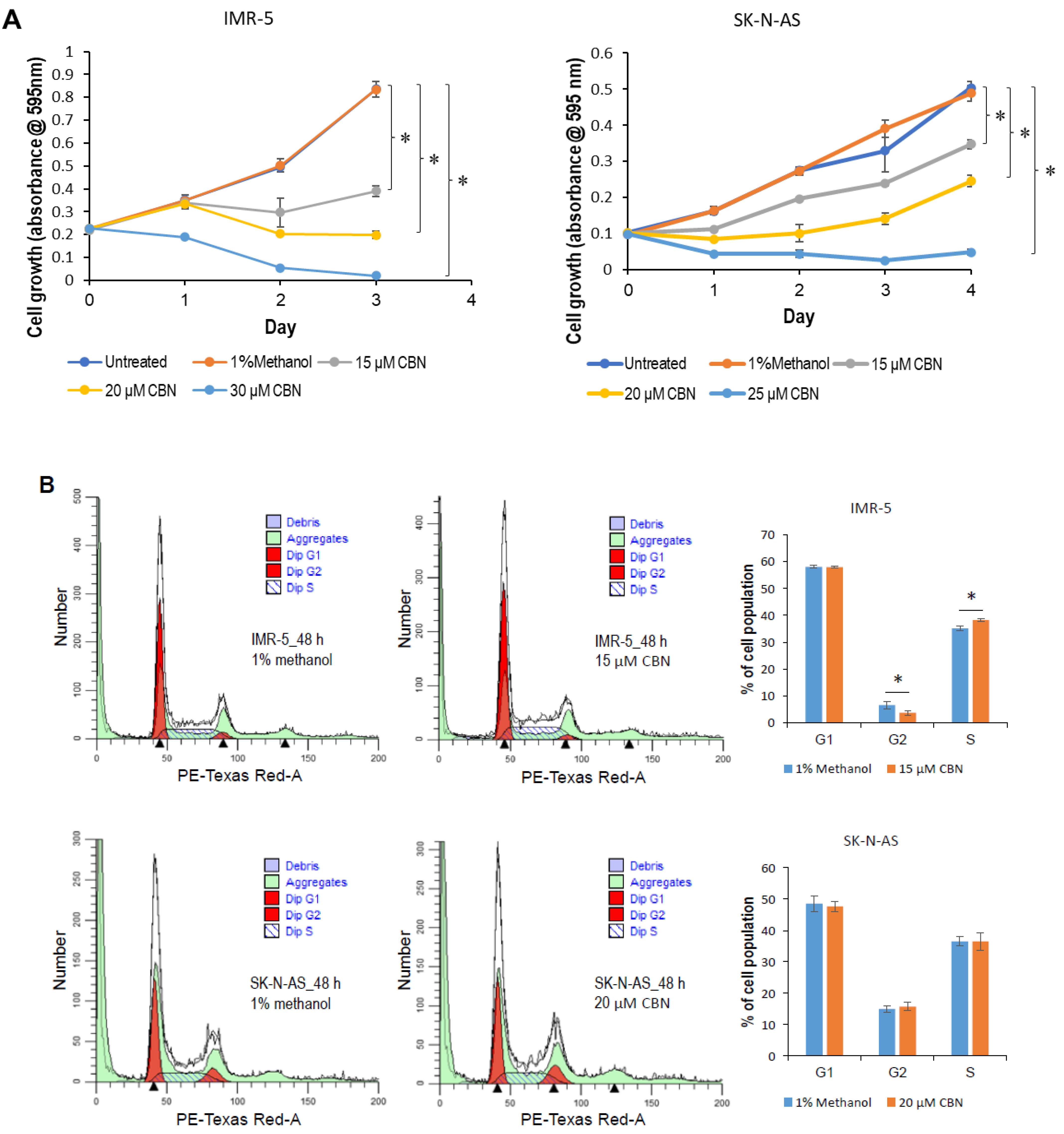
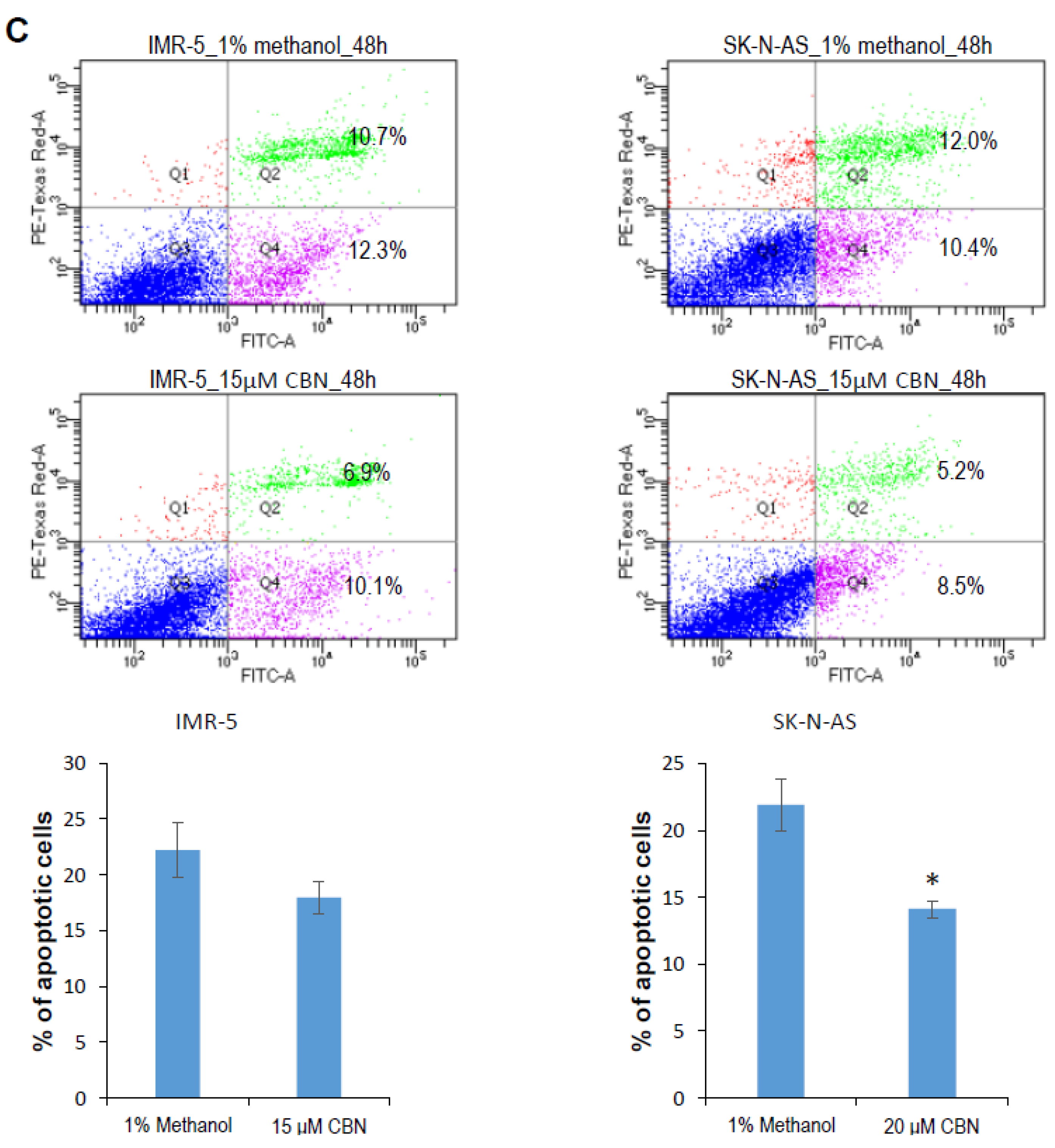
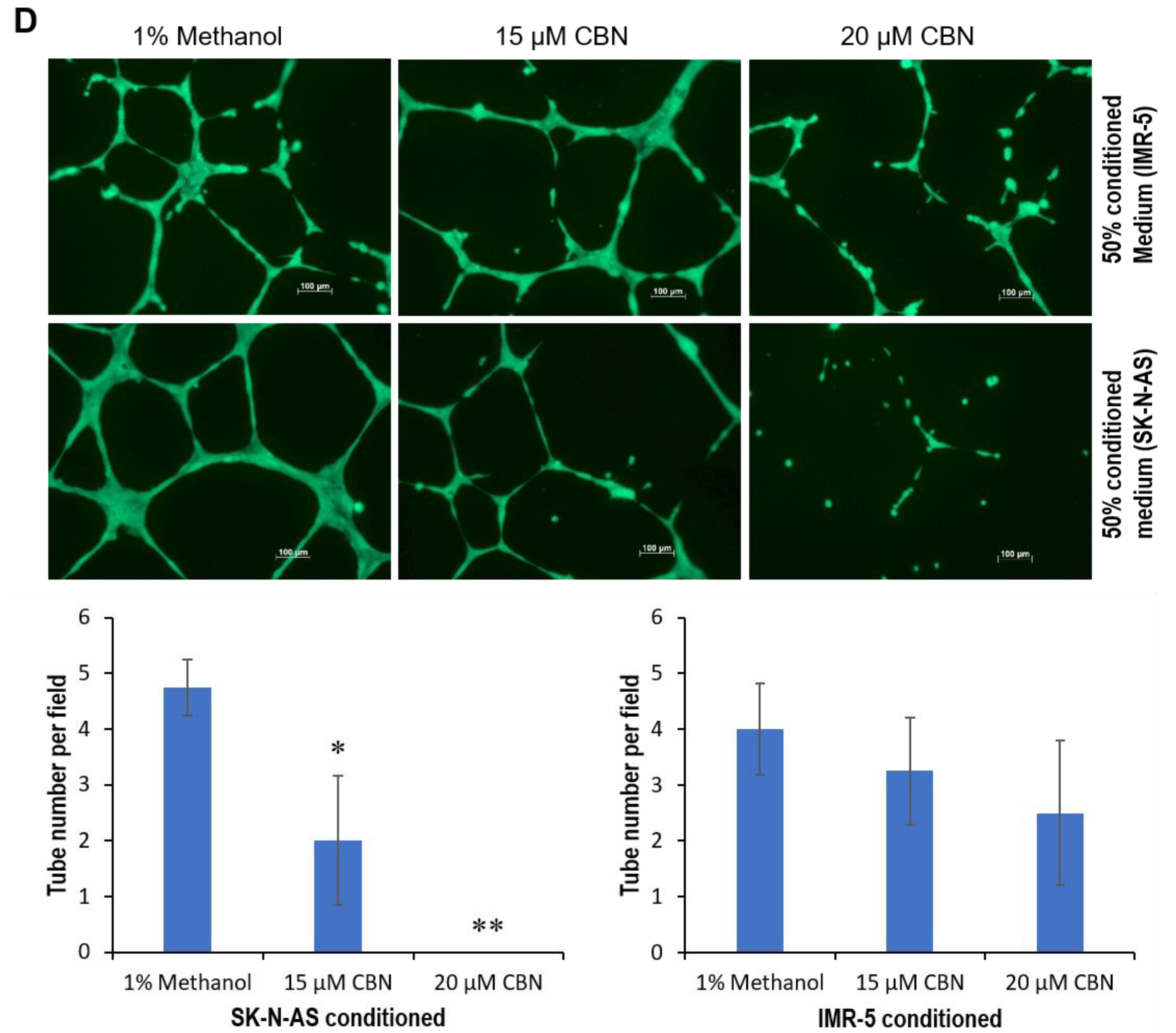
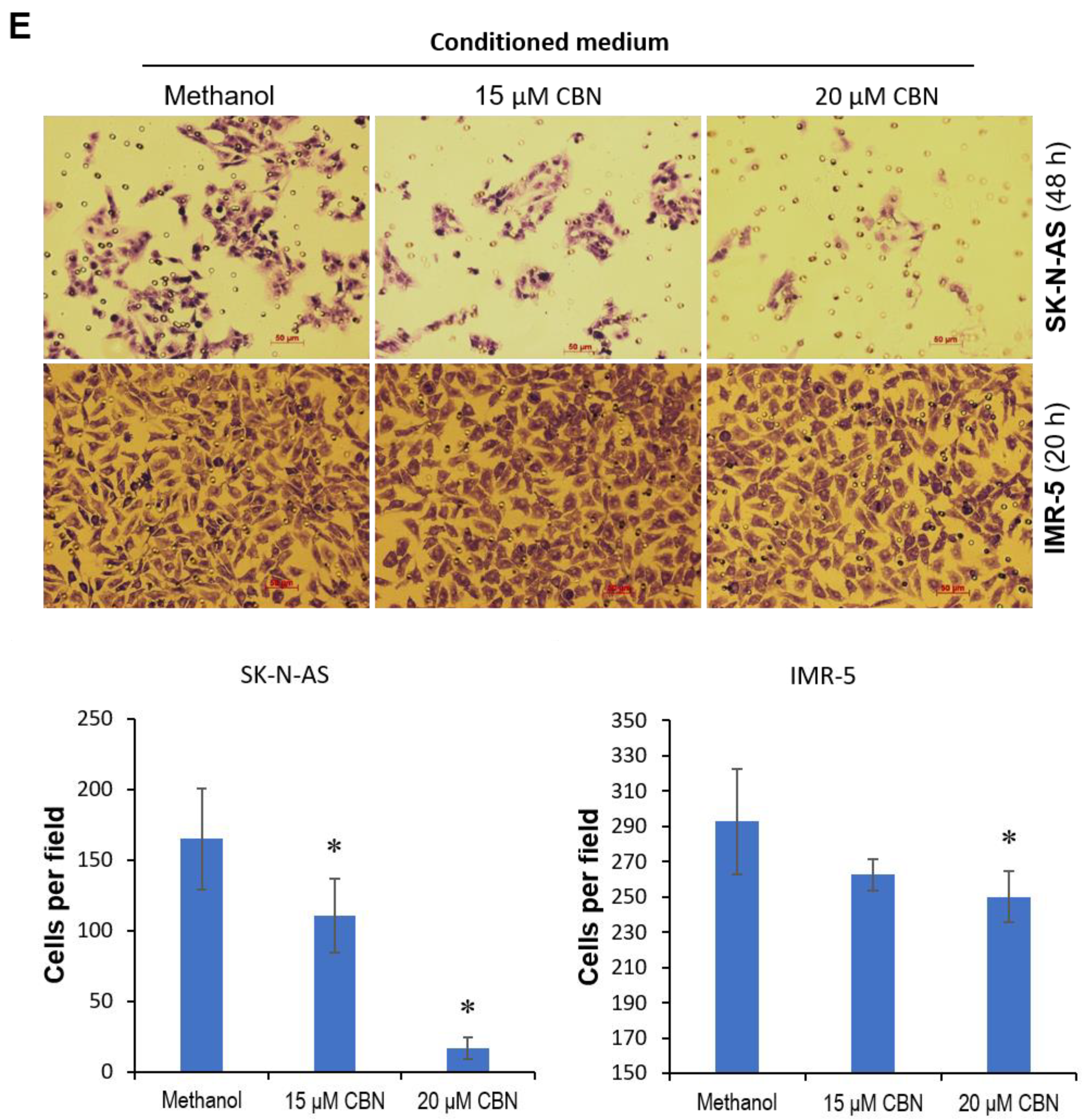
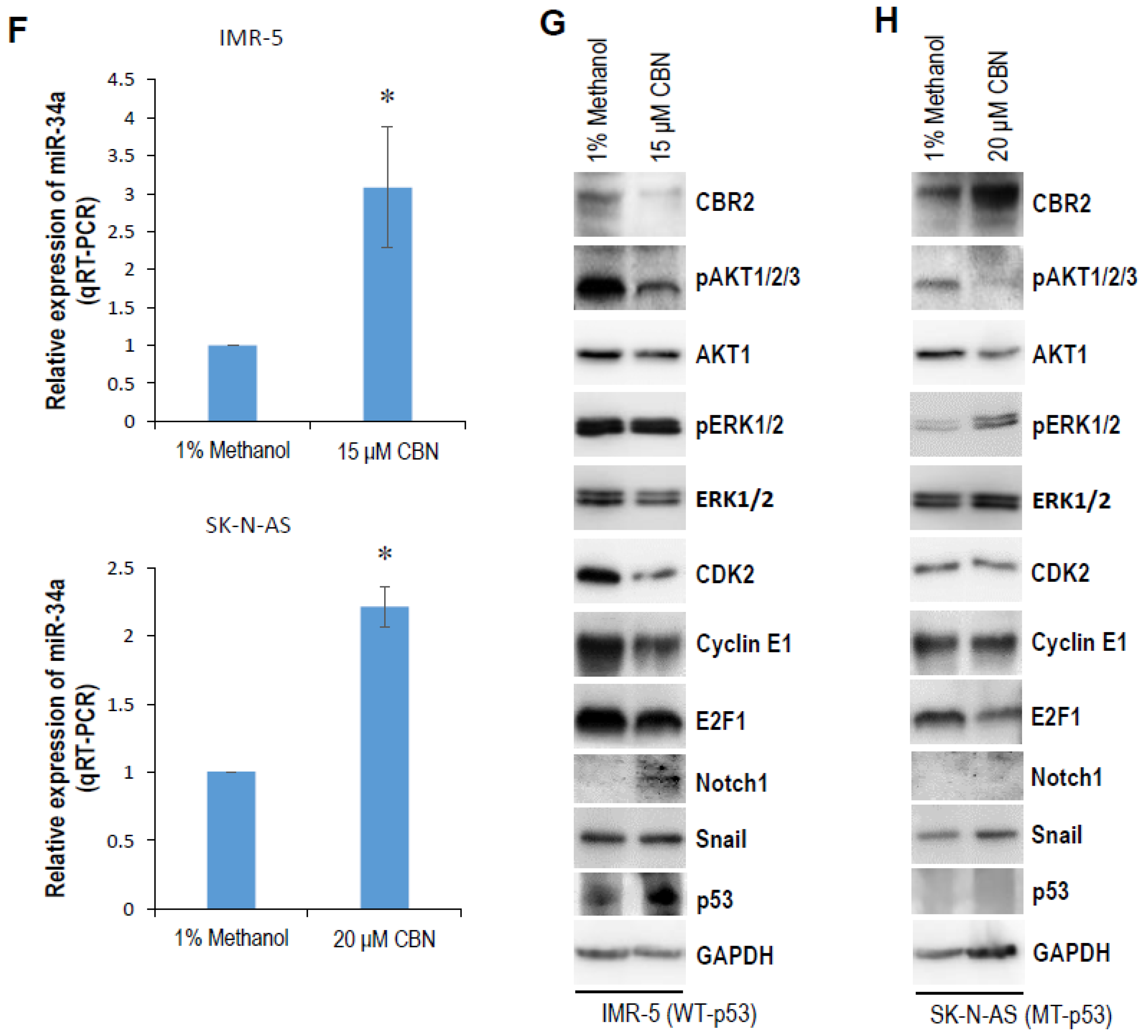
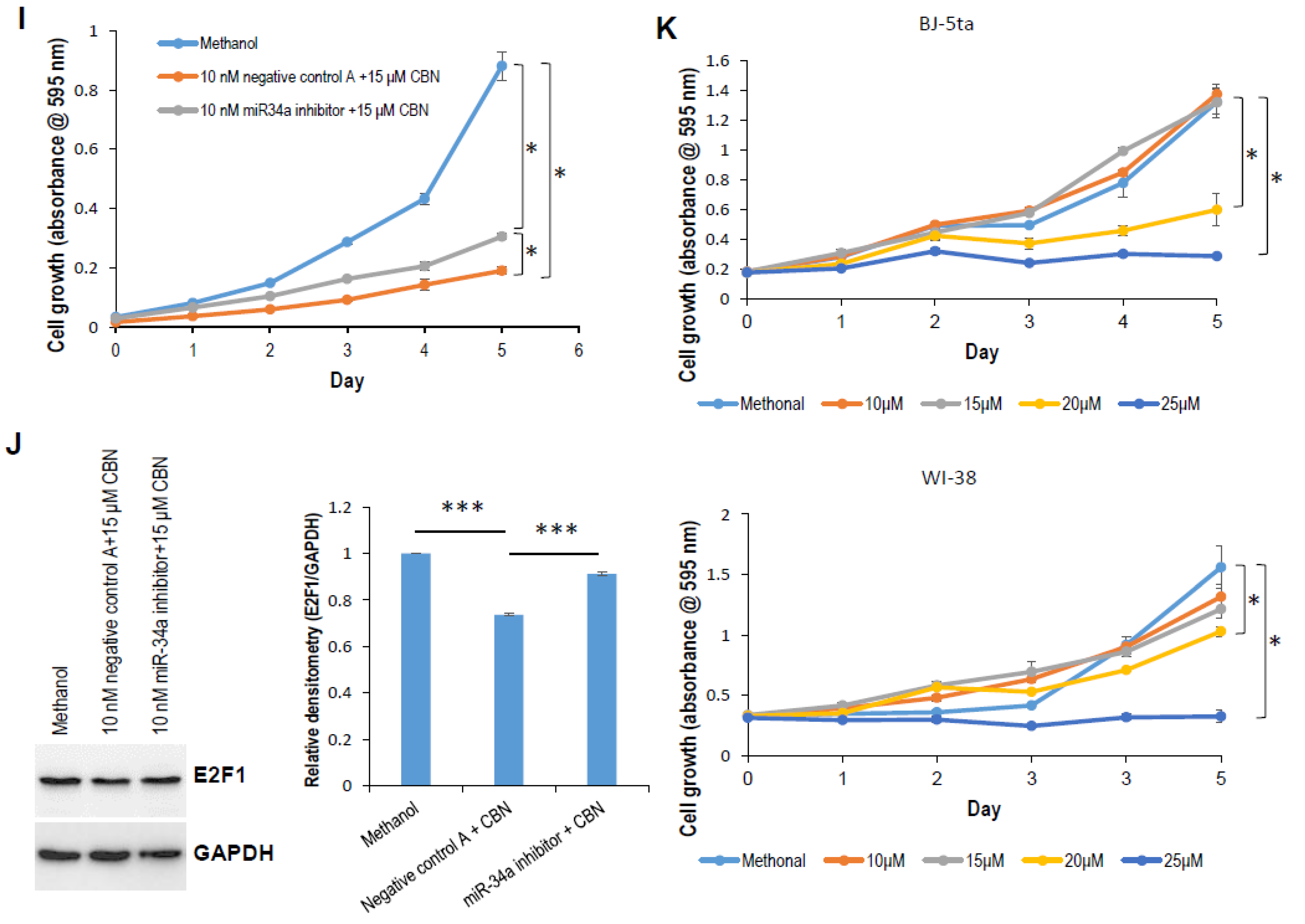
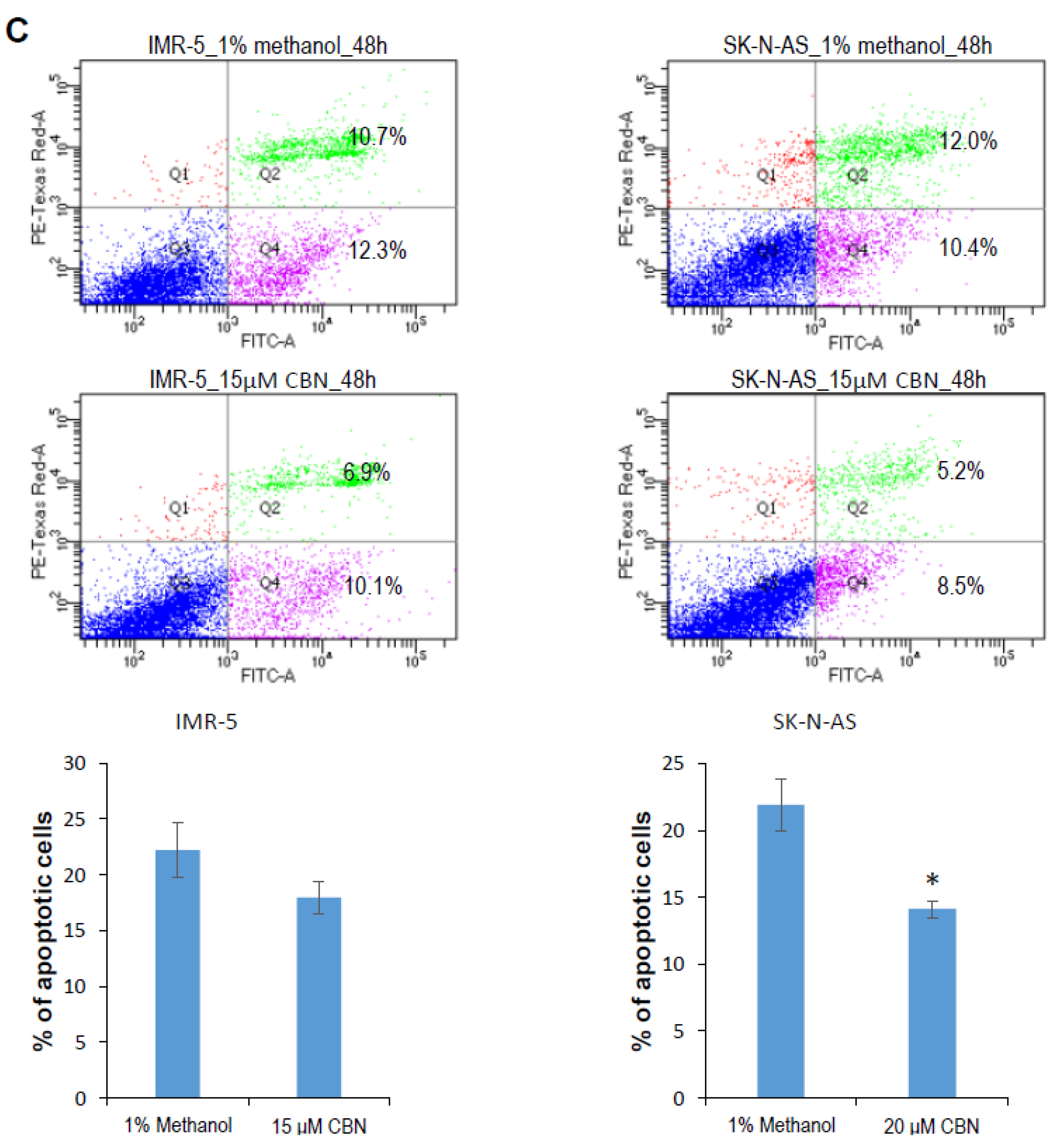
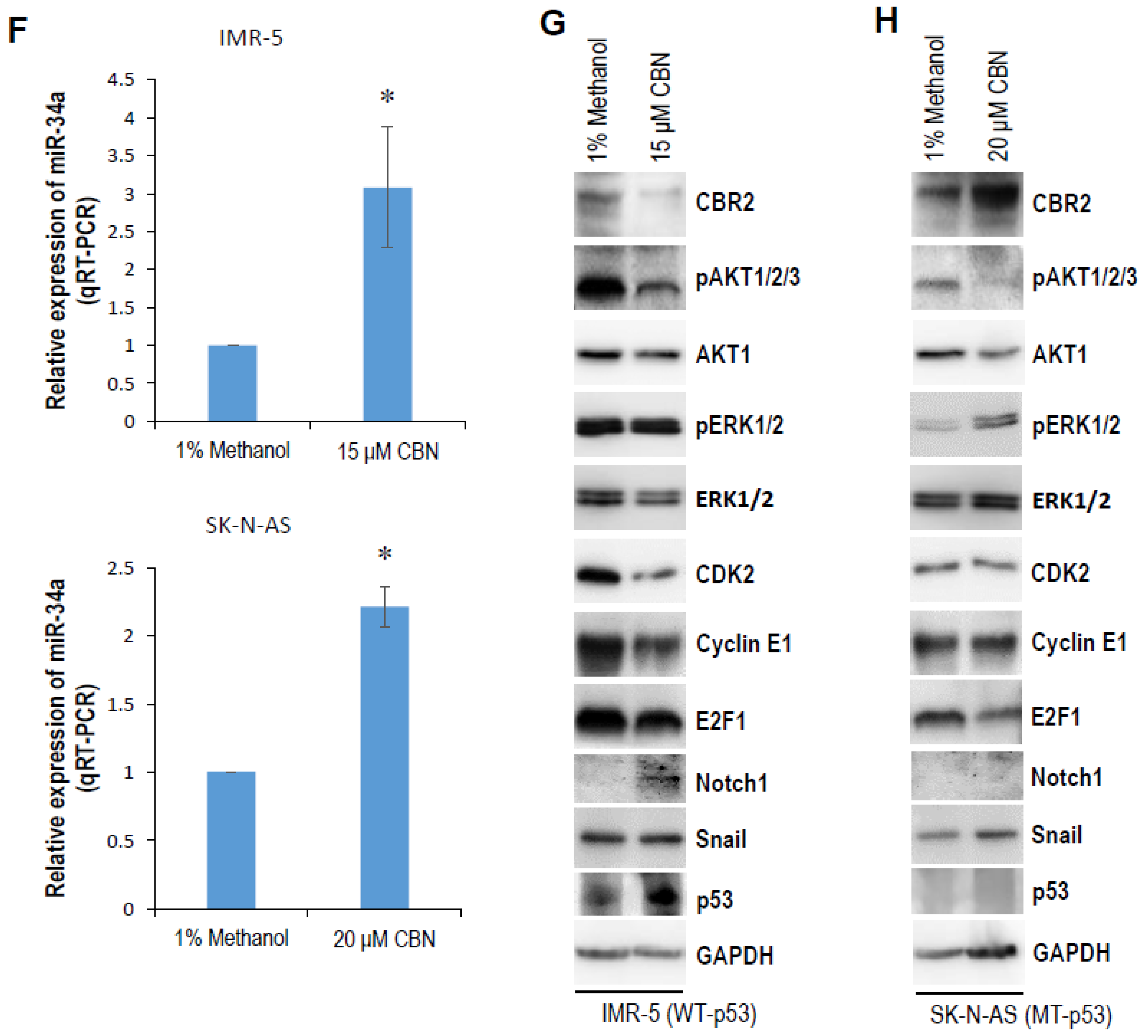
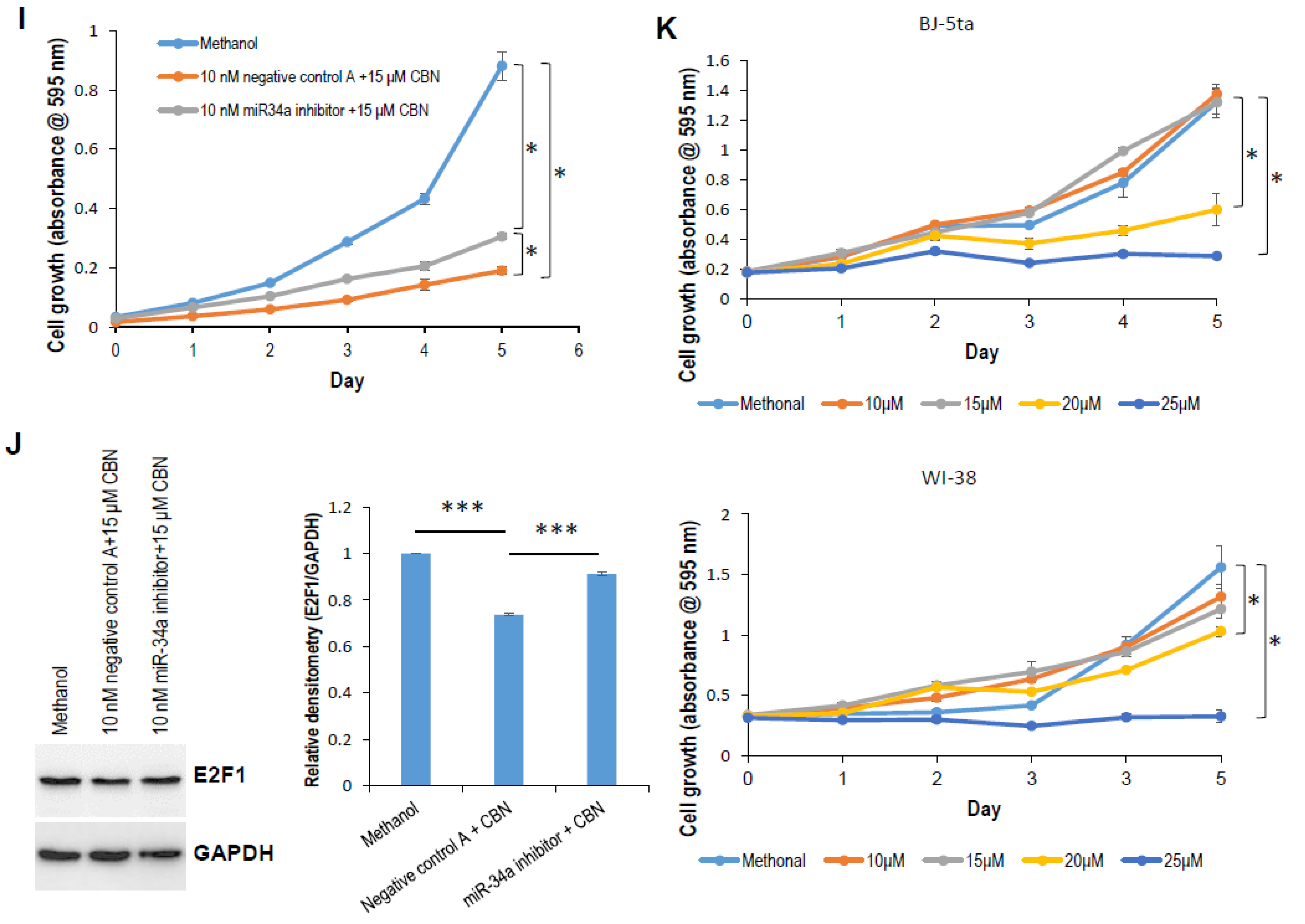
2.1. CBN Inhibits Cell Proliferation, Invasion, and Angiogenesis of Neuroblastoma via Inhibiting AKT Pathway and Upregulating miR-34a
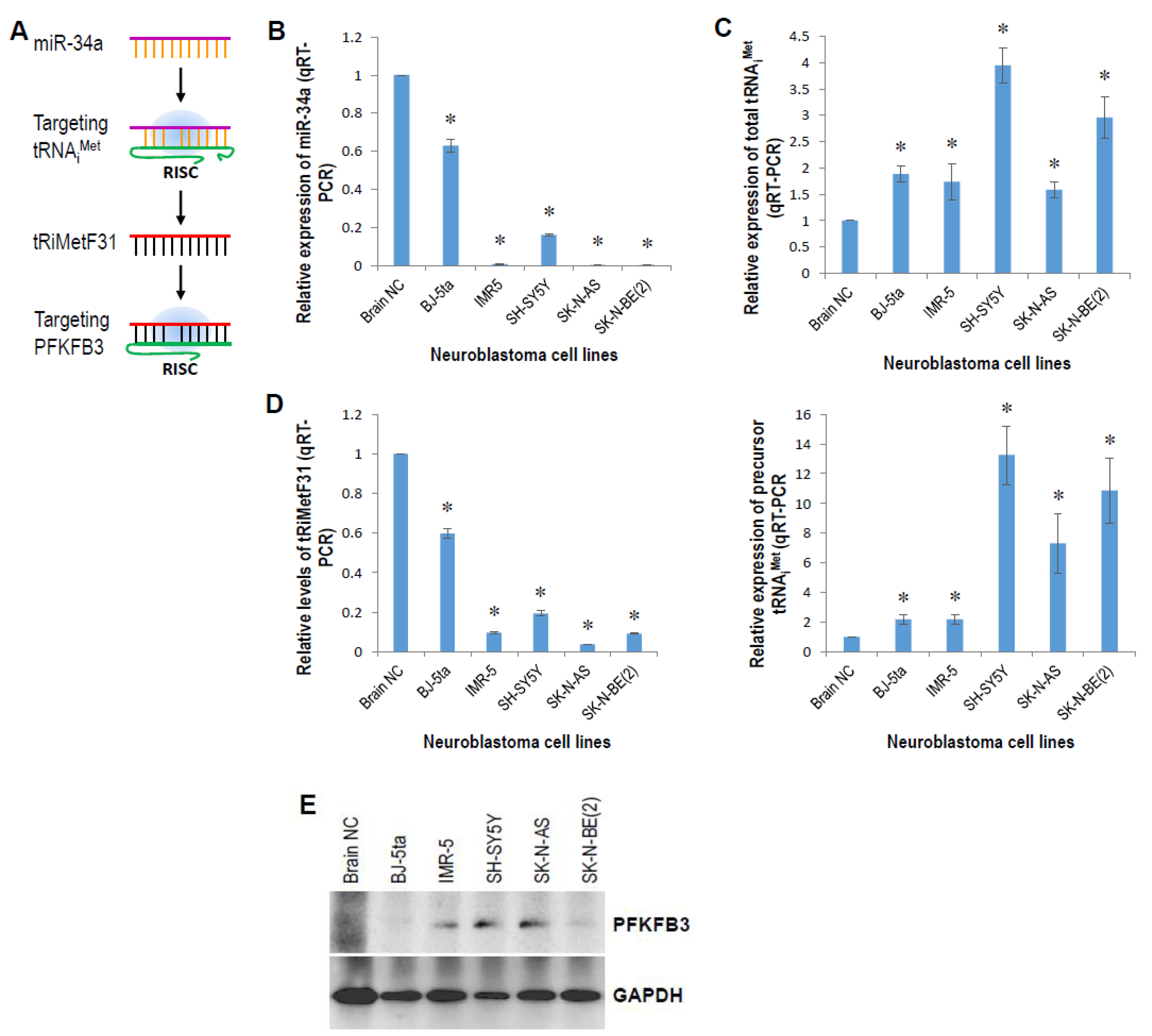
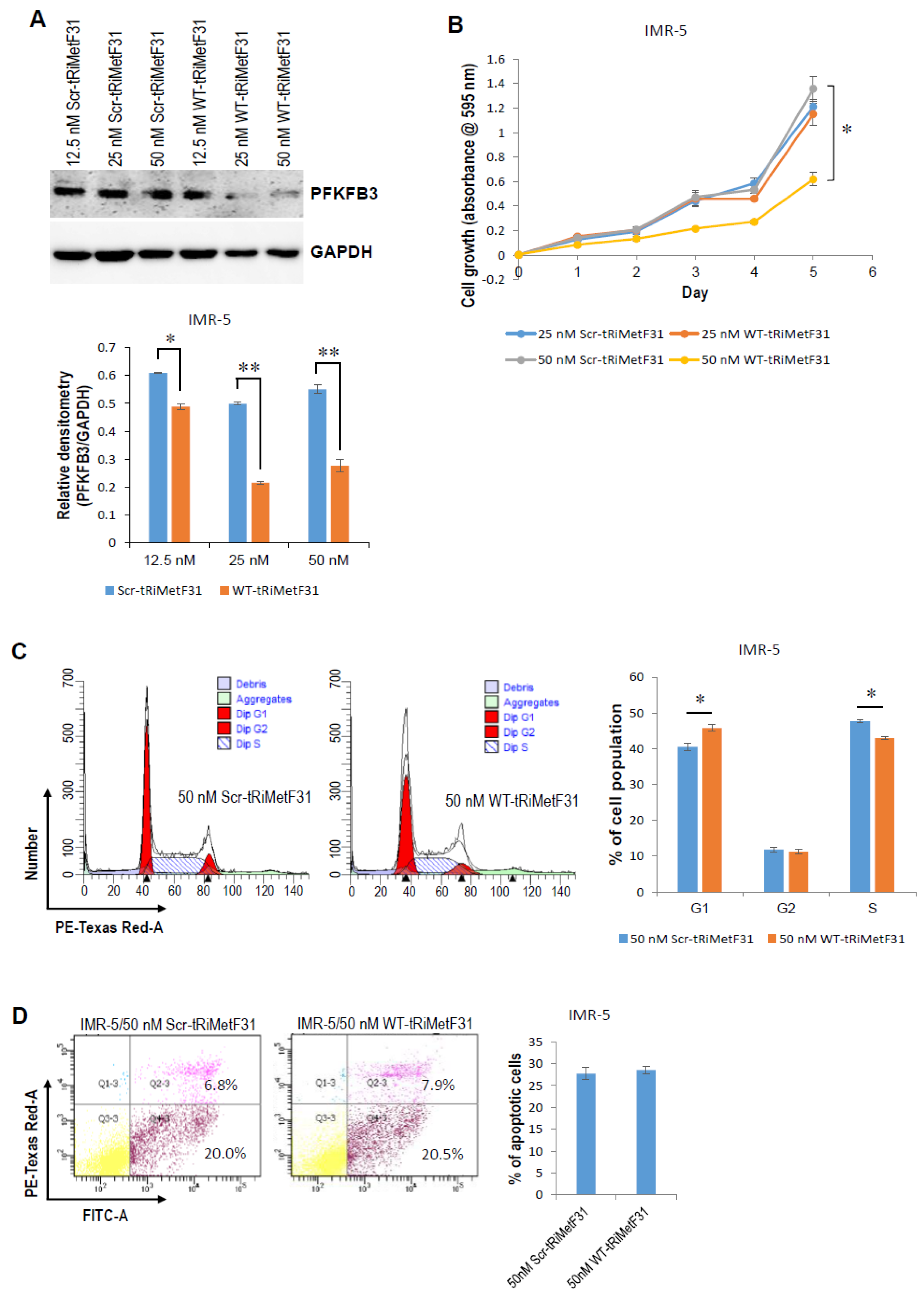
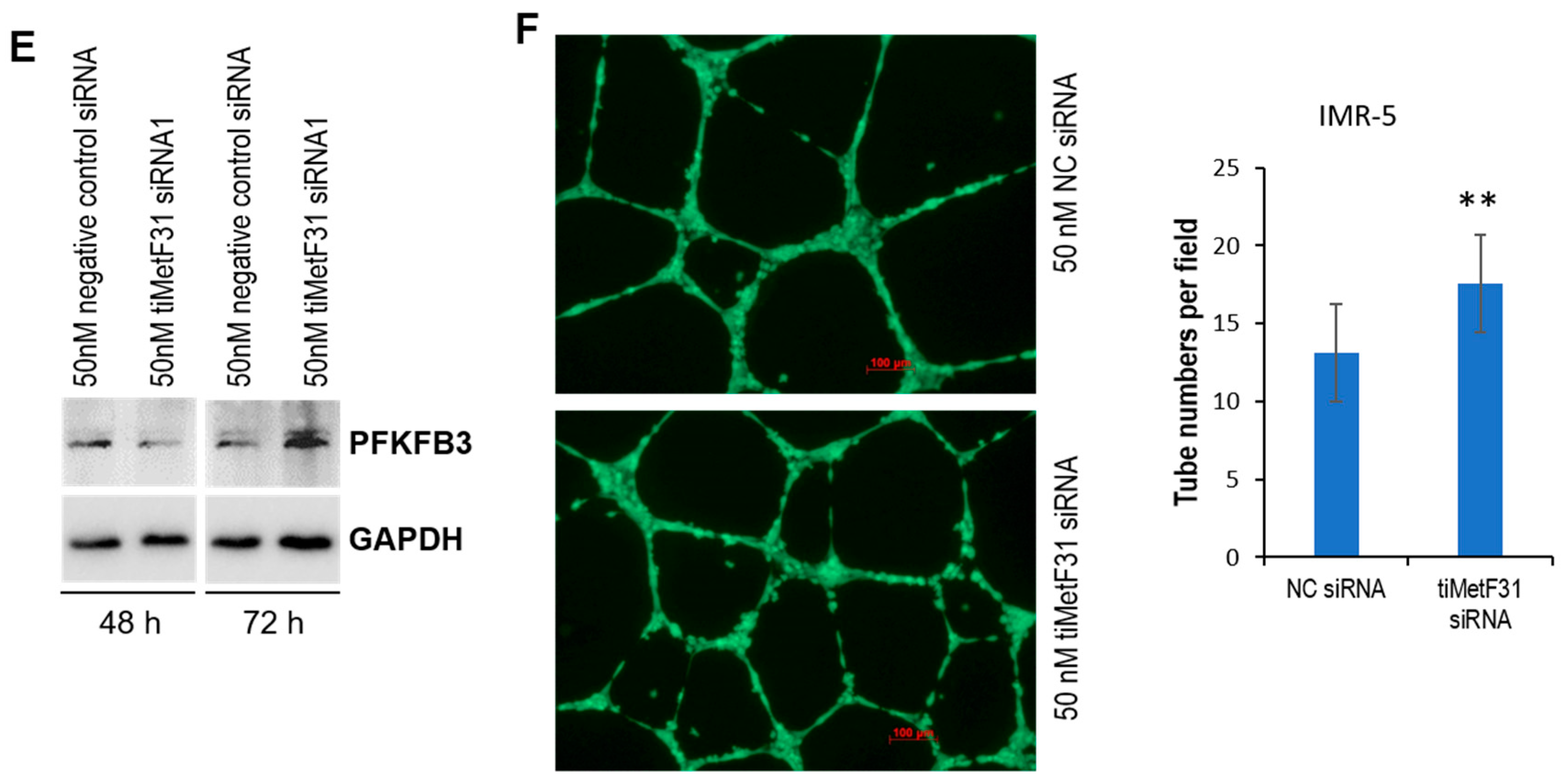
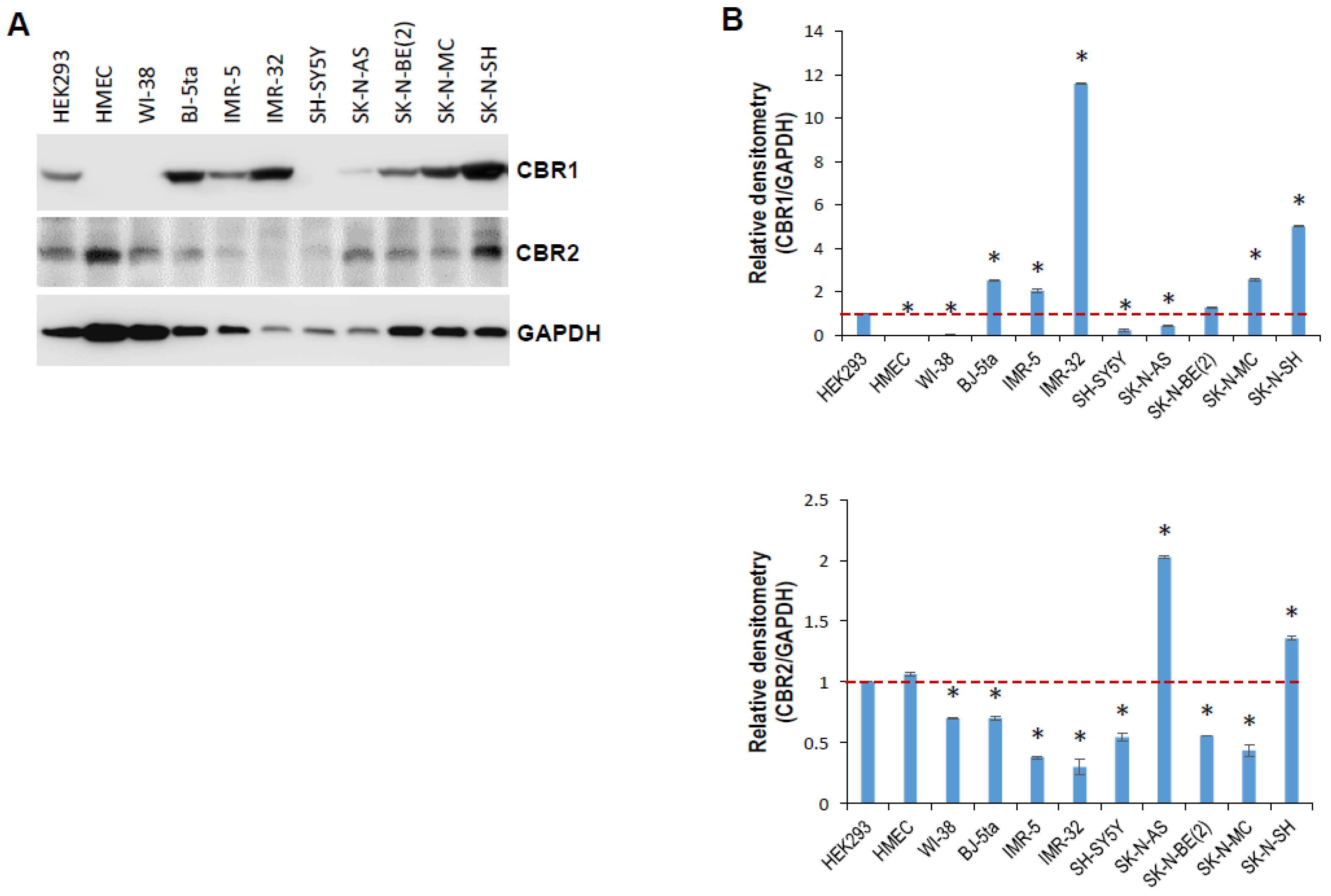
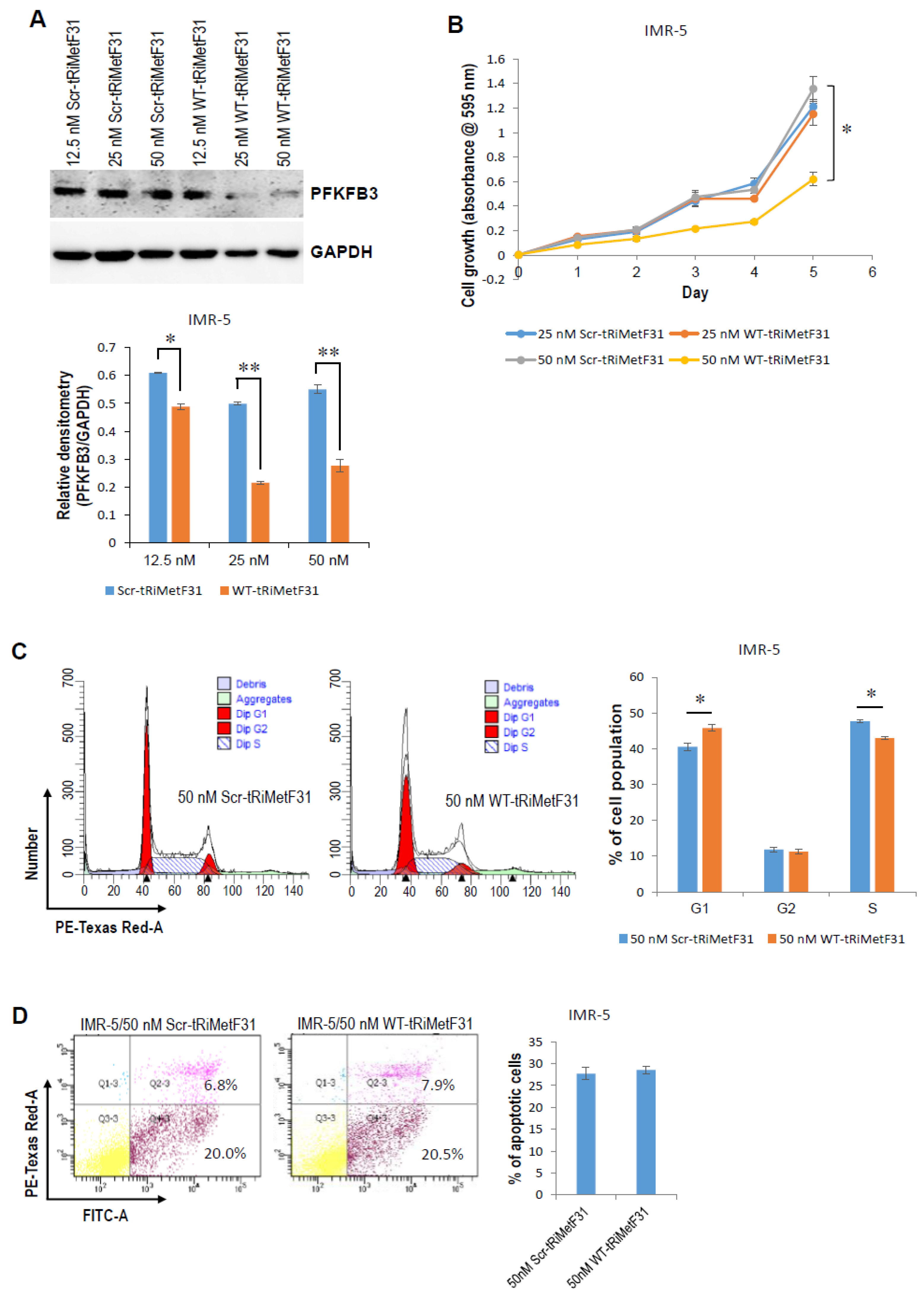
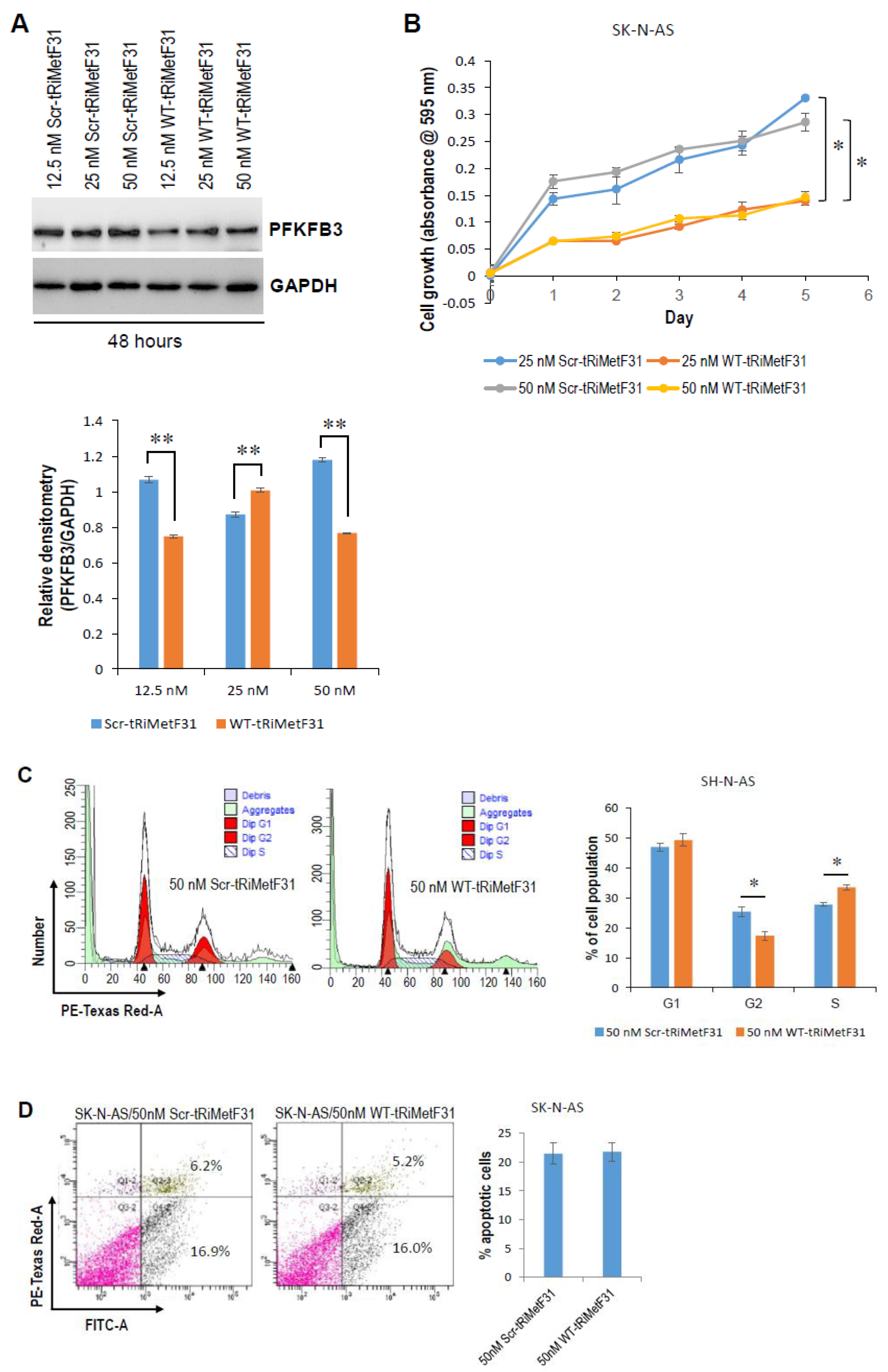
2.2. tRiMetF31 Is Downregulated, whereas Its Direct Target PFKFB3 Is Overexpressed in Neuroblastoma Cells
2.3. tRiMetF31 Inhibits Cell Proliferation and Induces Cell Cycle Arrest
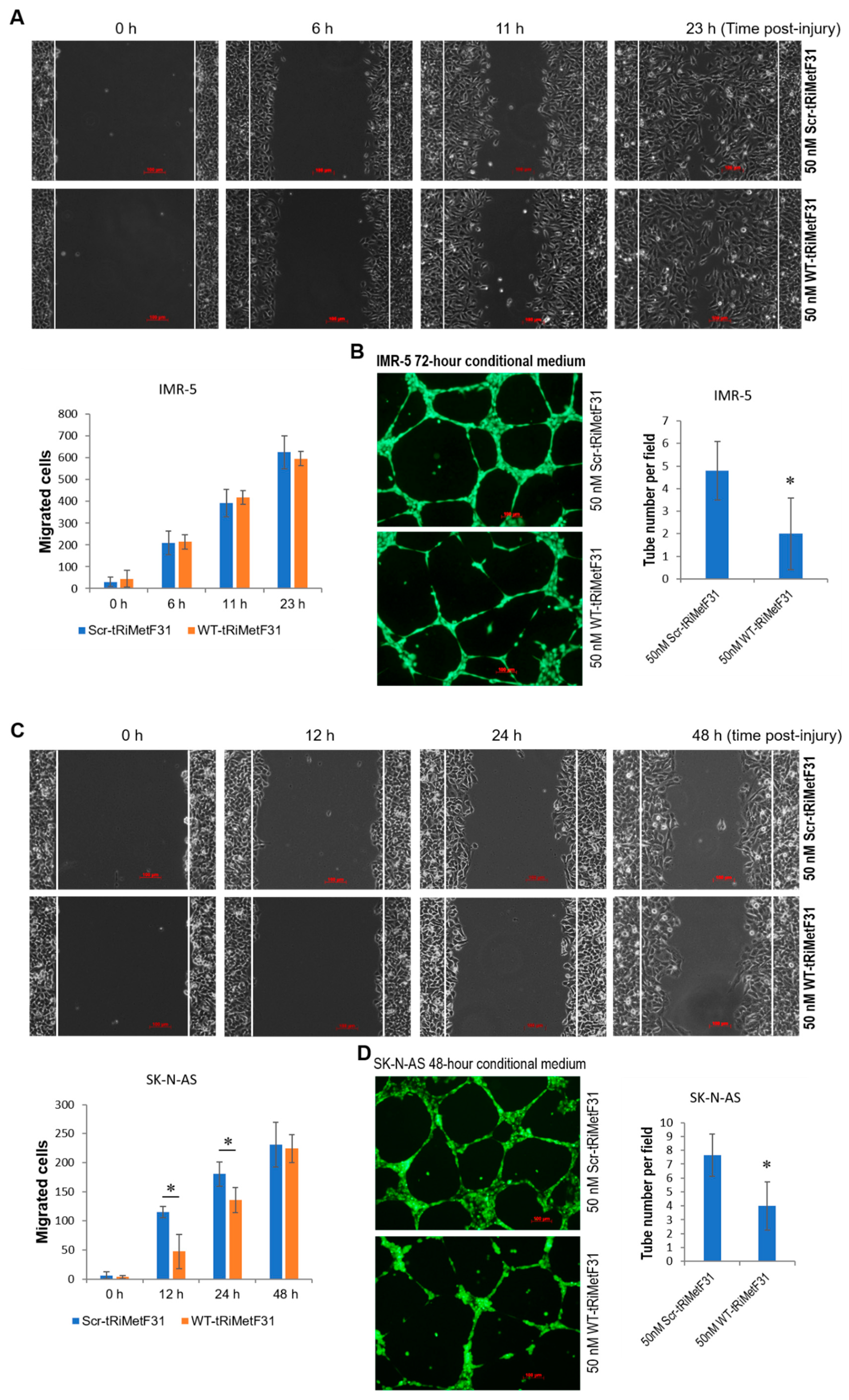
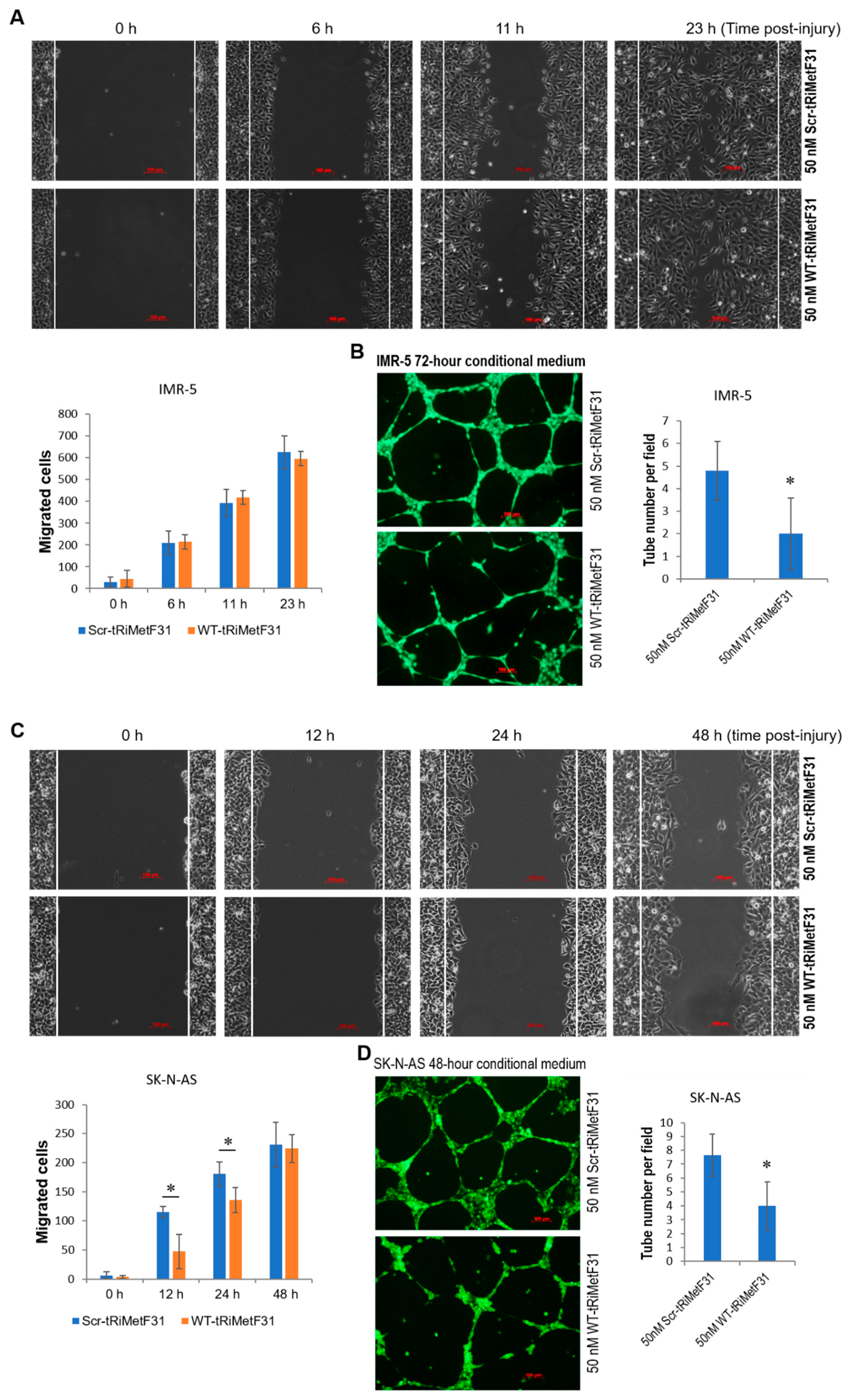
2.4. tRiMetF31 Suppresses Migration and Angiogenesis
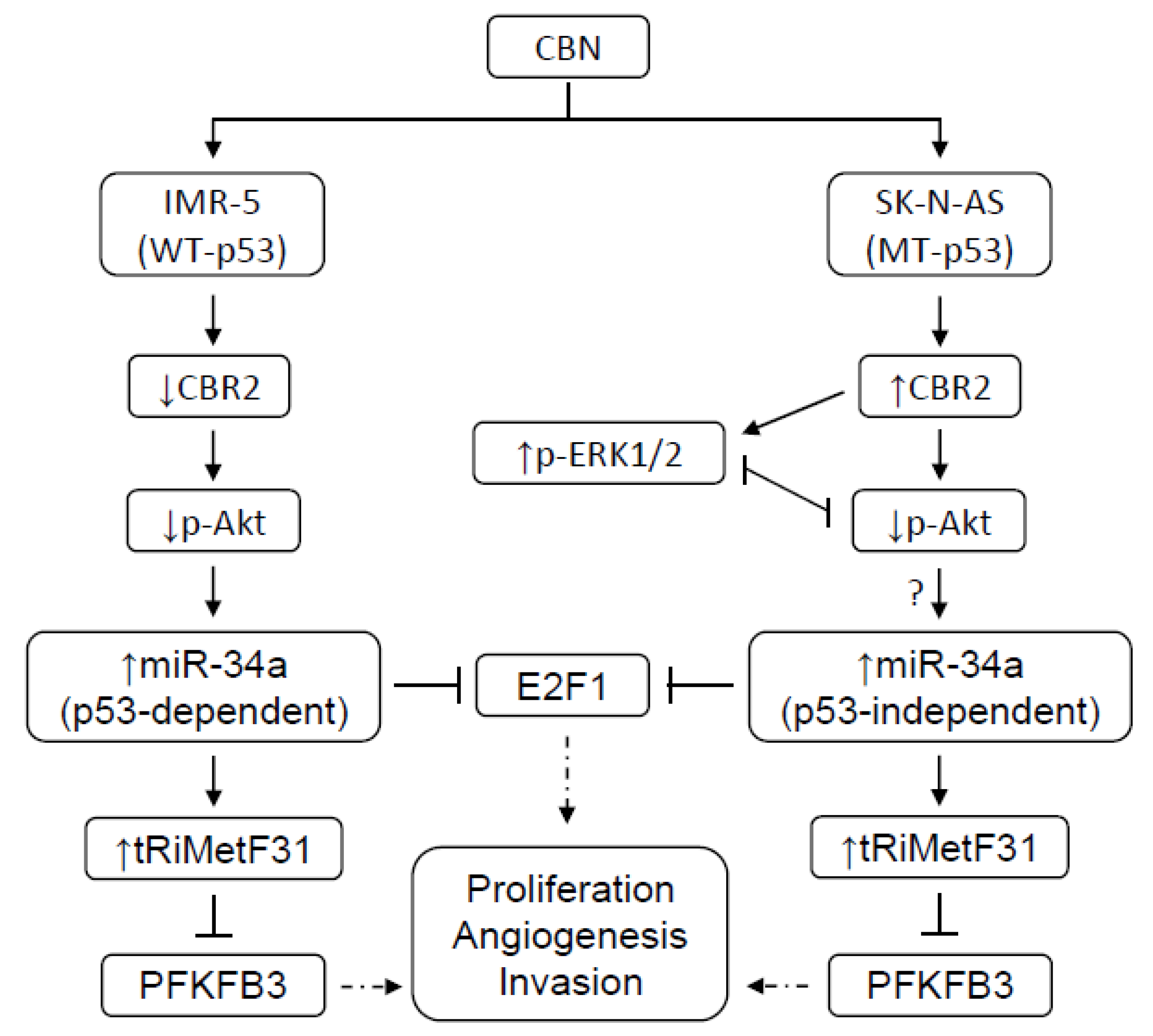
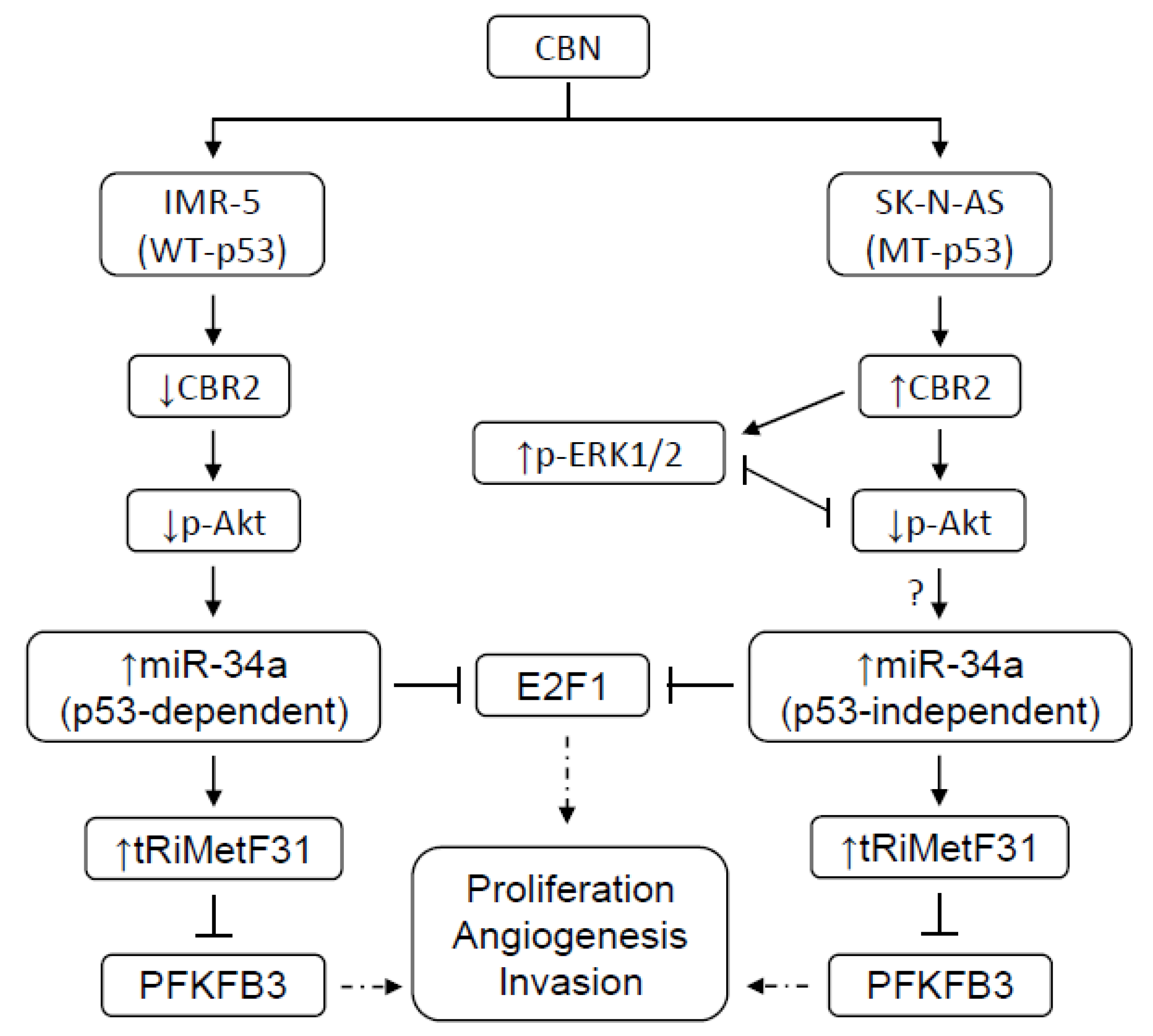
3. Discussion
4. Materials and Methods
4.1. Human Cell Lines
4.2. Western Blot Analysis
4.3. IC50 Determination
4.4. MTT Assay
4.5. Cell Cycle and Apoptosis Analyses
4.6. Tube Formation Assay
4.7. Cell Invasion Assay
4.8. Quantitative Real-Time RT-PCR (qRT-PCR)
4.9. Wound Healing Assay
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Steliarova-Foucher, E.; Colombet, M.; Ries, L.A.; Moreno, F.; Dolya, A.; Bray, F.; Tretarre, B. International incidence of childhood cancer, 2001–2010: A population-based registry study. Lancet Oncol. 2017, 18, 719–731. [Google Scholar] [CrossRef]
- Park, J.R.; Eggert, A.; Caron, H. Neuroblastoma: Biology, prognosis, and treatment. Hematol./Oncol. Clin. N. Am. 2010, 24, 65–86. [Google Scholar] [CrossRef] [PubMed]
- Davidoff, A.M. Neuroblastoma. Semin. Pediatr. Surg. 2012, 21, 2–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [Green Version]
