Simple Summary
This review summarizes gene-expression profiling insights into the background and origination of diffuse large B-cell lymphomas (DLBCL). To further unravel the molecular biology of these lymphomas, a consortium panel called BLYM-777 was designed including genes important for subtype classifications, genetic pathways, tumor-microenvironment, immune response and resistance to targeted therapies. This review proposes to combine this transcriptomic method with genomics, proteomics, and patient characteristics to facilitate diagnostic classification, prognostication, and the development of new targeted therapeutic strategies in DLBCL.
Abstract
Gene-expression profiling (GEP) is used to study the molecular biology of lymphomas. Here, advancing insights from GEP studies in diffuse large B-cell lymphoma (DLBCL) lymphomagenesis are discussed. GEP studies elucidated subtypes based on cell-of-origin principles and profoundly changed the biological understanding of DLBCL with clinical relevance. Studies integrating GEP and next-generation DNA sequencing defined different molecular subtypes of DLBCL entities originating at specific anatomical localizations. With the emergence of high-throughput technologies, the tumor microenvironment (TME) has been recognized as a critical component in DLBCL pathogenesis. TME studies have characterized so-called “lymphoma microenvironments” and “ecotypes”. Despite gained insights, unexplained chemo-refractoriness in DLBCL remains. To further elucidate the complex biology of DLBCL, we propose a novel targeted GEP consortium panel, called BLYM-777. This knowledge-based biology-driven panel includes probes for 777 genes, covering many aspects regarding B-cell lymphomagenesis (f.e., MYC signature, TME, immune surveillance and resistance to CAR T-cell therapy). Regarding lymphomagenesis, upcoming DLBCL studies need to incorporate genomic and transcriptomic approaches with proteomic methods and correlate these multi-omics data with patient characteristics of well-defined and homogeneous cohorts. This multilayered methodology potentially enhances diagnostic classification of DLBCL subtypes, prognostication, and the development of novel targeted therapeutic strategies.
1. Introduction
The main challenge for diffuse large B-cell lymphoma (DLBCL), not otherwise specified (NOS), the most common lymphoid malignancy, is to improve survival outcomes. Approximately 40% of patients die or relapse within 3 years from diagnosis after standard one-size-fits-all immunochemotherapy R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) [1,2]. As an explanation, DLBCL is generally assumed to be a complex disease with significant genetic heterogeneity resulting in different biological behavior and drug-refractoriness. Many studies examined the molecular background to understand the various mechanisms of lymphomagenesis and therapy resistance in DLBCL. Recurrently mutated genes corresponding to multiple pathways have been discovered demonstrating the intricate molecular background of DLBCL [3,4,5]. Despite these insights, an in-depth understanding of this biological heterogeneity is still lacking.
Over the past decades, analysis of the molecular background of DLBCL has advanced through gene-expression profiling (GEP) studies allowing for the investigation of cell-of-origin (COO), MYC expression and tumor microenvironment (TME). This review focuses on the emerging role of GEP studies in elucidating the biological heterogeneity of DLBCL, thereby improving diagnostic classification, prognosis, and ultimately the development of targeted treatment. Finally, to facilitate subsequent molecular studies in DLBCL, we propose a knowledge-based biology-driven and ready-to-use targeted GEP consortium panel, named BLYM-777, including probes targeting 777 genes, covering many aspects of lymphoma B cells and the TME.
2. Technical Approaches of Gene-Expression Profiling
Several GEP methodologies have been applied in DLBCL studies, as summarized in Table 1 and Figure 1. The most conventional technique is reverse-transcription quantitative polymerase chain reaction (RT-qPCR), in which mRNA is converted into complementary (c)DNA using reverse transcriptase and this cDNA is subsequently amplified using dyes or specific probes for quantification of the PCR product after each amplification cycle. This used to be a monogenic, labor-intensive method that was unable to screen multiple high-throughput transcripts, but major improvements in throughput have been made over the years allowing simultaneous amplification of multiple genes in parallel [6].

Table 1.
Literature overview of relevant DLBCL studies with their respective GEP methods, number of included cases and genes, cluster targets and clinical relevance. COO = cell-of-origin, TME = tumor microenvironment, N.A. = not available, complete gene lists of these studies were not available.
Table 1.
Literature overview of relevant DLBCL studies with their respective GEP methods, number of included cases and genes, cluster targets and clinical relevance. COO = cell-of-origin, TME = tumor microenvironment, N.A. = not available, complete gene lists of these studies were not available.
| First Author(s) | Year | GEP Method | No. of Cases | No. of Genes | No. of Genes in BLYM-777 | Clusters | Clinical Relevance |
|---|---|---|---|---|---|---|---|
| Alizadeh, Elsen, et al. [7] | 2000 | Microarrays | 47 | 2984 | N.A. | COO | COO classified DLBCL into GCB or ABC with prognostic impact, possible benefit from different treatment options |
| Rosenwald, et al. [8] | 2002 | Microarrays | 240 | 100 | N.A. | GEP subgroups | COO classification into GCB and non-GCB (ABC and type 3), molecular predictor of survival after treatment |
| Monti, Savage, et al. [9] | 2005 | Microarrays | 176 | 2118 | 97 | Consensus clustering | Three identified DLBCL clusters; oxidative phosphorylation, BCR/proliferation or host response, no relation with survival |
| Lenz, et al. [10] | 2008 | Microarrays | 414 | 382 | 60 | Stromal signatures | Consensus clustering identified two stromal signatures predictive for survival and one GCB cluster |
| Alizadeh, Gentles, et al. [6] | 2011 | RT-qPCR | 787 | 2 | 2 | LMO2 and TNFRSF9 | Two survival-correlated biomarkers and associated with TME |
| Scott, et al. [11] | 2014 | NanoString | 119 | 20 | 20 | COO | Validation of COO classification into GCB or ABC, reflecting survival, possible benefit from different treatment options |
| Carey, et al. [12] | 2015 | NanoString | 55 | 200 | 33 | MYC high- and low-risk clusterss | Classification and stratification of MYC-driven, aggressive BCL |
| Dybkær, Bøgsted, et al. [13] | 2015 | Microarrays | 1139 | 223 | 37 | B-cell associated gene signature (BAGS) | Further discrimination of COO in centrocytes, centroblasts, plasmablasts, or memory B cells, with survival outcomes |
| Ciavarella, Vegliante, Fabbri, et al. [14] | 2018 | Publicly available GEP-data and NanoString | 482 | 45 | 45 | TME clusters | TME classification presenting high prevalence of myofibroblasts, dendritic cells, or CD4 T cells related to survival outcomes |
| Michaelsen, et al. [15] | 2018 | NanoString | 1058 | 128 | 53 | BAGS2Clinic(expanded BAGS) | Intensified BAGS classification in centrocytes, centroblasts, plasmablasts, or memory B cells, predictive for survival |
| Davies, et al. [16] | 2019 | Illumina HiSeq sequencing | 1076 | N.A. | N.A. | COO | Molecular characterization for prospective stratification, randomization and analysis of DLBCL subgroups |
| Ennishi, et al. [17] | 2019 | RNA-seq | 157 | 104 | 43 | DHITsig | Defined GEP signature high-grade B-cell lymphoma double or triple hit with BCL2 translocation |
| Staiger, Altenbuchinger, Ziepert, et al. [18] | 2020 | NanoString | 466 | 145 | 17 | Lymphoma-associated macrophage interaction signature (LAMIS) | Signature indicating the presence of macrophages and associated with poor survival |
| Tripodo, Zanardi, Ianelli, Mazzara, et al. [19] | 2020 | NanoString | 551 | 87 | 52 | Spatial dark- versus light-zone microenvironment signature | Distinguishing COO GCB subtype into dark or light zone with prognostic significance |
| Kotlov, et al. [20] | 2021 | Publicly available GEP-data | 4580 | 203 | 144 | Functional gene signatures and TME clusters | Four TME specific categories associated with survival and with opportunities for novel targeted treatment |
| Steen, et al. [21] | 2021 | Bulk/single-cell RNA sequencing | 1584 | 20380 | 192 | Cell states and ecotypes of the TME | Discrimination into cell types and cell states within the TME, correlated with survival, and facilitating development of new targeted treatment strategies |
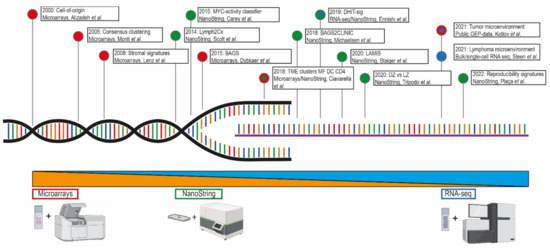
Figure 1.
