
Cutting-Edge Platforms for Analysis of Immune Cells in the Hepatic Microenvironment—Focus on Tumor-Associated Macrophages in Hepatocellular Carcinoma

 , ,
, , 
.png)
Abstract
Simple Summary
Abstract
1. Introduction
2. Multispectral Imaging
2.1. Traditional Methods for Phenotyping Immune Cells In Situ
2.2. Multiplex Immunofluorescen Staining Technique Followed by Multispectral Imaging Analysis
2.3. Vectra 3, Vectra Polaris and CODEX
2.4. Phenotyping Tumor-Associated Macrophages in the Tumor Microenvironment Using Multispectral Imaging
2.5. Limitations of Multispectral Imaging and Future Directions
3. Cytometry by Time-of-Flight
3.1. Cytometry by Time-of-Flight: A Fusion of Multiple Techniques
3.2. Cytometry by Time-of-Flight Workflow
3.3. Use of Cytometry by Time-of-Flight for Characterization of Multiple Macrophage Phenotypes in the Hepatic Microenvironment
3.4. Cytometry by Time-of-Flight Limitations and Future Directions
4. Digital Spatial Profiling
4.1. Digital Spatial Profiling: From DNA Microarrays to Spatial Genomics
4.2. Digital Spatial Profiling Workflow
4.3. Digital Spatial Profiling of Biomarkers and Fetal-like TAMs
4.4. Digital Spatial Profiling Limitations and Future Directions
5. Single-Cell RNA Sequencing
5.1. Single-Cell RNA Sequencing: Genomic Characterization of Individual Cells
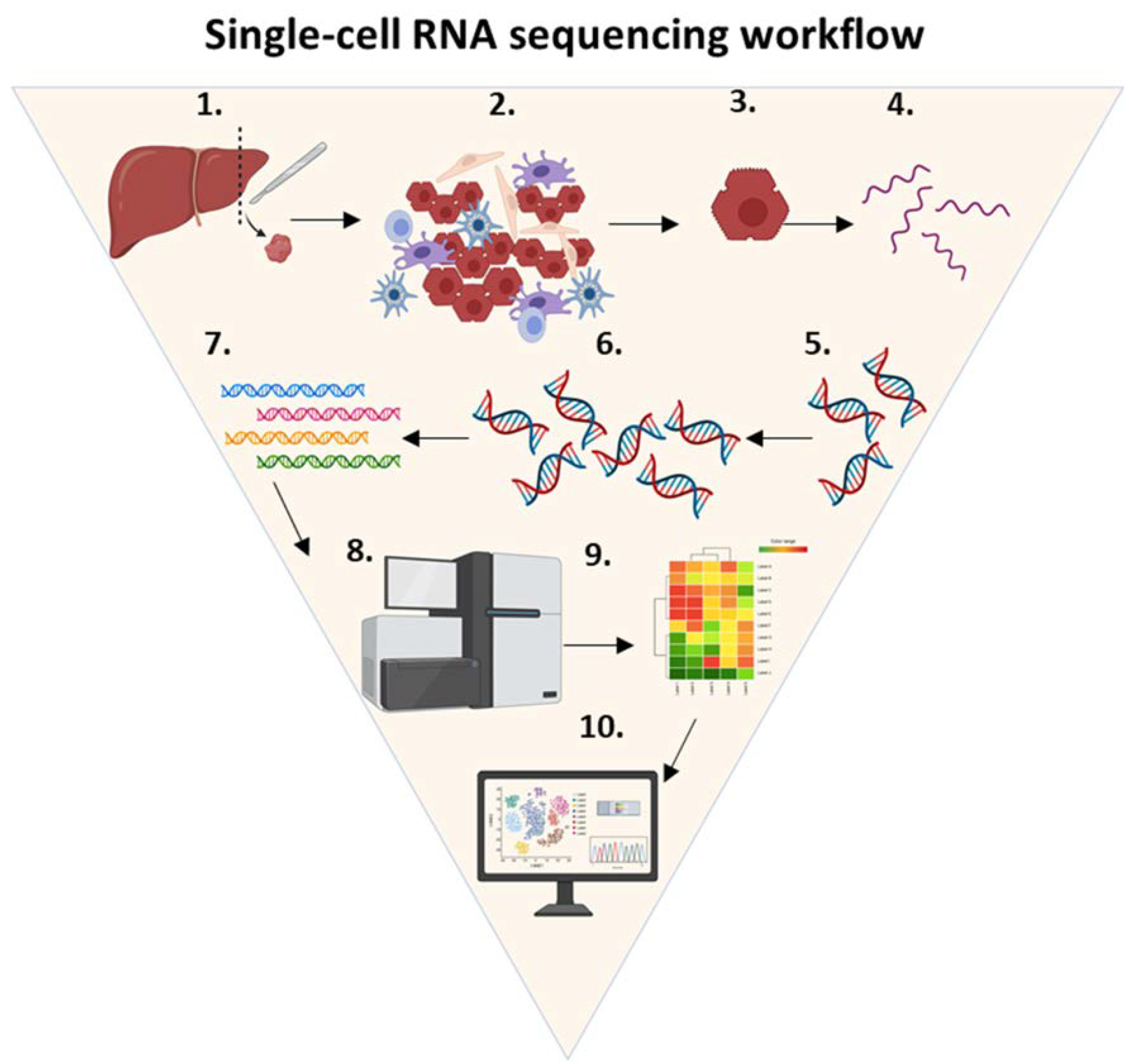
5.2. Single-Cell RNA Sequencing Workflow
5.3. Decoding the Tumor Microenvironment of Hepatocellular Carcinoma One Cell at a Time
5.4. Single Cell RNA Sequencing Limitations and Future Directions
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef]
- Kim, E.; Viatour, P. Hepatocellular carcinoma: Old friends and new tricks. Exp. Mol. Med. 2020, 52, 1898–1907. [Google Scholar] [CrossRef] [PubMed]
- Poczobutt, J.M.; De, S.; Yadav, V.K.; Nguyen, T.T.; Li, H.; Sippel, T.R.; Weiser-Evans, M.C.; Nemenoff, R.A. Expression Profiling of Macrophages Reveals Multiple Populations with Distinct Biological Roles in an Immunocompetent Orthotopic Model of Lung Cancer. J. Immunol. 2016, 196, 2847–2859. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Hou, X.; Liu, W.; Han, Z.; Wei, L. Macrophages and hepatocellular carcinoma. Cell Biosci. 2019, 9, 79. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ge, W.; Zhou, J.; Gao, B.; Qian, X.; Wang, W. The Role of Tumor Associated Macrophages in Hepatocellular Carcinoma. J. Cancer 2021, 12, 1284–1294. [Google Scholar] [CrossRef] [PubMed]
- Capece, D.; Fischietti, M.; Verzella, D.; Gaggiano, A.; Cicciarelli, G.; Tessitore, A.; Zazzeroni, F.; Alesse, E. The inflammatory microenvironment in hepatocellular carcinoma: A pivotal role for tumor-associated macrophages. Biomed. Res. Int. 2013, 2013, 187204. [Google Scholar] [CrossRef]