- Cohen, L.E.; Gordon, J.H.; Popovsky, E.Y.; Gunawardene, S.; Duffey-Lind, E.; Lehmann, L.E.; Diller, L.R. Late effects in children treated with intensive multimodal therapy for high-risk neuroblastoma: High incidence of endocrine and growth problems. Bone Marrow Transplant. 2014, 49, 502–508. [Google Scholar] [CrossRef] [Green Version]
- Mosse, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Janoueix-Lerosey, I.; Lequin, D.; Brugieres, L.; Ribeiro, A.; de Pontual, L.; Combaret, V.; Raynal, V.; Puisieux, A.; Schleiermacher, G.; Pierron, G.; et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008, 455, 967–970. [Google Scholar] [CrossRef]
- Mosse, Y.P.; Laudenslager, M.; Khazi, D.; Carlisle, A.J.; Winter, C.L.; Rappaport, E.; Maris, J.M. Germline PHOX2B mutation in hereditary neuroblastoma. Am. J. Hum. Genet. 2004, 75, 727–730. [Google Scholar] [CrossRef] [Green Version]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Oehme, I.; Deubzer, H.E.; Wegener, D.; Pickert, D.; Linke, J.P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S.M.; von Deimling, A.; et al. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin. Cancer Res. 2009, 15, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Klein, S.; Abraham, M.; Bulvik, B.; Dery, E.; Weiss, I.D.; Barashi, N.; Abramovitch, R.; Wald, H.; Harel, Y.; Olam, D.; et al. CXCR4 Promotes Neuroblastoma Growth and Therapeutic Resistance through miR-15a/16-1-Mediated ERK and BCL2/Cyclin D1 Pathways. Cancer Res. 2018, 78, 1471–1483. [Google Scholar] [CrossRef] [Green Version]
- Challagundla, K.B.; Wise, P.M.; Neviani, P.; Chava, H.; Murtadha, M.; Xu, T.; Kennedy, R.; Ivan, C.; Zhang, X.; Vannini, I.; et al. Exosome-mediated transfer of microRNAs within the tumor microenvironment and neuroblastoma resistance to chemotherapy. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neviani, P.; Wise, P.M.; Murtadha, M.; Liu, C.W.; Wu, C.H.; Jong, A.Y.; Seeger, R.C.; Fabbri, M. Natural Killer-Derived Exosomal miR-186 Inhibits Neuroblastoma Growth and Immune Escape Mechanisms. Cancer Res. 2019, 79, 1151–1164. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in the Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnsen, J.I.; Segerstrom, L.; Orrego, A.; Elfman, L.; Henriksson, M.; Kagedal, B.; Eksborg, S.; Sveinbjornsson, B.; Kogner, P. Inhibitors of mammalian target of rapamycin downregulate MYCN protein expression and inhibit neuroblastoma growth in vitro and in vivo. Oncogene 2008, 27, 2910–2922. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Zhou, D.; Chen, L.; Tian, Y.; Zhong, B.; Cao, Y.; Dong, Q.; Zhou, M.; Yan, J.; Wang, Y.; et al. Polo-like kinase 4 mediates epithelial-mesenchymal transition in neuroblastoma via PI3K/Akt signaling pathway. Cell Death Dis. 2018, 9, 54. [Google Scholar] [CrossRef]
- Liu, X.H.; Yu, E.Z.; Li, Y.Y.; Rollwagen, F.M.; Kagan, E. RNA interference targeting Akt promotes apoptosis in hypoxia-exposed human neuroblastoma cells. Brain Res. 2006, 1070, 24–30. [Google Scholar] [CrossRef]
- Wickstrom, M.; Dyberg, C.; Shimokawa, T.; Milosevic, J.; Baryawno, N.; Fuskevag, O.M.; Larsson, R.; Kogner, P.; Zaphiropoulos, P.G.; Johnsen, J.I. Targeting the hedgehog signal transduction pathway at the level of GLI inhibits neuroblastoma cell growth in vitro and in vivo. Int. J. Cancer 2013, 132, 1516–1524. [Google Scholar] [CrossRef]
- Hewson, Q.D.; Lovat, P.E.; Corazzari, M.; Catterall, J.B.; Redfern, C.P. The NF-kappaB pathway mediates fenretinide-induced apoptosis in SH-SY5Y neuroblastoma cells. Apoptosis 2005, 10, 493–498. [Google Scholar] [CrossRef]
- Loschmann, N.; Michaelis, M.; Rothweiler, F.; Zehner, R.; Cinatl, J.; Voges, Y.; Sharifi, M.; Riecken, K.; Meyer, J.; von Deimling, A.; et al. Testing of SNS-032 in a Panel of Human Neuroblastoma Cell Lines with Acquired Resistance to a Broad Range of Drugs. Transl. Oncol. 2013, 6, 685–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovalchuk, O.; Kovalchuk, I. Cannabinoids as anticancer therapeutic agents. Cell Cycle 2020, 19, 961–989. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.; Hurst, D.P.; Reggio, P.H. Molecular Targets of the Phytocannabinoids: A Complex Picture. Prog. Chem. Org. Nat. Prod. 2017, 103, 103–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zurier, R.B.; Burstein, S.H. Cannabinoids, inflammation, and fibrosis. FASEB J. 2016, 30, 3682–3689. [Google Scholar] [CrossRef] [Green Version]
- Juknat, A.; Gao, F.; Coppola, G.; Vogel, Z.; Kozela, E. miRNA expression profiles and molecular networks in resting and LPS-activated BV-2 microglia-Effect of cannabinoids. PLoS ONE 2019, 14, e0212039. [Google Scholar] [CrossRef]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microRNA component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Li, D.; Kovalchuk, I.; Apel, I.J.; Chinnaiyan, A.M.; Woycicki, R.K.; Cantor, C.R.; Kovalchuk, O. miR-34a directly targets tRNAi(Met) precursors and affects cellular proliferation, cell cycle, and apoptosis. Proc. Natl. Acad. Sci. USA 2018, 115, 7392–7397. [Google Scholar] [CrossRef] [Green Version]
- Munson, A.E.; Harris, L.S.; Friedman, M.A.; Dewey, W.L.; Carchman, R.A. Antineoplastic activity of cannabinoids. J. Natl. Cancer Inst. 1975, 55, 597–602. [Google Scholar] [CrossRef]
- Granja, A.G.; Carrillo-Salinas, F.; Pagani, A.; Gomez-Canas, M.; Negri, R.; Navarrete, C.; Mecha, M.; Mestre, L.; Fiebich, B.L.; Cantarero, I.; et al. A cannabigerol quinone alleviates neuroinflammation in a chronic model of multiple sclerosis. J. Neuroimmune Pharm. 2012, 7, 1002–1016. [Google Scholar] [CrossRef]
- Upham, B.L.; Rummel, A.M.; Carbone, J.M.; Trosko, J.E.; Ouyang, Y.; Crawford, R.B.; Kaminski, N.E. Cannabinoids inhibit gap junctional intercellular communication and activate ERK in a rat liver epithelial cell line. Int. J. Cancer 2003, 104, 12–18. [Google Scholar] [CrossRef]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef]
