
Deciphering HER2-HER3 Dimerization at the Single CTC Level: A Microfluidic Approach

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Microfluidic Device Fabrication
2.2. Temperature Control in the System
2.3. Cell Culture
2.4. Gene Knockdown
2.5. RNA Extractions and Reverse Transcription
2.6. Real Time-Quantitative PCR
2.7. Patient Recruitment and Blood Sample Processing
2.8. In-Situ Indirect and Direct Proximity Ligation Assay on Glass Slides
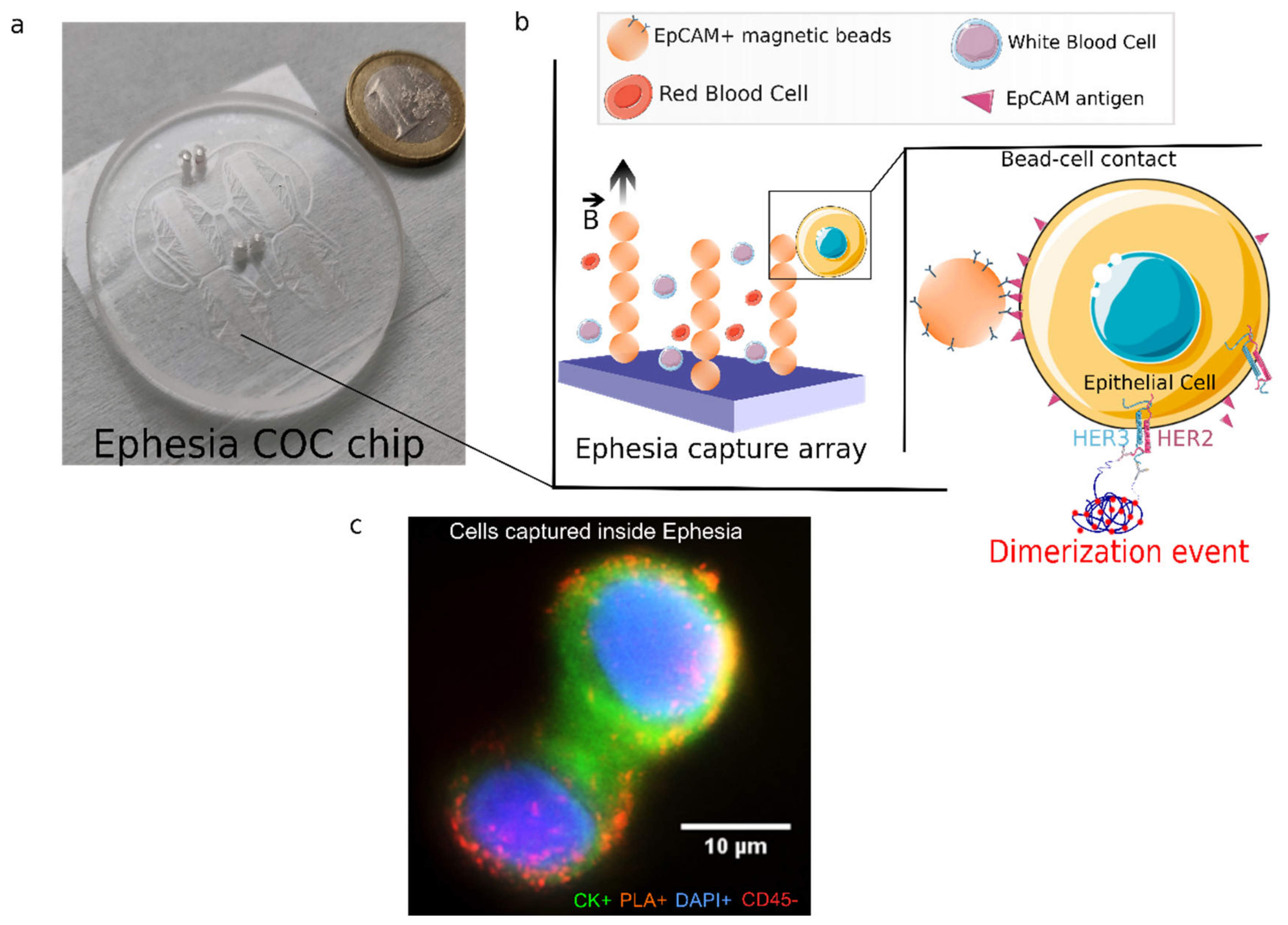
2.9. In-Situ Proximity Ligation Assay in Ephesia Microfluidic Device
2.10. Imaging and Signal Quantification
3. Results
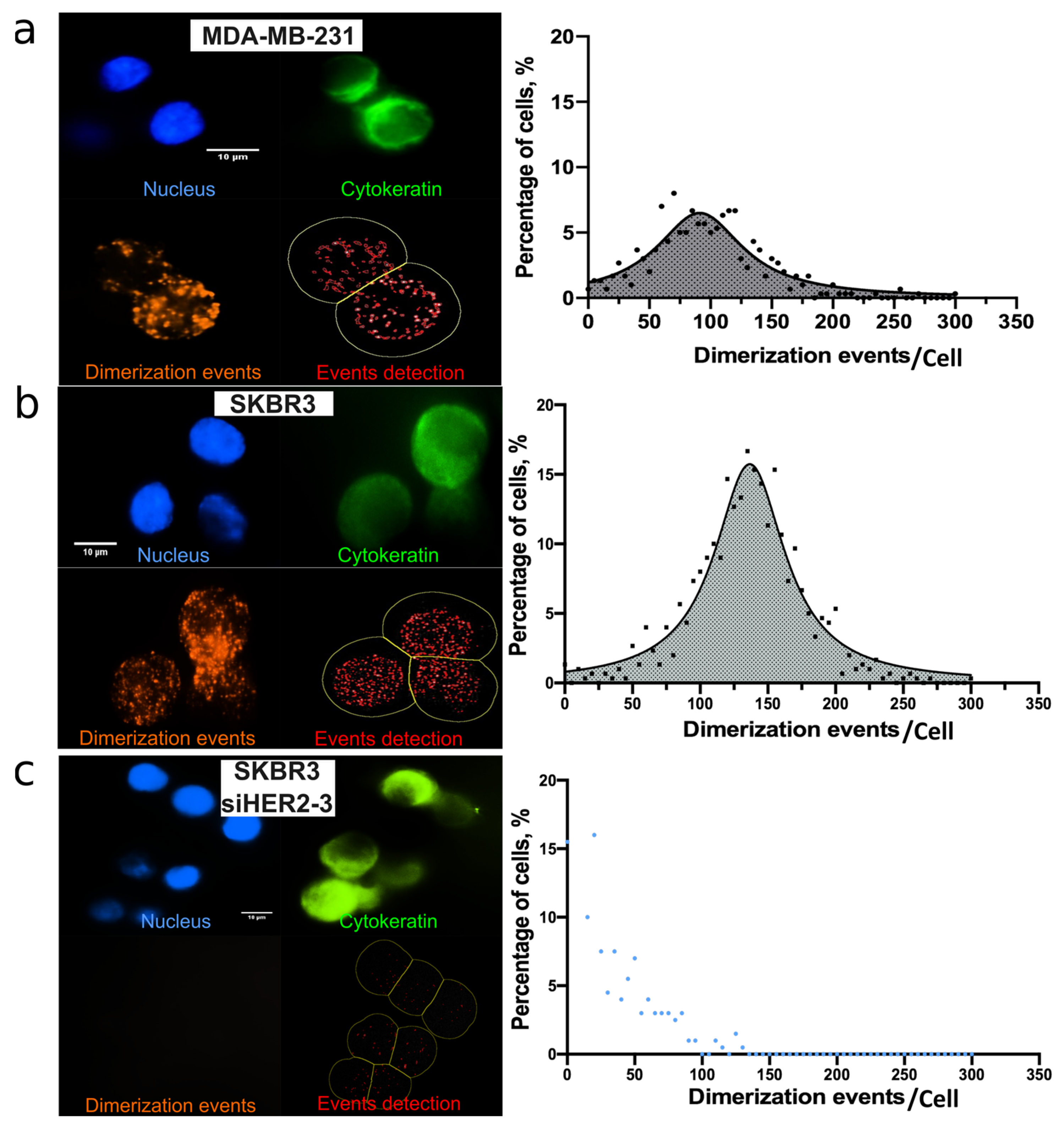
3.1. HER2-HER3 PLA Optimization on Coverslip
3.2. HER2-HER3 Dimerization of Single CTC: On-Chip PLA Integration
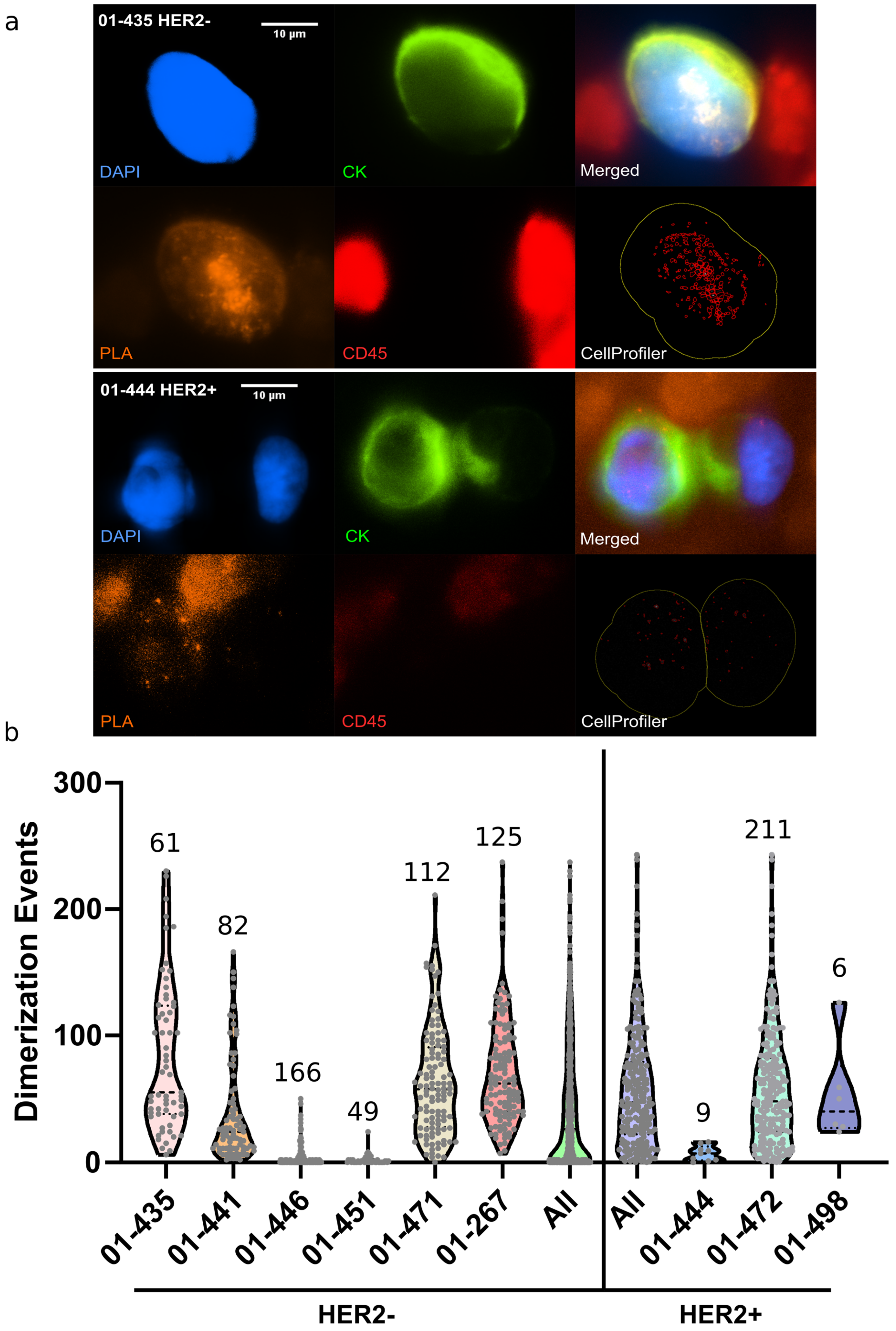
3.3. On-Chip Single CTC PLA Analysis from Metastatic Breast Cancer Patients’ Blood
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Goutsouliak, K.; Veeraraghavan, J.; Sethunath, V.; De Angelis, C.; Osborne, C.K.; Rimawi, M.F.; Schiff, R. Towards personalized treatment for early stage HER2-positive breast cancer. Nat. Rev. Clin. Oncol. 2019, 17, 233–250. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.C.; Hammond, M.E.H.; Hicks, D.G.; Dowsett, M.; McShane, L.M.; Allison, K.H.; Allred, D.C.; Bartlett, J.M.; Bilous, M.; Fitzgibbons, P.; et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J. Clin. Oncol. 2013, 31, 3997–4013. [Google Scholar] [CrossRef] [PubMed]
- Yamashita-Kashima, Y.; Shu, S.; Yorozu, K.; Moriya, Y.; Harada, N. Mode of action of pertuzumab in combination with trastuzumab plus docetaxel therapy in a HER2-positive breast cancer xenograft model. Oncol. Lett. 2017, 14, 4197–4205. [Google Scholar] [CrossRef] [Green Version]