The meaningful arrival of GEP in DLBCL. This timeline presents the implementation of GEP strategies in DLBCL studies throughout the past two decades and marks the relevant findings with their corresponding techniques [7,9,10,11,12,13,14,15,17,18,19,20,21]. Within the lymphoma research field, technological advances shifted the approach from microarrays (red) to NanoString (green) and ultimately (single cell) RNA sequencing (blue).
Over two decades ago, the application of a new gene expression profiling technique resulted in a hallmark study of DLBCL [7]. This microarray-based technology allows the simultaneous assessment of thousands of gene-transcripts. These microarrays contain probes that are complementary to fluorescently labelled cDNA produced by reverse transcription of mRNA from the genes of interest. After hybridization, digital cameras measure fluorescence intensity and translate this to gene-transcript counts. This method requires mRNA input from preferentially fresh frozen material over formalin-fixed paraffin-embedded (FFPE) samples. However, in routine diagnostics, the material is generally preserved in FFPE rather than fresh frozen due to practical considerations which impeded a broad implementation of studies using microarray-based technologies. Nowadays special FFPE kits (f.e., Agilent and Illumina) are available that allow the routine analysis of RNA isolated from FFPE.
The NanoString nCounter system (Seattle, WA 98109, USA) is an alternative hybridization-based gene expression profiling method. This technique detects and counts several hundreds of mRNA transcripts by using probe specific molecular “barcodes” combined with fluorescent-microscopic imaging. This system is efficient for targeted GEP strategies with (partially) degraded RNA samples (i.e., FFPE). After entering the lymphoma research field in 2014, the NanoString nCounter system has been widely used in studies to identify lymphoma subtypes.
With the advent of next-generation sequencing (NGS) techniques, RNA sequencing (RNA-seq) was developed as an alternative approach for GEP, enabling the analysis of entire transcriptomes. Besides the generation of gene-expression profiles, RNA-seq enables the analysis of gene fusions, mutations, single nucleotide polymorphisms, or even copy number alterations. For the generation of sequencing libraries, RNA is reverse transcribed to cDNA, and subsequently fragmented. Like microarray-based GEP assays, methodologies for RNA-seq using RNA isolated from FFPE has been developed and allows generation of reliable gene-expression profiles also from poor quality RNA. A derivative of RNA-seq is single-cell RNA-seq (scRNA-seq) for examination of the transcriptome of each individual nucleus as opposed to (tumor) bulk analysis. The main drawback of scRNA-seq is that massive data is produced that needs extensive bioinformatic procedures for appropriate analysis.
More comprehensive overviews of (dis)advantages of currently available technologies have been reviewed extensively by Narrandes et al. and Jiang et al. [22,23]. All of the above-described techniques, RT-qPCR, microarrays, NanoString, and (sc)RNA-seq, have been applied in DLBCL studies and are discussed below for their relevance to DLBCL pathogenesis.
3. The Arrival of Gene-Expression Profiling
As presented in Table 1 and Figure 1, several relevant DLBCL studies reported on the use of gene-expression assays with different platforms. As a cornerstone, Alizadeh et al. [7] were the first in 2000 to demonstrate a large diversity between DLBCL cases in a microarray-based gene-expression study. This study defined two molecularly distinct DLBCL subtypes with either germinal center B-cell (GCB) or activated B-cell like (ABC) phenotypes, as shown in Figure 2. Tumors classified as GCB showed a significantly superior overall survival (OS) compared to ABC DLBCL cases. Accordingly, Rosenwald et al. [8] independently reported similar results with an additional third discriminating DLBCL subtype (designated as type 3), which has a similar survival rate as ABC DLBCL, and grouped together are generally referred to as non-GCB subtypes. These results were at the basis of identifying the COO to better understand lymphomagenesis. Several other studies aimed to optimize the GCB/non-GCB COO classification, explore other potential signatures for DLBCL, and validate previous findings [6,24,25,26,27].
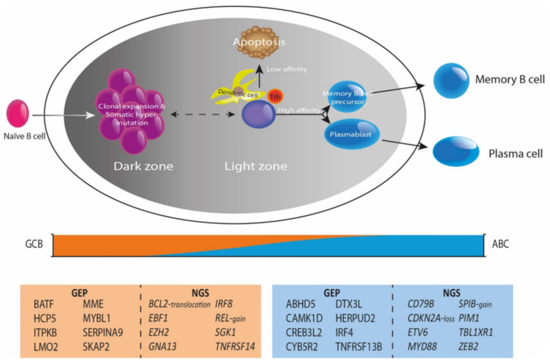
Figure 2.
Genetic perspectives of B-cell lymphomagenesis. Under normal physiological circumstances, the germinal center is crucial for B-cell development and maturation, defining different cellular subtypes and states throughout this continuing process. DLBCL lymphomagenesis shows a GCB subtype in the earlier stages of development and an ABC subtype in later stages, representing COO classification. The COO classification is substantiated by distinct characteristic GEP and mutational profiles between GCB and ABC. This insight shows the importance of combining DNA NGS and GEP in a more multidimensional approach that improves classification and prognostication of DLBCL.
The advent of the NanoString nCounter platform optimized gene-expression analysis of FFPE samples. Scott et al. [11] were the first to generate a COO classification using the NanoString technology in 2014. This approach utilized a targeted panel (Lymph2Cx), including 20 genes, and presented high intra-institutional concordance and overlap with microarray-based GEP. Subsequently, Dybkær et al. [13] aimed to further subdivide the classified COO subtypes of GCB and ABC into centrocytes, centroblasts, memory B-cells and plasmablasts. This initially led to the design of a microarray-based assay called B-cell associated gene signature (BAGS) including 223 genes, demonstrating a significantly different progression-free survival (PFS) and OS between the four cellular subtypes. Subsequently, in 2018, Michaelsen et al. [15] modified the BAGS assay to a new BAGS2CLINIC panel for the NanoString platform, including 128 genes, enabling fast and easy-to-use GEP with high overlap with the original BAGS classifier. Compared to the Lymph2Cx panel, the BAGS2CLINIC panel is more comprehensive and provides a more detailed stratification. Survival analyses using the COO assignment by BAGS2CLINIC indicated an inferior PFS and OS for the memory B-cell subtype compared to the plasmablast subtype, both originally classified as ABC subtypes. Although the centroblast and centrocyte subtypes were both classified as GCB subtypes, an inferior PFS was identified for the centroblast subtype, with no difference in OS.
In 2020, Tripodo et al. [19] generated a spatial signature including 87 genes that discriminates between the dark and light zone of the germinal center, with similarities to COO and BAGS(2CLINIC) classifications. The subtypes identified by this panel showed prognostic significance, as a light-zone-like phenotype was associated with superior OS compared to a dark-zone-like phenotype.
With the advancing insight into COO, interest and understanding of the TME have increased. In 2005 Monti et al. [9] analyzed transcriptional signatures in DLBCL and reported a so-called “consensus clustering” classification. This study implemented microarrays and sequential consensus cluster analysis to assess the stability of clusters in gene-expression data after different clustering methods. Three distinct DLBCL clusters were identified, two of which contained predominantly B-cell expression profiles characterized by oxidative phosphorylation and B-cell receptor/proliferation. In contrast, the third cluster was enriched for T-cell-mediated immune response and classical complement pathway and as such reflected the interaction of the microenvironment with the tumor. In contrast to COO, no correlation was found between the consensus clusters and survival [9].