- Udoh, M.; Santiago, M.; Devenish, S.; McGregor, I.S.; Connor, M. Cannabichromene is a cannabinoid CB2 receptor agonist. Br. J. Pharm. 2019, 176, 4537–4547. [Google Scholar] [CrossRef] [PubMed]
- Rosenthaler, S.; Pohn, B.; Kolmanz, C.; Huu, C.N.; Krewenka, C.; Huber, A.; Kranner, B.; Rausch, W.D.; Moldzio, R. Differences in receptor binding affinity of several phytocannabinoids do not explain their effects on neural cell cultures. Neurotoxicol. Teratol. 2014, 46, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Pisanti, S.; Picardi, P.; D’Alessandro, A.; Laezza, C.; Bifulco, M. The endocannabinoid signaling system in cancer. Trends Pharmacol. Sci. 2013, 34, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Arkun, Y. Dynamic Modeling and Analysis of the Cross-Talk between Insulin/AKT and MAPK/ERK Signaling Pathways. PLoS ONE 2016, 11, e0149684. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Mo, Y.Y. The Akt-associated microRNAs. Cell Mol. Life Sci. 2012, 69, 3601–3612. [Google Scholar] [CrossRef] [Green Version]
- Hermeking, H. MicroRNAs in the p53 network: Micromanagement of tumour suppression. Nat. Rev. Cancer 2012, 12, 613–626. [Google Scholar] [CrossRef]
- Li, X.J.; Ren, Z.J.; Tang, J.H. MicroRNA-34a: A potential therapeutic target in human cancer. Cell Death Dis. 2014, 5, e1327. [Google Scholar] [CrossRef]
- Nalls, D.; Tang, S.N.; Rodova, M.; Srivastava, R.K.; Shankar, S. Targeting epigenetic regulation of miR-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PLoS ONE 2011, 6, e24099. [Google Scholar] [CrossRef]
- Han, Z.; Zhang, Y.; Yang, Q.; Liu, B.; Wu, J.; Zhang, Y.; Yang, C.; Jiang, Y. miR-497 and miR-34a retard lung cancer growth by co-inhibiting cyclin E1 (CCNE1). Oncotarget 2015, 6, 13149–13163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christoffersen, N.R.; Shalgi, R.; Frankel, L.B.; Leucci, E.; Lees, M.; Klausen, M.; Pilpel, Y.; Nielsen, F.C.; Oren, M.; Lund, A.H. p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ. 