- Sawyers, C.L. Herceptin: A fist assault on oncongenes that launched a revolution. Cell 2019, 197, 8–12. [Google Scholar] [CrossRef] [Green Version]
- Gala, K.; Chandarlapaty, S. Molecular pathways: HER3 targeted therapy. Clin. Cancer Res. 2014, 20, 1410–1416. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Huo, X.; Wang, M.; Liu, Z.; Zhao, Y.; Ren, D.; Xie, Q.; Liu, Z.; Li, Z.; Du, F.; et al. Comparing Biomarkers for Predicting Pathological Responses to Neoadjuvant Therapy in HER2-Positive Breast Cancer: A Systematic Review and Meta-Analysis. Front. Oncol. 2021, 11, 731148. [Google Scholar] [CrossRef]
- Weitsman, G.; Barber, P.R.; Nguyen, L.K.; Lawler, K.; Patel, G.; Woodman, N.; Kelleher, M.T.; Pinder, S.E.; Rowley, M.; Ellis, P.A.; et al. HER2-HER3 dimer quantification by FLIM-FRET predicts breast cancer metastatic relapse independently of HER2 IHC status. Oncotarget 2016, 7, 51012–51026. [Google Scholar] [CrossRef] [Green Version]
- Green, A.R.; Barros, F.F.; Abdel-Fatah, T.; Moseley, P.; Nolan, C.C.; Durham, A.C.; Rakha, E.A.; Chan, S.; Ellis, I.O. HER2/HER3 heterodimers and p21 expression are capable of predicting adjuvant trastuzumab response in HER2+ breast cancer. Breast Cancer Res. Treat. 2014, 145, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Mattox, A.K.; Bettegowda, C.; Zhou, S.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Applications of liquid biopsies for cancer. Sci. Transl. Med. 2019, 11, eaay1984. [Google Scholar] [CrossRef] [Green Version]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Revm Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Eslami-S, Z.; Cortés-Hernández, L.E.; Cayrefourcq, L.; Alix-Panabières, C. The different facets of liquid biopsy: A kaleidoscopic view. Cold Spring Harbor Persp. Med. 2019, 12, a037333. [Google Scholar] [CrossRef] [PubMed]