- Degroote, H.; Lefere, S.; Vandierendonck, A.; Vanderborght, B.; Meese, T.; Van Nieuwerburgh, F.; Verhelst, X.; Geerts, A.; Van Vlierberghe, H.; Devisscher, L. Characterization of the inflammatory microenvironment and hepatic macrophage subsets in experimental hepatocellular carcinoma models. Oncotarget 2021, 12, 562–577. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Heideveld, E.; Horcas-Lopez, M.; Lopez-Yrigoyen, M.; Forrester, L.M.; Cassetta, L.; Pollard, J.W. Methods for macrophage differentiation and in vitro generation of human tumor associated-like macrophages. Methods Enzymol. 2020, 632, 113–131. [Google Scholar] [CrossRef]
- Morrison, L.E.; Lefever, M.R.; Behman, L.J.; Leibold, T.; Roberts, E.A.; Horchner, U.B.; Bauer, D.R. Brightfield multiplex immunohistochemistry with multispectral imaging. Lab. Investig. 2020, 100, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Levenson, R.; Beechem, J.; McNamara, G. Spectral imaging in preclinical research and clinical pathology. Stud. Health Technol. Inform. 2013, 185, 43–75. [Google Scholar] [PubMed]
- Akhtar, M.; Rashid, S.; Al-Bozom, I.A. PD-L1 immunostaining: What pathologists need to know. Diagn. Pathol. 2021, 16, 94. [Google Scholar] [CrossRef] [PubMed]
- Levenson, R.M.; Fornari, A.; Loda, M. Multispectral imaging and pathology: Seeing and doing more. Expert Opin. Med. Diagn. 2008, 2, 1067–1081. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.C.C.; Nerurkar, S.N.; Cai, H.Y.; Ng, H.H.M.; Wu, D.; Wee, Y.T.F.; Lim, J.C.T.; Yeong, J.; Lim, T.K.H. Overview of multiplex immunohistochemistry/immunofluorescence techniques in the era of cancer immunotherapy. Cancer Commun. 2020, 40, 135–153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hubbard, A.; Jones, T.; Racolta, A.; Bhaumik, S.; Cummins, N.; Zhang, L.; Garsha, K.; Ventura, F.; Lefever, M.R.; et al. Fully automated 5-plex fluorescent immunohistochemistry with tyramide signal amplification and same species antibodies. Lab. Investig. 2017, 97, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Hennrick, K.; Drew, S. A colorful future of quantitative pathology: Validation of Vectra technology using chromogenic multiplexed immunohistochemistry and prostate tissue microarrays. Hum. Pathol. 2013, 44, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.A.; Morrison, L.E.; Behman, L.J.; Draganova-Tacheva, R.; O’Neill, R.; Solomides, C.C. Chromogenic immunohistochemical quadruplex provides accurate diagnostic differentiation of non-small cell lung cancer. Ann. Diagn. Pathol. 2020, 45, 151454. [Google Scholar] [CrossRef] [PubMed]
- Saldarriaga, O.A.; Freiberg, B.; Krishnan, S.; Rao, A.; Burks, J.; Booth, A.L.; Dye, B.; Utay, N.; Ferguson, M.; Akil, A.; et al. Multispectral Imaging Enables Characterization of Intrahepatic Macrophages in Patients With Chronic Liver Disease. Hepatol. Commun. 2020, 4, 708–723. [Google Scholar] [CrossRef] [PubMed]
- Zielinski, J.M.; Luke, J.J.; Guglietta, S.; Krieg, C. High Throughput Multi-Omics Approaches for Clinical Trial Evaluation and Drug Discovery. Front. Immunol. 2021, 12, 590742. [Google Scholar] [CrossRef]