2010, 17, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.; Kovacs, M.; Chellappan, S. Regulation of vascular endothelial growth factor receptors by Rb and E2F1: Role of acetylation. Cancer Res. 2010, 70, 4931–4940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollern, D.P.; Honeysett, J.; Cardiff, R.D.; Andrechek, E.R. The E2F transcription factors regulate tumor development and metastasis in a mouse model of metastatic breast cancer. Mol. Cell Biol. 2014, 34, 3229–3243. [Google Scholar] [CrossRef] [Green Version]
- Cole, K.A.; Attiyeh, E.F.; Mosse, Y.P.; Laquaglia, M.J.; Diskin, S.J.; Brodeur, G.M.; Maris, J.M. A functional screen identifies miR-34a as a candidate neuroblastoma tumor suppressor gene. Mol. Cancer Res. 2008, 6, 735–742. [Google Scholar] [CrossRef] [Green Version]
- Di Paolo, D.; Pastorino, F.; Brignole, C.; Corrias, M.V.; Emionite, L.; Cilli, M.; Tamma, R.; Priddy, L.; Amaro, A.; Ferrari, D.; et al. Combined Replenishment of miR-34a and let-7b by Targeted Nanoparticles Inhibits Tumor Growth in Neuroblastoma Preclinical Models. Small 2020, 16, e1906426. [Google Scholar] [CrossRef]
- Eichmann, A.; Simons, M. Need glucose to sprout: Local metabolic control of angiogenesis. EMBO Mol. Med. 2013, 5, 1459–1461. [Google Scholar] [CrossRef]
- Espinoza, I.; Miele, L. Notch inhibitors for cancer treatment. Pharm. Ther. 2013, 139, 95–110. [Google Scholar] [CrossRef] [Green Version]
- Kamba, T.; McDonald, D.M. Mechanisms of adverse effects of anti-VEGF therapy for cancer. Br. J. Cancer 2007, 96, 1788–1795. [Google Scholar] [CrossRef]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquiere, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [Green Version]
- O’Neal, J.; Clem, A.; Reynolds, L.; Dougherty, S.; Imbert-Fernandez, Y.; Telang, S.; Chesney, J.; Clem, B.F. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) suppresses glucose metabolism and the growth of HER2+ breast cancer. Breast Cancer Res. Treat. 