In 2008, Lenz et al. [10,28] identified a GCB cluster and two stromal signatures, characterized by their TME association. The first stromal signature (stromal-1) reflected the extracellular matrix and histiocyte infiltration and was associated with a favorable PFS and OS in comparison to the second stromal signature (stromal-2) which represented tumor angiogenesis. Over the following years, other independent studies investigated these stromal signatures and other biological markers for their relevance to survival, reporting similar findings [21,26,29]. The identification of these stromal signatures emphasized the importance of studying the TME to improve the biological understanding of DLBCL [10].
Carey et al. [12] performed targeted GEP and identified a molecular classifier of MYC activity including 80 genes that stratified DLBCL patients into high- (MYC score > 0.5) and low-risk (MYC score < 0.5) groups. Patients with low MYC scores showed significantly better OS. This classification was further optimized by Ennishi et al. [17] who generated a double-hit gene-expression signature (DHITsig) including 104 genes. DHITsig positivity was determined by overexpression of genes of high-grade B-cell lymphoma double hit or triple hit with BCL2 translocations. DHITsig-positive cases showed strong cell-autonomous survival and proliferation signals and reduced dependence on the TME. Using this DHITsig, approximately twice as many tumors were classified as high-grade B-cell lymphoma than with conventional fluorescence in situ hybridization (FISH). PFS and OS were significantly worse in DHITsig-positive patients in comparison to DHITsig negative patients. Plaça et al. [30] have successfully reproduced the MYC classifier of Carey et al. [12] and the consensus clustering of Monti et al. [9] in 175 samples of the HOVON-84 trial on a panel of 117 genes using the NanoString platform. These GEP signatures can facilitate the search for optimization of treatment algorithms, for example, which patients would benefit from the addition of lenalidomide to standard R-CHOP treatment (as described by Chamuleau et al. [31]) or to common intensive chemotherapy regimens.
In summary, along with technological advancements over time, several GEP signatures of lymphoma cells have been identified which have significantly augmented the biological knowledge of DLBCL, distinguishing several COO and TME-based molecular subtypes with prognostic relevance.
4. Integrating Gene-Expression Profiling and Mutational Profiles
DLBCL belongs to the spectrum of cancers with high mutational burden, reporting 7.8 driver mutations per case and a mean number of 23.5 mutations in ABC and 31 in GCB DLBCL patients, respectively [3,4,32,33,34,35]. Using (targeted) DNA (t)NGS technologies, from now on referred to as NGS, multiple studies identified the involvement of various intracellular signaling cascades (f.e., apoptosis, DNA damage response, JAK/STAT, MAPK, NF-κB, NOTCH, PI3K) in DLBCL lymphomagenesis. Karube et al. [36] defined the relevance of genomic alterations in genes involved in the NOTCH pathway in DLBCL suggesting that analysis of aberrations in defined pathways may be more instructive than independent genes alone. Recently, several large NGS studies have shown that various molecular subgroups informative for prognosis can be distinguished in DLBCL [4,5,37,38,39,40]. In 2018, Chapuy et al. [4] identified five robust molecular DLBCL subgroups, C1-C5. Similarly, Schmitz et al. [5] identified four distinct subtypes, MCD (co-occurrence of MYD88L265P and CD79B mutations), BN2 (BCL6 fusions or NOTCH2 mutations), N1 (NOTCH1 mutations), and EZB (EZH2 mutations or BCL2 translocations). Wright et al. [37] revealed six genetic subtypes, including the four subtypes that Schmitz et al. already reported, supplemented by the A53 (TP53 mutations) and ST2 (SGK1 and TET2 mutations) subtypes, known as the LymphGen profiles. Similarly, Lacy et al. [38] identified five molecular subgroups, MYD88, BCL2, SOCS1/SGK1, TET2/SGK1, NOTCH2 and an unclassified group. From these large sequencing studies at least five distinct molecular subgroups have been defined, partially representing COO subtypes (Figure 2); MYD88/CD79B (NF-κB pathway), TP53, BCL2/NOTCH2, SOCS1/SGK1 (JAK/STAT pathway) and MYC.
While the pathogenicity of most aberrations on lymphomagenesis is well understood, for a deeper understanding of DLBCL biology it remains of major importance to complement these molecular profiles with gene-expression profiles. An elegant example is a key study by Steen et al. [21], that identified different B-cell states by GEP and integrated this with genomic data, by comparing the B-cell states to the results of the genomic LymphGen profiles and the C1-C5 subtypes [4,37]. This comparison resulted in a partial overlap between these two different subtyping methods and the identified B-cell states, but also revealed significant differences between these mutational and gene-expression classifications. These differences showed that tumors within similar mutational profiles differed in their transcriptional profile and depend on different effects on downstream pathways.
This concept was underscored by Shouval et al. [41], who identified two complementary mechanisms in TP53-mutated DLBCL using transcriptomic profiling. The first mechanism was the downregulation of IFN signaling and the second was characterized by a reduced tumor infiltration of CD8-positive T cells. Both mechanisms contributed to treatment resistance and thereby to inferior survival. This approach demonstrated that TP53-mutated DLBCL could be further subdivided by transcriptomic profiling improving the understanding of clinical behavior or treatment responses of these tumors.
As summarized above, Figure 2 shows that GEP and NGS data complement each other and provide clear added value in understanding the complicated molecular subtypes of DLBCL, for example, for further subtyping of COO classes. This strategy has shifted the field of research towards a more multidimensional approach connecting NGS and GEP data from individual DLBCL cases across the entire study cohort.
5. The Tumor Microenvironment as Defined by Gene-Expression Profiling
The development of GEP technologies has offered the possibility to study the TME more extensively. To address various biological and clinical questions, GEP approaches have primarily focused on the role of fibroblasts, macrophages, or T cells in DLBCL lymphomagenesis. Targeted panels with probe sets covering genes encompassing discriminatory aspects of fibroblasts, macrophages, T cells, or other cells, and their activation and differentiation states have been utilized. With this TME-directed GEP strategy, several TME signatures have been defined in DLBCL, as presented in Figure 3.
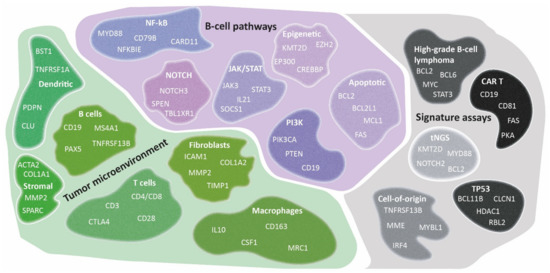
Figure 3.
Diversity of TME signatures in DLBCL. Several GEP signatures of lymphoma cells have been identified that have significantly augmented the biological knowledge of DLBCL. As presented, these signatures could be subdivided into three categories: tumor microenvironment, B-cell pathways, and signature assays. A relevant gene selection of potential pathways related to B-cell lymphomagenesis (purple), cell types within the TME (green), and other specific signature assays (grey) are depicted. GEP studies have demonstrated its added value in characterizing the DLBCL microenvironment and the discovery of early principles of their intriguing mechanisms. However biological issues remain, and further research is needed to determine the true clinical benefit.
Analysis of GEP data of 414 untreated DLBCL samples identified three distinct gene-expression signatures; GCB, stromal-1, and stromal-2 [10]. The stromal-1 signature was associated with favorable survival. A computational CIBERSORT method incorporating 17 immune and stromal cytotypes into a 1028-gene matrix was applied to the previously produced data [10,14,42]. This analysis revealed that a high prevalence of myofibroblasts, dendritic cells, or CD4-positive T cells was associated with superior PFS and OS as compared to an abundance of activated natural killer cells and plasma cells. Subsequently, a 45-gene set developed for NanoString-based profiling demonstrated a favorable survival of DLBCL with a high prevalence of these similar cell types [14].