- Stachtea, X.; Loughrey, M.B.; Salvucci, M.; Lindner, A.U.; Cho, S.; McDonough, E.; Sood, A.; Graf, J.; Santamaria-Pang, A.; Corwin, A.; et al. Stratification of chemotherapy-treated stage III colorectal cancer patients using multiplexed imaging and single-cell analysis of T-cell populations. Mod. Pathol. 2021, 35, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Kaldjian, E.P.; Ramirez, A.B.; Costandy, L.; Ericson, N.G.; Malkawi, W.I.; George, T.C.; Kasi, P.M. Beyond Circulating Tumor Cell Enumeration: Cell-Based Liquid Biopsy to Assess Protein Biomarkers and Cancer Genomics Using the RareCyte. Front. Pharmacol. 2022, 13, 835727. [Google Scholar] [CrossRef] [PubMed]
- Schürch, C.M.; Bhate, S.S.; Barlow, G.L.; Phillips, D.J.; Noti, L.; Zlobec, I.; Chu, P.; Black, S.; Demeter, J.; McIlwain, D.R.; et al. Coordinated Cellular Neighborhoods Orchestrate Antitumoral Immunity at the Colorectal Cancer Invasive Front. Cell 2020, 182, 1341–1359.e1319. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; He, K.; Pan, Y.; Wang, H.; Luo, Y.; Xia, Q. The role of tumor-associated macrophages in primary hepatocellular carcinoma and its related targeting therapy. Int. J. Med. Sci. 2021, 18, 2109–2116. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Zhang, J.; Wang, L.; Tano, V.; Tang, J.; Wang, X.; Wu, J.; Song, J.; Zhao, Y.; Rong, J.; et al. Topological analysis of hepatocellular carcinoma tumour microenvironment based on imaging mass cytometry reveals cellular neighbourhood regulated reversely by macrophages with different ontogeny. Gut 2021. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.H.; Ng, H.H.M.; Lim, C.J.; Sim, X.N.; Malavasi, F.; Li, H.; Loh, J.J.H.; Sabai, K.; Kim, J.K.; Ong, C.C.H.; et al. Expression of CD38 on Macrophages Predicts Improved Prognosis in Hepatocellular Carcinoma. Front. Immunol. 2019, 10, 2093. [Google Scholar] [CrossRef] [PubMed]
- Ortega, S.; Halicek, M.; Fabelo, H.; Callico, G.M.; Fei, B. Hyperspectral and multispectral imaging in digital and computational pathology: A systematic review [Invited]. Biomed. Opt. Express 2020, 11, 3195–3233. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.M.; Roman, K.; Engle, E.L.; Wang, C.; Ballesteros-Merino, C.; Jensen, S.M.; McGuire, J.; Jiang, M.; Coltharp, C.; Remeniuk, B.; et al. Multi-institutional TSA-amplified Multiplexed Immunofluorescence Reproducibility Evaluation (MITRE) Study. J. Immunother. Cancer 2021, 9, e002197. [Google Scholar] [CrossRef] [PubMed]
- Hoyt, C.C. Multiplex Immunofluorescence and Multispectral Imaging: Forming the Basis of a Clinical Test Platform for Immuno-Oncology. Front. Mol. Biosci. 2021, 8, 674747. [Google Scholar] [CrossRef]
- Pasquereau, S.; Al Moussawi, F.; Karam, W.; Diab Assaf, M.; Kumar, A.; Herbein, G. Cytomegalovirus, Macrophages and Breast Cancer. Open Virol. J. 2017, 11, 15–27. [Google Scholar] [CrossRef]
- Schlotter, C.M.; Tietze, L.; Vogt, U.; Heinsen, C.V.; Hahn, A. Ki67 and lymphocytes in the pretherapeutic core biopsy of primary invasive breast cancer: Positive markers of therapy response prediction and superior survival. Horm. Mol. Biol. Clin. Investig. 2017, 32. [Google Scholar] [CrossRef] [PubMed]