2016, 160, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Li, H.M.; Yang, J.G.; Liu, Z.J.; Wang, W.M.; Yu, Z.L.; Ren, J.G.; Chen, G.; Zhang, W.; Jia, J. Blockage of glycolysis by targeting PFKFB3 suppresses tumor growth and metastasis in head and neck squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2017, 36, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, K.; Noda, T.; Kobayashi, S.; Sakano, Y.; Yokota, Y.; Iwagami, Y.; Yamada, D.; Tomimaru, Y.; Akita, H.; Gotoh, K.; et al. Inhibition of glycolytic activator PFKFB3 suppresses tumor growth and induces tumor vessel normalization in hepatocellular carcinoma. Cancer Lett. 2021, 500, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Bando, H.; Atsumi, T.; Nishio, T.; Niwa, H.; Mishima, S.; Shimizu, C.; Yoshioka, N.; Bucala, R.; Koike, T. Phosphorylation of the 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase/PFKFB3 family of glycolytic regulators in human cancer. Clin. Cancer Res. 2005, 11, 5784–5792. [Google Scholar] [CrossRef] [Green Version]
- Trojan, S.E.; Markiewicz, M.J.; Leskiewicz, K.; Kocemba-Pilarczyk, K.A. The influence of PFK-II overexpression on neuroblastoma patients’ survival may be dependent on the particular isoenzyme expressed, PFKFB3 or PFKFB4. Cancer Cell Int. 2019, 19, 292. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Qi, X.M.; Wertz, R.; Mortensen, M.; Hagen, C.; Evans, J.; Sheinin, Y.; James, M.; Liu, P.; Tsai, S.; et al. p38gamma MAPK Is Essential for Aerobic Glycolysis and Pancreatic Tumorigenesis. Cancer Res. 2020, 80, 3251–3264. [Google Scholar] [CrossRef]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef]
- Fischer, M.; Berthold, F. Characterization of the gene expression profile of neuroblastoma cell line IMR-5 using serial analysis of gene expression. Cancer Lett. 2003, 190, 79–87. [Google Scholar] [CrossRef]
- Tsutsumimoto, T.; Williams, P.; Yoneda, T. The SK-N-AS human neuroblastoma cell line develops osteolytic bone metastases with increased angiogenesis and COX-2 expression. J. Bone Oncol. 2014, 3, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Feng, C.; Zhou, L.Y.; Yu, T.; Xu, G.; Tian, H.L.; Xu, J.J.; Xu, H.X.; Luo, K.Q. A new anticancer compound, oblongifolin C, inhibits tumor growth and promotes apoptosis in HeLa cells through Bax activation. Int. J. Cancer 2012, 131, 1445–1454. [Google Scholar] [CrossRef] [Green Version]