Furthermore, a lymphoma-associated macrophage interaction signature (called LAMIS, including 145 genes) was developed that specifically targets macrophages with the M2 phenotype that are immunosuppressive and promote tumor progression [18]. High expression of the LAMIS-signature indicated poor PFS and OS, independent of COO subtype and International Prognostic Index (IPI) score [14,18]. Accordingly, Marcelis et al. [43] characterized the TME of primary central nervous system lymphoma and reported that an increased M1-like/M2-like macrophage ratio was associated with superior OS. Keane et al. [44] quantified the TME independent of the revised IPI and COO by evaluating the ratios of immune effectors with potential implications for the selection of patients in clinical trials.
Two novel corner stone studies investigated the TME through GEP and correlated these data with clinical and NGS data. Initially, in 2021, Kotlov et al. [20] performed clustering analysis on a large dataset retrieved from several publicly available datasets (n = 25, 4580 DLBCL cases). Based on functional gene signatures, four different cellular subtypes of the lymphoma microenvironment were clustered: germinal center-like, mesenchymal, inflammatory, and depleted. The first cluster was characterized by germinal center features, the second cluster showed a high abundance of stromal cells and extracellular matrix pathways, the third cluster was associated with inflammatory pathways, while the depleted cluster lacked markers of these three defined GEP signatures. These lymphoma microenvironment clusters showed an impact on PFS and OS regardless of COO or genetic subtype, underlining its independent contribution to lymphomagenesis and clinical presentations. These clusters also demonstrated large similarities with previously discussed COO, consensus clustering, stromal signatures, and genetically defined entities (LymphGen profiles and C1-C5 subtypes) [4,5,20,37].
Simultaneously, as another landmark, Steen et al. reported on their so-called “Ecotyper” -algorithm that was generated for either solid tumors or lymphomas (mainly DLBCL). The landscapes of (tumor) cell states and lymphoma ecosystems were examined by means of bulk or single-cell RNA-sequencing. In this study, the B-cell states represented the previously discussed COO including GCB and ABC subtypes, as well as the subdivision into centrocytes, centroblasts, memory B-cells, and plasmablasts established by the BAGS2CLINIC. Accordingly, these B-cell states were associated with survival, corresponding to the COO or BAGS2CLINIC classification, and molecular classification with the LymphGen and C1-C5 clusters [4,37]. Furthermore, other cell-type states were identified along with a total of 9 lymphoma ecotypes that congregated multiple cell-type states and were equally associated with survival outcomes [21]. Thus, Kotlov et al. [20] and Steen et al. [21] individually identified distinct microenvironment subtypes associated with molecular profiles and survival outcomes.
As summarized in Figure 3, complementing GEP studies focusing on tumor cell subtyping and the lymphoma TME contributed to the insight that several cellular subtypes within DLBCL phenotypes were related to survival. Conclusively, an association is shown between the presence of high numbers of M2-type macrophages, natural killer cells, plasma cells, or increased angiogenesis with inferior survival. In contrast, abundant infiltration of myofibroblasts, dendritic cells, CD4-positive T cells, CD14-positive monocytes, extracellular matrix deposition and histiocytes demonstrated superior survival. However, results should be interpreted cautiously because validation studies are lacking.
The prognostic impact of the TME signatures does not answer the question of whether these microenvironmental features indicate an underlying interaction of infiltrating cells with tumor cells that promote tumor growth or, in contrast, represent an ultimate consequence of cellular damage and is thus initiated by the tumor itself. GEP studies have demonstrated their value in mapping the DLBCL microenvironment. Nevertheless, unresolved biological issues on the interaction and activation status of diverse cell types in the TME remain and further research is needed to determine the true clinical effect of the interaction between lymphoma cells and the TME.
6. Clinical Impact and Future Perspectives of Gene-Expression Profiling Studies
As depicted in Figure 4, GEP analysis has the potential to elucidate the phenotype of the tumor, the composition of the TME, and the presence of immune surveillance mechanisms. Consequently, GEP studies have refined current DLBCL categorization towards a more biologically driven classification with different COO subtypes. Besides a more general consensus clustering, the GEP COO classification into ABC and GCB subtypes has changed the view of DLBCL’s biological behavior and is steadily regaining its place in diagnostic procedures above the surrogate Hans algorithm for COO based on immunohistochemical staining [27]. In addition, GEP COO classification is used to allocate patients with particular DLBCL subtypes to novel targeted clinical trials although a true clinical benefit has not yet been established [16,45,46,47].
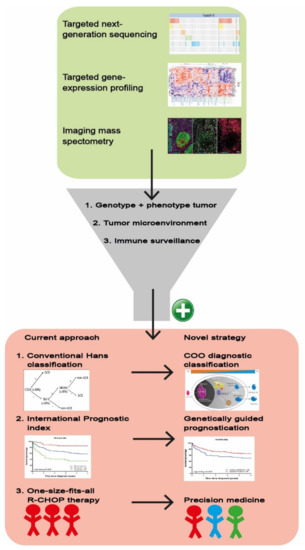
Figure 4.
Schematic overview of a multilayered research strategy. Combining targeted NGS, targeted GEP and imaging mass spectrometry allows for inclusive analysis of genotype, phenotype, TME and immune surveillance of the DLBCL. This methodology substantiates the conversion from the current approach towards a novel strategy including (1) Hans classification to COO diagnostic classification, (2) a general clinical prognostic score (International Prognostic Index) towards a biology-guided prognostication and ultimately (3) facilitating development from a one-size-fits-all R-CHOP treatment towards more precision medicine.
COO classification has been further developed into dark-zone-like and light-zone-like phenotypes or even more specific cell types, such as centrocytes, centroblasts, memory B-cells and plasmablasts, all harboring a prognostic impact. Other independently identified GEP clustering studies in DLBCL demonstrated predictive significance, such as stromal, immune-related, LAMIS, lymphoma microenvironment, “Ecotyper”, and other cellular-specific signatures (Table 1). However, despite the significant progress made in the refinement of these biological and predictive classifications, validation studies for these prognostic signatures are currently lacking and hinder direct implementation in routine clinical settings to optimize patient management and counseling. Besides that, GEP techniques are time consuming, many pathology departments are not equipped with these technologies, lack biostatistical tools needed for examining these signatures, and costs are currently not covered by most health insurances. Although optimizing the specificity of DLBCL classification is crucial to improving patient care, the challenge is to arrive at a consortium-oriented panel for discriminatory subtyping that is clinically relevant, easily accessible, has a short turnaround time, and is affordable for routine use.
GEP results can also be used to initiate a new era of therapeutic trials. Given the intermediate response to R-CHOP in DLBCL, better subtype classification of mainly high-risk DLBCL subgroups such as high-grade B-cell lymphoma could improve the effectiveness of targeted therapeutic strategies. Targeted therapies that complement or replace standard treatment have been investigated. For example, Davies et al. (2019) [16] studied the effect of adding bortezomib, a proteasome inhibitor, to R-CHOP treatment and reported no significant improvement in survival. Data retrieved from this trial, have been re-examined, identifying a DLBCL subgroup characterized by the prevalence of a distinct CD8 T-cell state that benefited from the addition of bortezomib to R-CHOP [21]. Therefore, further patient classification depending on COO status or particular signatures could improve the efficacy of bortezomib by more efficient upfront patient selection [48,49].
Kuo et al. [45] revealed that ibrutinib was less effective in ABC subtype DLBCL patients with high BCL2 expression. Wilson et al. [50] investigated this in more detail and reported that for patients of <60 years the event-free survival after treatment with ibrutinib and R-CHOP was 100% in the MCD (co-occurrence of MYD88L265P and CD79B mutations) and N1 (NOTCH1 mutations) subtype. Hartert et al. [51] described the favorable effect of adding lenalidomide to R-CHOP on event-free survival in patients with mutations in PIM1, SPEN or MYD88 or expression signatures including NF-κB, IRF4 and JAK-STAT. The addition of venetoclax to R-CHOP treatment reported worse outcomes than expected in patients overexpressing BCL2, underlining the necessity of analyzing involved pathways [52,53]. These examples show that intensifying molecular analysis is needed for the optimization of personalized treatment [54]. Consequently, this paves the way to re-evaluate previous clinical trials adopting targeted therapies, potentially providing new insights into the current conclusions.