- Dianat-Moghadam, H.; Azizi, M.; Eslami-S, Z.; Cortés-Hernández, L.E.; Heidarifard, M.; Nouri, M.; Alix-Panabières, C. The role of circulating tumor cells in the metastatic cascade: Biology, technical challenges, and clinical relevance. Cancers 2020, 12, 867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloten, V.; Lampignano, R.; Krahn, T.; Schlange, T. Circulating Tumor Cell PD-L1 Expression as Biomarker for Therapeutic Efficacy of Immune Checkpoint Inhibition in NSCLC. Cells 2019, 8, 809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasseur, A.; Kiavue, N.; Bidard, F.C.; Pierga, J.Y.; Cabel, L. Clinical utility of circulating tumor cells: An update. Mol. Oncol. 2021, 15, 1647–1666. [Google Scholar] [CrossRef]
- Bidard, F.C.; Proudhon, C.; Pierga, J.Y. Circulating tumor cells in breast cancer. Mol. Oncol. 2016, 10, 418–430. [Google Scholar] [CrossRef] [Green Version]
- Saias, L.; Autebert, J.; Malaquin, L.; Viovy, J.L. Desig, modeling and characterization of microfluidic architecture for high flow rate, small footprint microfluidic systems. Lab Chip 2011, 11, 822–832. [Google Scholar] [CrossRef]
- Autebert, J.; Coudert, B.; Champ, J.; Saias, L.; Tulukcuoglu Guneri, E.; Lebofsky, R.; Bidard, F.C.; Pierga, J.Y.; Farace, F.; Descroix, S.; et al. High Purity Microfluidic Sorting and Analysis of Circulating Tumor Cells: Towards Routine Mutations Detection. Lab Chip 2015, 15, 2090–2101. [Google Scholar] [CrossRef]