- Ro, S.; Park, C.; Jin, J.; Sanders, K.M.; Yan, W. A PCR-based method for detection and quantification of small RNAs. Biochem. Biophys. Res. Commun. 2006, 351, 756–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, T.; Cho, S.G.; Yi, Z.; Pang, X.; Rodriguez, M.; Wang, Y.; Sethi, G.; Aggarwal, B.B.; Liu, M. Thymoquinone inhibits tumor angiogenesis and tumor growth through suppressing AKT and extracellular signal-regulated kinase signaling pathways. Mol. Cancer Ther. 2008, 7, 1789–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, B.; Li, D.; Cherkasova, V.; Gerasymchuk, M.; Narendran, A.; Kovalchuk, I.; Kovalchuk, O. Cannabinol Inhibits Cellular Proliferation, Invasion, and Angiogenesis of Neuroblastoma via Novel miR-34a/tRiMetF31/PFKFB3 Axis. Cancers 2022, 14, 1908. https://doi.org/10.3390/cancers14081908
Wang B, Li D, Cherkasova V, Gerasymchuk M, Narendran A, Kovalchuk I, Kovalchuk O. Cannabinol Inhibits Cellular Proliferation, Invasion, and Angiogenesis of Neuroblastoma via Novel miR-34a/tRiMetF31/PFKFB3 Axis. Cancers. 2022; 14(8):1908. https://doi.org/10.3390/cancers14081908
Chicago/Turabian StyleWang, Bo, Dongping Li, Viktoriia Cherkasova, Marta Gerasymchuk, Aru Narendran, Igor Kovalchuk, and Olga Kovalchuk. 2022. "Cannabinol Inhibits Cellular Proliferation, Invasion, and Angiogenesis of Neuroblastoma via Novel miR-34a/tRiMetF31/PFKFB3 Axis" Cancers 14, no. 8: 1908. https://doi.org/10.3390/cancers14081908
APA StyleWang, B., Li, D., Cherkasova, V., Gerasymchuk, M., Narendran, A., Kovalchuk, I., & Kovalchuk, O. (2022). Cannabinol Inhibits Cellular Proliferation, Invasion, and Angiogenesis of Neuroblastoma via Novel miR-34a/tRiMetF31/PFKFB3 Axis. Cancers, 14(8), 1908. https://doi.org/10.3390/cancers14081908