Another important application of GEP results is in the management of resistance and efficacy of CAR T-cells and bispecific antibodies in DLBCL patients. These treatments have shown remarkable efficacy in chemorefractory DLBCL patients; however, in a significant proportion of patients, these therapies are still not effective [55,56,57,58,59]. Critical analysis of the TME and its influence on the effectivity of CAR T-cell and bispecific antibody therapy in DLBCL is lacking. GEP studies in DLBCL patients focusing on the TME will facilitate further evaluation of the disparate response to these novel treatments.
Kahle et al. [60] published a review on the contribution of molecular imaging to the understanding of the biology of lymphoma. Adding immunohistochemistry, proteomics, or imaging mass spectrometry to NGS and GEP data enables a multi-dimensional analysis of tumors related to the TME. For example, de Miranda et al. [61,62,63] performed imaging mass cytometry on tonsil and colorectal cancer tissues, thereby enhancing the understanding of the heterogeneous and intricate tumor-specific immune landscape of TME. Such molecular imaging analysis can complement current molecular evaluations in DLBCL to a more three-dimensional analysis, facilitating the identification of new mechanistic concepts. In addition, analysis of paired samples before and after an intervention will deepen the analysis of the TME, subclones and (acquired) therapeutic resistance [64]. The application of machine learning tools such as artificial intelligence will further enhance the utility of multi-omics data to better define distinct molecular and prognostic DLBCL subtypes [65].
In summary, to improve the understanding of the intricate molecular biology of DLBCL and the interaction with its TME, future studies should adopt a multi-dimensional strategy including immunohistochemistry, NGS, GEP and proteomics. Figure 4 shows such a multilayered approach and emphasizes that in addition to appropriate equipment, it also requires a diverse and well-trained team of molecular biologists, pathologists, hematologists, bioinformatics, and biostatisticians. Relevant findings of this multilayered analysis will ultimately be translated into manageable diagnostics that can be implemented by multiple medical centers.
7. Anatomical Localization and Age Matter
Together with other NGS studies, we demonstrated unique mutational profiles for DLBCL with a preferred localization, f.e., in primary central nervous system B-cell lymphoma, primary testicular lymphomas, intravascular large B-cell lymphoma, primary bone DLBCL, and primary cutaneous DLBCL leg-type [66,67,68,69,70,71,72,73]. These preferred anatomical localized DLBCLs were significantly associated with specific COO subtypes variably determined by GEP and Hans classification (Figure 5). Primary central nervous system B-cell lymphoma, primary breast DLBCL, intravascular large B-cell lymphoma, and primary testicular large B-cell lymphoma are mainly classified as ABC type lymphomas. In contrast, craniofacial, primary mediastinal (thymic) large B-cell lymphoma, primary ovarian DLBCL and primary bone DLBCL are mainly classified as GCB [67,74,75]. In addition, the Lymph3Cx GEP panel was developed as an update of the Lymph2Cx, which has distinguished primary mediastinal (thymic) large B-cell lymphoma from other DLBCL subtypes [75]. By applying a targeted NanoString panel we recently demonstrated that primary bone DLBCL mainly constitutes a centrocyte-like GCB-profile, while non-osseous DLBCL with a GCB subtype principally has a centroblast-like phenotype [67]. This conceptualizes that anatomical DLBCL localization is relevant for specific COO subtypes and even more for unique cellular phenotypes. In short, for COO subtypes, localization matters.
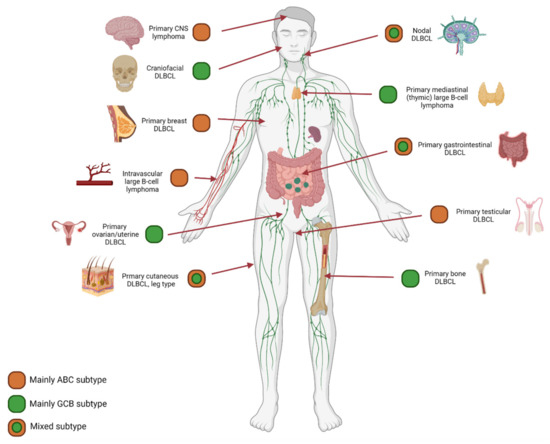
Figure 5.
COO subtype: Anatomical localization matters. The results of diverse studies using GEP or Hans classification for COO determination demonstrated an evident association between anatomical preferred localization and COO subtype. For example, primary central nervous system lymphoma, primary testicular lymphoma, and intravascular large B-cell lymphoma harbor predominantly an ABC subtype. In contrast, we recently demonstrated a GCB subtype for primary bone DLBCL, that could be specified even further to unique cellular phenotypes [67]. This concept calls for additional investigation of well-annotated homogeneous cohorts of preferred localization DLBCL, including in-depth molecular studies.
Another correlation was seen between age and COO subtype, as in the elderly an ABC subtype was predominant, indicating that COO follows the physiology of senescence and alteration of the T-cell repertoire [76,77]. Altogether, these concepts have further broadened the molecular view of DLBCL, as these techniques allow COO to even be considered down to the individual cell level. As we have mentioned earlier, these results confirm the additional value of exploring well-annotated homogeneous cohorts and appeal to the need for in-depth molecular studies of DLBCL with preferred localization [78].
8. A Proposal for a Consortium Gene-Expression Profiling Panel: BLYM-777
From a biological point of view, it is important to apply GEP analysis as broadly as possible. In practice, this is often not feasible, for instance if only limited amounts of archived FFPE material are available. For this reason, a targeted GEP approach is frequently used because it is clinically applicable and allows the analysis of a limited set of genes of interest. Based on reviewing more than 45 studies and considering the maximum number of 800 genes that can be analyzed using NanoString technology, we generated a targeted knowledge-based biology-driven (t)GEP consortium panel, called BLYM-777 (Figure 3 and Figure 6 and Appendix A). This BLYM-777 panel includes 777 genes involved in the NF-κB (f.e. MYD88, CD79B and CARD11), JAK/STAT (f.e., SOCS1, JAK1 and STAT1), MAPK (f.e., BCL2 and MEK2), NOTCH (f.e., NOTCH3 and TBL1XR1), PI3K (f.e., PTEN and PI3K) pathways that are known to be important in lymphomagenesis of DLBCL. In addition, the BLYM-777 includes genes relevant for COO identification, such as the original COO-classification NanoString tool Lymph2Cx (n = 20 genes) (f.e., IRF4, ITPKB, MME and MYBL1), the BAGS2CLINIC (n = 53) (f.e., STAT3 and IL16), and dark-/light-zone signature (n = 52) (f.e., B2M, CTLA4, KI67 and AICDA), all of which have individually been shown to facilitate DLBCL subtype classification [11,15,19]. As the interest in the TME increases in DLBCL, BLYM-777 additionally includes genes related to TME-focused signatures, such as the consensus clustering classification (n = 86) (f.e., CD37, TNFRSF1A and PDL1), LAMIS signature (n = 63) (f.e., CCND2 and CXCR4), a 45-gene TME assay (Ciavarella et al. n = 45) (f.e., COL1A1 and MMP2), lymphoma microenvironments (n = 155) and ecotypes (n = 314) [9,14,18,20,21]. This BLYM-777 design also covers other signatures relevant for DLBCL, such as the DHITsig (n = 35) (f.e., ETV6 and RGCC) since DHIT lymphomas show an inferior survival, genes relevant in MYC driven B-cell lymphomas (n = 80) (f.e., RFC3 and TRAP1), genes upregulated in wildtype-TP53 DLBCL with high mutational burden (n = 37) (f.e., HDAC1 and BBC3), and genes relevant for the identification of resistance to CAR T-cell or bispecific antibody treatment (n = 35) (f.e., CD58 and FOXP1) [12,17,55,56,57,79,80,81,82,83,84,85,86,87,88]. To evaluate the influence of mutations on gene expression, 95 genes important for current molecular classification based on NGS results have also been included [67]. Supplementary Table S1: BLYM-777 included genes per author, lists all genes belonging to this BLYM-777 tGEP consortium panel. To approach multiple biologically and clinically relevant questions on the lymphomagenesis of DLBCL, such as the relevance of certain mutations, the interaction of malignant cells with different components of the TME, immune surveillance and effectivity of new treatment strategies, the proposed BLYM-777 panel can be deployed in combination with other molecular characterization techniques. In addition, BLYM-777 is ready to use with NanoString technology and benefits from low-threshold accessibility and good performance using RNA isolated from FFPE material. However, analysis of the proposed panel of 777 genes is also possible by selective bioinformatic analysis of whole transcriptome data or targeted expression data generated by other platforms. The advantages and disadvantages of different gene-expression detection technologies have been extensively described by Narrandes et al. and Jiang et al. [22,23] and are beyond the scope of this clinical translational review.