- García-Teijido, P.; Cabal, M.L.; Fernández, I.P.; Pérez, Y.F. Tumor-Infiltrating Lymphocytes in Triple Negative Breast Cancer: The Future of Immune Targeting. Clin. Med. Insights Oncol. 2016, 10, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Herzenberg, L.A.; Parks, D.; Sahaf, B.; Perez, O.; Roederer, M. The history and future of the fluorescence activated cell sorter and flow cytometry: A view from Stanford. Clin. Chem. 2002, 48, 1819–1827. [Google Scholar] [CrossRef] [PubMed]
- Melamed, M.R. A brief history of flow cytometry and sorting. Methods Cell. Biol. 2001, 63, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Cheung, R.K.; Utz, P.J. Screening: CyTOF-the next generation of cell detection. Nat. Rev. Rheumatol. 2011, 7, 502–503. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, M.H.; Nolan, G.P. Mass Cytometry: Single Cells, Many Features. Cell 2016, 165, 780–791. [Google Scholar] [CrossRef]
- Bjornson, Z.B.; Nolan, G.P.; Fantl, W.J. Single-cell mass cytometry for analysis of immune system functional states. Curr. Opin. Immunol. 2013, 25, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Bendall, S.C.; Simonds, E.F.; Qiu, P.; Amir, e.-A.; Krutzik, P.O.; Finck, R.; Bruggner, R.V.; Melamed, R.; Trejo, A.; Ornatsky, O.I.; et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 2011, 332, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Bodenmiller, B. Multiplexed Epitope-Based Tissue Imaging for Discovery and Healthcare Applications. Cell Syst. 2016, 2, 225–238. [Google Scholar] [CrossRef]
- Park, L.M.; Lannigan, J.; Jaimes, M.C. OMIP-069: Forty-Color Full Spectrum Flow Cytometry Panel for Deep Immunophenotyping of Major Cell Subsets in Human Peripheral Blood. Cytom. Part A 2020, 97, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Subrahmanyam, P.B.; Maecker, H.T. CyTOF Measurement of Immunocompetence Across Major Immune Cell Types. Curr. Protoc. Cytom. 2017, 82, 9.54.1–9.54.12. [Google Scholar] [CrossRef] [PubMed]
- Chew, V.; Lai, L.; Pan, L.; Lim, C.J.; Li, J.; Ong, R.; Chua, C.; Leong, J.Y.; Lim, K.H.; Toh, H.C.; et al. Delineation of an immunosuppressive gradient in hepatocellular carcinoma using high-dimensional proteomic and transcriptomic analyses. Proc. Natl. Acad. Sci. USA 2017, 114, E5900–E5909. [Google Scholar] [CrossRef] [PubMed]
- Gong, R.; Ren, H. Targeting chemokines/chemokine receptors: A promising strategy for enhancing the immunotherapy of pancreatic ductal adenocarcinoma. Signal. Transduct. Target. Ther. 2020, 5, 149. [Google Scholar] [CrossRef] [PubMed]
- Tu, M.M.; Abdel-Hafiz, H.A.; Jones, R.T.; Jean, A.; Hoff, K.J.; Duex, J.E.; Chauca-Diaz, A.; Costello, J.C.; Dancik, G.M.; Tamburini, B.A.J.; et al. Inhibition of the CCL2 receptor, CCR2, enhances tumor response to immune checkpoint therapy. Commun. Biol. 2020, 3, 720. [Google Scholar] [CrossRef] [PubMed]
- Strazza, M.; Mor, A. The Complexity of Targeting Chemokines to Promote a Tumor Immune Response. Inflammation 2020, 43, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Letourneur, D.; Danlos, F.X.; Marabelle, A. Chemokine biology on immune checkpoint-targeted therapies. Eur. J. Cancer 2020, 137, 260–271. [Google Scholar] [CrossRef]