- Delapierre, F.-D.; Mottet, G.; Taniga, V.; Boisselier, J.; Viovy, J.; Malaquin, L. High Throughput Micropatterning of Interspersed Cell Arrays Using Capillary Assembly. Biofabrication 2017, 9, 015015. [Google Scholar] [CrossRef]
- Kraus, T.; Malaquin, L.; Schmid, H.; Riess, W.; Spencer, N.D.; Wolf, H. Nanoparticle Printing with Single-Particle Resolution. Nat. Nanotechnol. 2007, 2, 570–576. [Google Scholar] [CrossRef]
- Söderberg, O.; Leuchowius, K.J.; Gullberg, M.; Jarvius, M.; Weibrecht, I.; Larsson, L.G.; Landegren, U. Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods 2008, 45, 227–232. [Google Scholar] [PubMed]
- Fredriksson, S.; Gullberg, M.; Jarvius, J.; Olsson, C.; Pietras, K.; Gústafsdóttir, S.M.; Östman, A.; Landegren, U. Protein detection using proximity-dependent DNA ligation assays. Nat. Biotechnol. 2002, 20, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Subik, K.; Lee, J.F.; Baxter, L.; Strzepek, T.; Costello, D.; Crowley, P.; Xing, L.; Hung, M.C.; Bonfiglio, T.; Hicks, D.G.; et al. The Expression Patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by Immunohistochemical Analysis in Breast Cancer Cell Lines. Breast Cancer 2010, 4, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Miserere, S.; Mottet, G.; Taniga, V.; Descroix, S.; Viovy, J.-L.; Malaquin, L. Fabrication of Thermoplastics Chips through Lamination Based Techniques. Lab Chip 2012, 12, 1849–1856. [Google Scholar] [CrossRef] [PubMed]
- Kujur, P.K.; Flores, B.C.T.; Ramalingam, N.; Chinen, L.T.D.; Jeffrey, S.S. Advances in the Characterization of Circulating Tumor Cells in Metastatic Breast Cancer: Single Cell Analyses and Interactions, and Patient-Derived Models for Drug Testing. Adv. Exp. Med. Biol. 2020, 1220, 61–80. [Google Scholar]