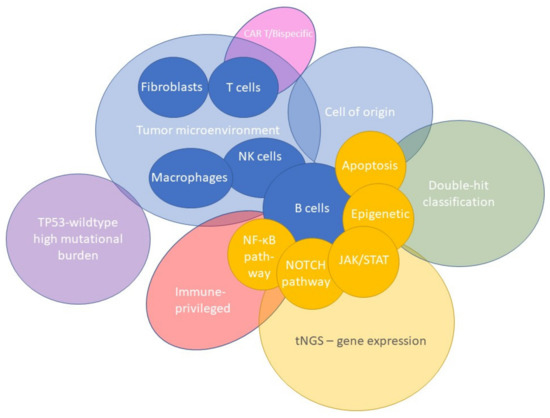
Figure 6.
A proposal for a targeted BLYM-777 consortium panel. Based on 45 studies, we propose a knowledge-based, biology-driven targeted (t)GEP consortium panel, called BLYM-777. This BLYM-777 panel primarily focuses on DLBCL and covers 777 B-cell lymphoma relevant genes, including their involved pathways (f.e., NF-κB, NOTCH, PI3K). Accordingly, genes were included for COO identification, TME-focused signatures, ecotypes, DHITsig, differentially expressed genes found in wildtype-TP53 DLBCL, and genes relevant for resistance to CAR T-cell or bispecific antibody therapy. Moreover, 95 genes important for current molecular classification based on NGS results have been included.
In summary, the BLYM-777 panel covers many aspects of B-cell lymphomagenesis, COO classification, therapeutic efficacy and TME-focused signatures and can facilitate subsequent molecular investigations. The authors of this publication are currently in discussion with NanoString with the goal to create this consortium gene-expression BLYM-777 panel capturing the biology mentioned in this work. Such a panel could bring value to the hematological field by providing a standardized tool to facilitate collaboration and shared learnings throughout the community. If you are interested in joining this consortium effort, please respond to the corresponding author for more information.
9. Conclusion
This review provides a comprehensive overview of current molecular insights into the biological background of DLBCL obtained by several GEP technologies. These methods utilized in DLBCL studies identified several GEP signatures including cell-of-origin discrimination in GCB and ABC subtypes and an in-depth analysis of the TME regarding the exact cell type and state. Combining GEP with other NGS and proteomic-based methodologies will facilitate a multi-layered analysis and a next step forward in understanding biological principles and elucidating the genetic heterogeneity of DLBCL. The proposed novel knowledge-based biology-driven consortium tGEP panel, named BLYM-777, encompasses many aspects of B-cell lymphomagenesis, TME and immune surveillance and is thereby expected to gain new molecular concepts of DLBCL lymphomagenesis. Applying BLYM-777 in such a multilayered methodology potentially enhances diagnostic classification of DLBCL subtypes, prognostication, and ultimately the development of novel targeted therapeutic strategies improving patient survival.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cancers14081857/s1, Table S1: BLYM-777 included genes per author.
Author Contributions
Conceptualization, A.D., A.v.d.B., F.A.d.G., R.A.L.d.G., J.S.P.V., M.N.; original draft preparation, F.A.d.G. and R.A.L.d.G.; visualization, F.A.d.G. and R.A.L.d.G.; review and editing, P.M.J., K.H.L., P.G.N.J.M., C.J.M.v.N., M.E.D.C., W.B.C.S., J.R.P., R.M., M.J.K., M.M.W.v.d.P., T.T. and F.J.S.H.W.-a.-J.; supervision, J.S.P.V., M.N. and A.v.d.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
Table A1.
A proposal for a consortium gene-expression profiling panel: BLYM-777.
Table A1.
A proposal for a consortium gene-expression profiling panel: BLYM-777.
| ACTA2 | CKAP4 | HIST1H2BC | MPST | SBK1 |
| ACTB | CLCN1 | HLA-A | MRC1 | SCNN1D |
| ACTG1 | CLCN2 | HLA-B | MRC2 | SCOTIN |
| ACTG2 | CLU | HLA-C | MRPL15 | SAMD13 |
| ACTL7A | COL12A1 | HLA-DMA | MRPL3 | SELPLG |
| ADA | COL1A1 | HLA-DMB | MRPL33 | SEMA7A |
| ADHFE1 | COL1A2 | HLA-DPA1 | MRPS34 | SEP15 |
| AEBP1 | COL3A1 | HLA-DPB1 | MS4A1 | SERPINA1 |
| AEN | COL4A1 | HLA-DQA1 | MSH3 | SERPINA9 |
| AFMID | COL4A2 | HLA-DQB1 | MSR1 | SERPING1 |
| AGER | COL5A2 | HLA-DRA | MUC16 | SGK1 |
| AGR2 | COL6A3 | HLA-DRB1 | MXRA5 | SGPP2 |
| AHCY | COMMD8 | HLA-E | MYBBP1A | SH2D1A |
| AHR | COX7A2L | HMG20A | MYBL1 | SH2D1B |
| AICDA | CPD | HNRNPLL | MYBL2 | SH2D3C |
| AKAP1 | CPNE3 | HPDL | MYC | SH3PXD2A |
| AKAP5 | CPT1A | HRK | MYD88 | SHARPIN |
| AKR1D1 | CREB1 | HS3ST3A1 | NBR1 | SHISA8 |
| ALCAM | CREB3L2 | HSPBL2 | NCAM1 | SIGLEC9 |
| ALDH3B1 | CREBBP | HTR1A | NCR1 | SIK1 |
| ALOX5 | CSF1 | ICAM1 | NCR3 | SIPA1L3 |
| AMT | CSF1R | ICOS | NDUFB1 | SKAP2 |
| ANAPC16 | CTHRC1 | IDH1 | NEMF | SLAMF1 |
| ANGPT1 | CTLA4 | IDO1 | NFAM1 | SLAMF8 |
| ANGPT2 | CTNNA1 | IFITM1 | NFATC2 | SLC12A8 |
| ANO9 | CTNNB1 | IFNA16 | NFKB1 | SLC16A9 |
| ANTXR2 | CTPS1 | IFNAR1 | NFKB2 | SLC25A27 |
| AP1B1 | CTSB | IFNG | NFKBIE | SLC29A3 |
| APLP2 | CTSK | IGHM | NKG7 | SLC2A3 |
| APOL6 | CTSZ | IGLL3 | NME1 | SLC41A1 |
| APRIL | CX3CL1 | IGLL5 | NOD2 | SLFN5 |
| ARG1 | CX3CR1 | IGSF10 | NOLC1 | SMAD1 |
| ARHGAP17 | CXCL10 | IGSF6 | NOTCH3 | SMARCA5 |
| ARID1B | CXCL11 | IK | NPFF | SMIM14 |
| ARSI | CXCL12 | IKZF2 | NPFFR2 | SNHG19 |
| ASB13 | CXCL13 | IKZF4 | NR4A2 | SOCS1 |
| ASNSD1 | CXCL5 | IL10 | NRF1 | SOD1 |
| ASPH | CXCL8 | IL15 | NRN1L | SP3 |
| ATM | CXCL9 | IL16 | NSA2 | SPARC |
| ATP5D | CXCR2 | IL18BP | NSUN2 | SPEN |
| ATRAID | CXCR3 | IL1R1 | NSUN5 | SPI1 |
| AURKA | CXCR4 | IL2 | NTRK1 | SPIB |