- Elaldi, R.; Hemon, P.; Petti, L.; Cosson, E.; Desrues, B.; Sudaka, A.; Poissonnet, G.; Van Obberghen-Schilling, E.; Pers, J.O.; Braud, V.M.; et al. High Dimensional Imaging Mass Cytometry Panel to Visualize the Tumor Immune Microenvironment Contexture. Front. Immunol. 2021, 12, 666233. [Google Scholar] [CrossRef]
- Castella, B.; Mina, R.; Gay, F. CyTOF®: A New Tool to Decipher the Immunomodulatory Activity of Daratumumab. Cytom. Part A 2019, 95, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.; Ong, R.; Li, J.; Albani, S. A CD45-based barcoding approach to multiplex mass-cytometry (CyTOF). Cytom. Part A 2015, 87, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Gresham, D.; Dunham, M.J.; Botstein, D. Comparing whole genomes using DNA microarrays. Nat. Rev. Genet. 2008, 9, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Bumgarner, R. Overview of DNA microarrays: Types, applications, and their future. Curr. Protoc. Mol. Biol. 2013, 101, 22. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Bhatt, R.; Brown, C.; Brown, E.A.; Buhr, D.L.; Chantranuvatana, K.; Danaher, P.; Dunaway, D.; Garrison, R.G.; Geiss, G.; et al. High-plex multiomic analysis in FFPE at subcellular level by spatial molecular imaging. bioRxiv 2022. preprint. [Google Scholar] [CrossRef]
- Bhaduri, A.; Sandoval-Espinosa, C.; Otero-Garcia, M.; Oh, I.; Yin, R.; Eze, U.C.; Nowakowski, T.J.; Kriegstein, A.R. An atlas of cortical arealization identifies dynamic molecular signatures. Nature 2021, 598, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Zhang, C.; Wang, H.; Zhao, L. Exosome-mediated communication between tumor cells and tumor-associated macrophages: Implications for tumor microenvironment. Oncoimmunology 2021, 10, 1887552. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fan, L.; Yu, H.; Zhang, J.; He, Y.; Feng, D.; Wang, F.; Li, X.; Liu, Q.; Li, Y.; et al. Endoplasmic Reticulum Stress Causes Liver Cancer Cells to Release Exosomal miR-23a-3p and Up-regulate Programmed Death Ligand 1 Expression in Macrophages. Hepatology 2019, 70, 241–258. [Google Scholar] [CrossRef]
- Yi, M.; Jiao, D.; Xu, H.; Liu, Q.; Zhao, W.; Han, X.; Wu, K. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol. Cancer 2018, 17, 129. [Google Scholar] [CrossRef]
- Della Starza, I.; De Novi, L.A.; Santoro, A.; Salemi, D.; Tam, W.; Cavalli, M.; Menale, L.; Soscia, R.; Apicella, V.; Ilari, C.; et al. Digital droplet PCR and next-generation sequencing refine minimal residual disease monitoring in acute lymphoblastic leukemia. Leuk. Lymphoma 2019, 60, 2838–2840. [Google Scholar] [CrossRef]