- Gómez-Sjöberg, R.; Leyrat, A.A.; Pirone, D.M.; Chen, C.S.; Quake, S.R. Versatile, fully automated, microfluidic cell culture system. Anal. Chem. 2007, 79, 8557–8563. [Google Scholar] [CrossRef]
- Wu, Y.H.; Lai, M.Z. Measuring NLR Oligomerization V: In Situ Proximity Ligation Assay. Methods Mol. Biol. 2016, 1417, 185–195. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tulukcuoglu Guneri, E.; Lakis, E.; Hajji, I.; Martin, E.; Champ, J.; Rampanou, A.; Pierga, J.-Y.; Viovy, J.-L.; Proudhon, C.; Bidard, F.-C.; et al. Deciphering HER2-HER3 Dimerization at the Single CTC Level: A Microfluidic Approach. Cancers 2022, 14, 1890. https://doi.org/10.3390/cancers14081890
Tulukcuoglu Guneri E, Lakis E, Hajji I, Martin E, Champ J, Rampanou A, Pierga J-Y, Viovy J-L, Proudhon C, Bidard F-C, et al. Deciphering HER2-HER3 Dimerization at the Single CTC Level: A Microfluidic Approach. Cancers. 2022; 14(8):1890. https://doi.org/10.3390/cancers14081890
Chicago/Turabian StyleTulukcuoglu Guneri, Ezgi, Emile Lakis, Ismail Hajji, Elian Martin, Jerome Champ, Aurore Rampanou, Jean-Yves Pierga, Jean-Louis Viovy, Charlotte Proudhon, François-Clément Bidard, and et al. 2022. "Deciphering HER2-HER3 Dimerization at the Single CTC Level: A Microfluidic Approach" Cancers 14, no. 8: 1890. https://doi.org/10.3390/cancers14081890
APA StyleTulukcuoglu Guneri, E., Lakis, E., Hajji, I., Martin, E., Champ, J., Rampanou, A., Pierga, J.-Y., Viovy, J.-L., Proudhon, C., Bidard, F.-C., & Descroix, S. (2022). Deciphering HER2-HER3 Dimerization at the Single CTC Level: A Microfluidic Approach. Cancers, 14(8), 1890. https://doi.org/10.3390/cancers14081890