| AURKB | CXCR5 | IL21 | OAZ1 | SPP1 |
| B2M | CYB5R2 | IL21R | OPA1 | SRM |
| BATF | DAB2 | IL22 | OR13A1 | SSBP3 |
| BATF3 | DBI | IL2RA | OR4D5 | STAM |
| BAX | DBP | IL2RB | OSBPL10 | STAP1 |
| BBC3 | DDX11 | IL4 | OSMR | STAT1 |
| BCAS4 | DDX21 | IL4I1 | OTULIN | STAT2 |
| BCL10 | DDX6 | IL6 | OXTR | STAT3 |
| BCL11B | DHRS2 | IL6R | P2RY12 | STAT6 |
| BCL2 | DHX33 | IL6ST | P2RY14 | STAU1 |
| BCL2A1 | DKK3 | IL7R | PABPC3 | STC2 |
| BCL2L1 | DNAJB12 | INHBA | PAICS | SULF1 |
| BCL2L12 | DPP8 | INPP5D | PALLD | SYBU |
| BCL6 | DPYSL3 | INSM2 | PAPSS2 | SYNE1 |
| BCL7A | DTX1 | IQCD | PARP1 | TADA2B |
| BCLAF1 | DTX3L | IRF1 | PARP3 | TAP1 |
| BGLAP | DUSP2 | IRF2BP2 | PATL2 | TAP2 |
| BGN | DUSP4 | IRF4 | PAX5 | TBL1XR1 |
| BID | DUSP5 | IRG1 | PAX8-AS1 | TBP |
| BIRC2 | E2F1 | IRS2 | PCDH9 | TBX21 |
| BIRC3 | EARS2 | ISY1 | PCLAF | TCIRG1 |
| BLK | EBER1 | ITGA6 | PCNP | TCL1A |
| BRAF | EBER2 | ITGB2 | PCOLCE | TCP10 |
| BSG | EBF1 | ITGB8 | PDCD1 | TEDC2 |
| BST1 | EBI3 | ITK | PDCD10 | TEK |
| BTBD3 | EBNA1BP2 | ITM2A | PDCD1LG2 | TESPA1 |
| BTC | EEPD1 | ITPKB | PDE5A | TET2 |
| BTG1 | EGFR | ITPR2 | PDGFC | TGFBI |
| BTG2 | EGR1 | JAK1 | PDGFRB | THBS2 |
| BUB1 | EGR3 | JAK2 | PDPN | THPO |
| C10orf128 | ELL2 | JAK3 | PDXDC1 | TIGIT |
| C14orf70 | EMCN | JAKMIP1 | PECAM1 | TIM3 |
| C16orf54 | EOMES | JAML | PEG10 | TIMP1 |
| C17orf56 | EP300 | JCHAIN | PERP | TIMP2 |
| C19orf24 | EPHA4 | KCNA4 | PGF | TIMP3 |
| C2 | ERCC2 | KCNH4 | PHB2 | TINAGL1 |
| C3AR1 | ERN1 | KCNU1 | PHF23 | TJP1 |
| C3orf22 | ESCO2 | KDR | PIK3CA | TLR8 |
| C3orf37 | ETFA | KI67 | PILRA | TMEM119 |
| CA9 | ETS1 | KIAA1128 | PIM1 | TMEM127 |
| CABP2 | ETV6 | KIAA1462 | PIM2 | TMEM135 |
| CACNA1I | EZH2 | KIF14 | PKA | TMEM140 |
| CACNA2D2 | EZR | KIR2DL4 | PLCG2 | TMEM175 |
| CADM4 | F8A3 | KIT | PLCH2 | TMEM202 |
| CALR | FABP5 | KLF2 | PLD3 | TMEM219 |
| CAMK1D | FADD | KLHL14 | PLK1 | TMEM224 |
| CAPS | FAM108C1 | KLHL6 | PLOD2 | TMEM30A |
| CARD10 | FAM117B | KLRC2 | PMP22 | TMEM47 |
| CARD11 | FAM13AOS | KLRF1 | PMPCB | TMEM97 |
| CARD14 | FAM153A | KLRK1 | PMS2P2 | TMSB4X |
| CARD9 | FAM216A | KMT2D | PMS2P9 | TNF |
| CASP10 | FAM26F | KRAS | POLD2 | TNFAIP3 |
| CASP8 | FAS | KRT73 | POLH | TNFRSF10B |
| CBLB | FASLG | LAG3 | POLR1B | TNFRSF13B |
| CCDC154 | FASN | LAMB1 | POSTN | TNFRSF13C |
| CCDC50 | FAT4 | LAMP1 | POTEC | TNFRSF14 |
| CCDC6 | FBL | LDHB | POU6F1 | TNFRSF17 |
| CCL20 | FBLN2 | LGALS7 | PPAT | TNFRSF18 |
| CCL4 | FBLN7 | LGALS9 | PPP1R3B | TNFRSF1A |
| CCL5 | FBXW7 | LIMD1 | PPRC1 | TNFRSF1B |
| CCNB1 | FCER1G | LINC01215 | PRDM1 | TNFRSF4 |
| CCND1 | FCGR1B | LMO2 | PRDX5 | TNFRSF9 |
| CCND2 | FCRL5 | LOC100128071 | PRKCH | TNFSF13B |
| CCND3 | FDCSP | LOC100128682 | PRKCQ | TNFSF8 |
| CCNE1 | FEM1C | LOC100131225 | PRMT1 | TOX |
| CCR6 | FGD5 | LOC100131354 | PRNP | TP53 |
| CCR8 | FGFBP2 | LOC100287094 | PSAT1 | TPO |
| CD11C | FIBP | LOC100287259 | PSEN1 | TPT1 |
| CD160 | FLJ37307 | LOC100287308 | PSIMCT.1 | TRAC |
| CD163 | FLJ37786 | LOC100288639 | PSMA2 | TRAF1 |
| CD19 | FLT1 | LOC100288728 | PSMA5 | TRAF2 |
| CD2 | FN1 | LOC100289566 | PSMA6 | TRAP1 |
| CD20 | FNDC1 | LOC196415 | PSMB10 | TRAT1 |
| CD22 | FNDC3B | LOC284889 | PSMB9 | TRBC1 |
| CD226 | FOXJ3 | LOC391358 | PSMD14 | TRIAL-R1 |
| CD24 | FOXP1 | LOC401433 | PSMD3 | TRIM21 |
| CD244 | FOXP3 | LOC440311 | PTEN | TRIM56 |
| CD274 | FSTL1 | LOC729535 | PTGES2 | TRRAP |
| CD276 | FYB | LRP12 | PTPN11 | TSKU |
| CD28 | FYN | LRP1B | PTPN13 | TSPAN9 |
| CD300LF | GABRB1 | LRP8 | PTPRC | TTC8 |
| CD37 | GAMT | LSM1 | PTTG1IP | UBA1 |
| CD39 | GATA2 | LTB | QRSL1 | UBASH3A |
| CD3D | GATA3 | LTBR | R3HDM1 | UBE2D2 |
| CD3E | GATAD2B | LUM | RAB27A | UBXN4 |
| CD3G | GBP1 | LY6E | RAB29 | UBXN7 |
| CD4 | GBP5 | LY75 | RAB33A | UCHL3 |
| CD40 | GDF2 | LYAR | RAB3GAP2 | UCK2 |
| CD40LG | GEMIN4 | MAF | RAB7A | VASP |
| CD44 | GIT2 | MAFB | RABEPK | VCAM1 |
| CD47 | GLRX | MAG | RAD54L | VEGFA |
| CD58 | GNA13 | MALT1 | RAG2 | VEGFB |
| CD6 | GNAI2 | MAML3 | RANBP1 | VEGFC |
| CD68 | GNG12 | MAP2 | RASA1 | VISTA |
| CD70 | GNLY | MAP2K2 | RASGRF1 | VPS24 |
| CD79A | GOT2 | MAP4K1 | RASL11A | VRK3 |
| CD79B | GPNMB | MARCKSL1 | RBL2 | VTN |
| CD80 | GPR124 | MCL1 | RBPJL | VWF |
| CD81 | GPR137B | MCM2 | REL | WAC |
| CD83 | GPRIN3 | MCM6 | RELA | WASH2P |
| CD84 | GRHPR | MDFIC | RELB | WASL |
| CD8A | GRIN3A | MDM2 | RFC3 | WDR3 |
| CDC25A | GRN | MED23 | RFFL | WDR55 |
| CDCA7L | GSK3B | MEF2B | RGCC | XBP1 |
| CDH23 | GUK1 | MEX3C | RNF130 | XRCC3 |
| CDH5 | GZMB | MFAP2 | RNF213 | XRCC5 |
| CDK2 | GZMH | MFGE8 | ROCK1 | ZBTB4 |
| CDK4 | HDAC1 | MIF | RPLP0 | ZEB2 |
| CDK5R1 | HERPUD2 | MIR155HG | RRS1 | ZFAND4 |
| CDKN2A | HIF1A | MME | RUBCNL | ZNF22 |
| CDKN2B | HIST1H1C | MMP14 | RUNX3 | ZNF438 |
| CETN3 | HIST1H1D | MMP2 | S100A11 | |
| CFLAR | HIST1H1E | MMP9 | S100Z | |
| CIITA | HIST1H2AC | MPEG1 | S1PR2 |
References
- Ernst, M.; Oeser, A.; Besiroglu, B.; Caro-Valenzuela, J.; Abd El Aziz, M.; Monsef, I.; Borchmann, P.; Estcourt, L.J.; Skoetz, N.; Goldkuhle, M. Chimeric antigen receptor (CAR) T-cell therapy for people with relapsed or refractory diffuse large B-cell lymphoma. Cochrane Database Syst. Rev. 2021, 9, Cd013365. [Google Scholar] [PubMed]