- Sharma, A.; Seow, J.J.W.; Dutertre, C.A.; Pai, R.; Blériot, C.; Mishra, A.; Wong, R.M.M.; Singh, G.S.N.; Sudhagar, S.; Khalilnezhad, S.; et al. Onco-fetal Reprogramming of Endothelial Cells Drives Immunosuppressive Macrophages in Hepatocellular Carcinoma. Cell 2020, 183, 377–394.e321. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Kim, J.K.; Svensson, V.; Marioni, J.C.; Teichmann, S.A. The technology and biology of single-cell RNA sequencing. Mol. Cell 2015, 58, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Ho, D.W.; Tsui, Y.M.; Chan, L.K.; Sze, K.M.; Zhang, X.; Cheu, J.W.; Chiu, Y.T.; Lee, J.M.; Chan, A.C.; Cheung, E.T.; et al. Single-cell RNA sequencing shows the immunosuppressive landscape and tumor heterogeneity of HBV-associated hepatocellular carcinoma. Nat. Commun. 2021, 12, 3684. [Google Scholar] [CrossRef] [PubMed]
- MacParland, S.A.; Liu, J.C.; Ma, X.Z.; Innes, B.T.; Bartczak, A.M.; Gage, B.K.; Manuel, J.; Khuu, N.; Echeverri, J.; Linares, I.; et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 2018, 9, 4383. [Google Scholar] [CrossRef] [PubMed]
- Avraham, R.; Hung, D.T. A perspective on single cell behavior during infection. Gut Microbes 2016, 7, 518–525. [Google Scholar] [CrossRef][Green Version]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Petitprez, F.; Meylan, M.; de Reyniès, A.; Sautès-Fridman, C.; Fridman, W.H. The Tumor Microenvironment in the Response to Immune Checkpoint Blockade Therapies. Front. Immunol. 2020, 11, 784. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Gloghini, A.; Pruneri, G.; Dolcetti, R. Optimizing checkpoint inhibitors therapy for relapsed or progressive classic Hodgkin lymphoma by multiplex immunohistochemistry of the tumor microenvironment. Cancer Med. 2019, 8, 3012–3016. [Google Scholar] [CrossRef] [PubMed]
- Onuma, A.E.; Zhang, H.; Huang, H.; Williams, T.M.; Noonan, A.; Tsung, A. Immune Checkpoint Inhibitors in Hepatocellular Cancer: Current Understanding on Mechanisms of Resistance and Biomarkers of Response to Treatment. Gene Expr. 2020, 20, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Moldogazieva, N.T.; Zavadskiy, S.P.; Sologova, S.S.; Mokhosoev, I.M.; Terentiev, A.A. Predictive biomarkers for systemic therapy of hepatocellular carcinoma. Expert Rev. Mol. Diagn. 2021, 21, 1147–1164. [Google Scholar] [CrossRef]
- Lou, J.; Zhang, L.; Lv, S.; Zhang, C.; Jiang, S. Biomarkers for Hepatocellular Carcinoma. Biomark. Cancer 2017, 9, 1–9. [Google Scholar] [CrossRef]
- Xiang, X.; Wang, J.; Lu, D.; Xu, X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 75. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Platform | Strengths | Weakness | Opportunities | Threats |
|---|---|---|---|---|
| MSI | Provides in situ visualization of multiple cell phenotypes, while preserving tissue architecture Data can be used for spatial and nearest-neighbor analyses Allows data collection across entire tissue section | Limited amount of markers can be analyzed Spectral overlap can hinder colocalization analysis and biomarker quantification | Presence of specific TAMs in the TME may allowed for personalized treatment Automated equipment for high throughput is available Can be incorporated into routine brightfield microscopy [10] | Additional standardization is needed prior to clinical implementation [11,27] Most pathologist lack training with MSI microscopy [11] |
| IMC | Allows for in situ visualization of more than 40 targets [35,39] No spectral overlapping The use of non-biologic markers increases signal-to-noise ratio [38] | Vaporization of cells and tissue ablation are irreversible steps | Provides high-dimensional analysis of various cellular features [35] Allows for stratification of patients into treatment responders and non-responders [41,47] | Targets with low exppression or specific populations of cells may be challenging to detect [35,48] Complexity of data analysis [35,48] |
| DSP | Spatial characterization of both RNA and protein expression using a limited amount of tissue Allows for morphological segmentation of a unique population of cells [57] Combines high-plex microscopy and spatial genomics | Analysis is limited to pre-selected proteins and RNA probes Gene and protein assays are evaluated on two sequential slide sections from a tissue block | Gene expression and protein profiling in a single ROI Single cell resolution is in development | High Cost Requires at least 150–200 cells per ROI for sufficient counts Rare target analysis is more costly and time consuming |
| ScRNA-seq | Single cell RNA resolution transcriptomics Accurate identification of specific phenotypes and subpopulations of rare cell types | Loss of tissue architecture [62] No spatial co-localization analysis | Characterization of cellular crosstalk at single cell resolution Profile potential therapeutic targets for rare phenotypes [60,61] | Cell isolation method, number of cells per experiment, cost per cell and sensitivity vary between scRNA-seq platforms [62] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Millian, D.E.; Saldarriaga, O.A.; Wanninger, T.; Burks, J.K.; Rafati, Y.N.; Gosnell, J.; Stevenson, H.L. Cutting-Edge Platforms for Analysis of Immune Cells in the Hepatic Microenvironment—Focus on Tumor-Associated Macrophages in Hepatocellular Carcinoma. Cancers 2022, 14, 1861. https://doi.org/10.3390/cancers14081861
Millian DE, Saldarriaga OA, Wanninger T, Burks JK, Rafati YN, Gosnell J, Stevenson HL. Cutting-Edge Platforms for Analysis of Immune Cells in the Hepatic Microenvironment—Focus on Tumor-Associated Macrophages in Hepatocellular Carcinoma. Cancers. 2022; 14(8):1861. https://doi.org/10.3390/cancers14081861
Chicago/Turabian StyleMillian, Daniel E., Omar A. Saldarriaga, Timothy Wanninger, Jared K. Burks, Yousef N. Rafati, Joseph Gosnell, and Heather L. Stevenson. 2022. "Cutting-Edge Platforms for Analysis of Immune Cells in the Hepatic Microenvironment—Focus on Tumor-Associated Macrophages in Hepatocellular Carcinoma" Cancers 14, no. 8: 1861. https://doi.org/10.3390/cancers14081861
APA StyleMillian, D. E., Saldarriaga, O. A., Wanninger, T., Burks, J. K., Rafati, Y. N., Gosnell, J., & Stevenson, H. L. (2022). Cutting-Edge Platforms for Analysis of Immune Cells in the Hepatic Microenvironment—Focus on Tumor-Associated Macrophages in Hepatocellular Carcinoma. Cancers, 14(8), 1861. https://doi.org/10.3390/cancers14081861