- Miyazaki, K. Treatment of Diffuse Large B-Cell Lymphoma. J. Clin. Exp. Hematop. 2016, 56, 79–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, A.; Zhang, J.; Davis, N.S.; Moffitt, A.B.; Love, C.L.; Waldrop, A.; Leppa, S.; Pasanen, A.; Meriranta, L.; Karjalainen-Lindsberg, M.L.; et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017, 171, 481–494.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Gentles, A.J.; Alencar, A.J.; Liu, C.L.; Kohrt, H.E.; Houot, R.; Goldstein, M.J.; Zhao, S.; Natkunam, Y.; Advani, R.H.; et al. Prediction of survival in diffuse large B-cell lymphoma based on the expression of 2 genes reflecting tumor and microenvironment. Blood 2011, 118, 1350–1358. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef]
- Rosenwald, A.; Wright, G.; Chan, W.C.; Connors, J.M.; Campo, E.; Fisher, R.I.; Gascoyne, R.D.; Muller-Hermelink, H.K.; Smeland, E.B.; Giltnane, J.M.; et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002, 346, 1937–1947. [Google Scholar] [CrossRef]
- Monti, S.; Savage, K.J.; Kutok, J.L.; Feuerhake, F.; Kurtin, P.; Mihm, M.; Wu, B.; Pasqualucci, L.; Neuberg, D.; Aguiar, R.C.; et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood 2005, 105, 1851–1861. [Google Scholar] [CrossRef] [Green Version]
- Lenz, G.; Wright, G.; Dave, S.S.; Xiao, W.; Powell, J.; Zhao, H.; Xu, W.; Tan, B.; Goldschmidt, N.; Iqbal, J.; et al. Stromal gene signatures in large-B-cell lymphomas. N. Engl. J. Med. 2008, 359, 2313–2323. [Google Scholar] [CrossRef] [Green Version]
- Scott, D.W.; Wright, G.W.; Williams, P.M.; Lih, C.J.; Walsh, W.; Jaffe, E.S.; Rosenwald, A.; Campo, E.; Chan, W.C.; Connors, J.M.; et al. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood 2014, 123, 1214–1217. [Google Scholar] [CrossRef] [PubMed]
- Carey, C.D.; Gusenleitner, D.; Chapuy, B.; Kovach, A.E.; Kluk, M.J.; Sun, H.H.; Crossland, R.E.; Bacon, C.M.; Rand, V.; Dal Cin, P.; et al. Molecular classification of MYC-driven B-cell lymphomas by targeted gene expression profiling of fixed biopsy specimens. J. Mol. Diagn. 2015, 17, 19–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dybkaer, K.; Bogsted, M.; Falgreen, S.; Bodker, J.S.; Kjeldsen, M.K.; Schmitz, A.; Bilgrau, A.E.; Xu-Monette, Z.Y.; Li, L.; Bergkvist, K.S.; et al. Diffuse large B-cell lymphoma classification system that associates normal B-cell subset phenotypes with prognosis. J. Clin. Oncol. 2015, 33, 1379–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciavarella, S.; Vegliante, M.C.; Fabbri, M.; De Summa, S.; Melle, F.; Motta, G.; De Iuliis, V.; Opinto, G.; Enjuanes, A.; Rega, S.; et al. Dissection of DLBCL microenvironment provides a gene expression-based predictor of survival applicable to formalin-fixed paraffin-embedded tissue. Ann. Oncol. 2018, 29, 2363–2370. [Google Scholar] [CrossRef] [Green Version]
- Michaelsen, T.Y.; Richter, J.; Brondum, R.F.; Klapper, W.; Johnsen, H.E.; Albertsen, M.; Dybkaer, K.; Bogsted, M. A B-cell-associated gene signature classification of diffuse large B-cell lymphoma by NanoString technology. Blood Adv. 2018, 2, 1542–1546. [Google Scholar] [CrossRef]
- Davies, A.; Cummin, T.E.; Barrans, S.; Maishman, T.; Mamot, C.; Novak, U.; Caddy, J.; Stanton, L.; Kazmi-Stokes, S.; McMillan, A.; et al. Gene-expression profiling of bortezomib added to standard chemoimmunotherapy for diffuse large B-cell lymphoma (REMoDL-B): An open-label, randomised, phase 3 trial. Lancet Oncol. 2019, 20, 649–662. [Google Scholar] [CrossRef] [Green Version]
- Ennishi, D.; Jiang, A.; Boyle, M.; Collinge, B.; Grande, B.M.; Ben-Neriah, S.; Rushton, C.; Tang, J.; Thomas, N.; Slack, G.W.; et al. Double-Hit Gene Expression Signature Defines a Distinct Subgroup of Germinal Center B-Cell-Like Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2019, 37, 190–201. [Google Scholar] [CrossRef]
- Staiger, A.M.; Altenbuchinger, M.; Ziepert, M.; Kohler, C.; Horn, H.; Huttner, M.; Huttl, K.S.; Glehr, G.; Klapper, W.; Szczepanowski, M.; et al. A novel lymphoma-associated macrophage interaction signature (LAMIS) provides robust risk prognostication in diffuse large B-cell lymphoma clinical trial cohorts of the DSHNHL. Leukemia 2020, 34, 543–552. [Google Scholar] [CrossRef]
- Tripodo, C.; Zanardi, F.; Iannelli, F.; Mazzara, S.; Vegliante, M.; Morello, G.; Di Napoli, A.; Mangogna, A.; Facchetti, F.; Sangaletti, S.; et al. A Spatially Resolved Dark- versus Light-Zone Microenvironment Signature Subdivides Germinal Center-Related Aggressive B Cell Lymphomas. iScience 2020, 23, 101562. [Google Scholar] [CrossRef]
- Kotlov, N.; Bagaev, A.; Revuelta, M.V.; Phillip, J.M.; Cacciapuoti, M.T.; Antysheva, Z.; Svekolkin, V.; Tikhonova, E.; Miheecheva, N.; Kuzkina, N.; et al. Clinical and Biological Subtypes of B-cell Lymphoma Revealed by Microenvironmental Signatures. Cancer Discov. 2021, 11, 1468–1489. [Google Scholar] [CrossRef]
- Steen, C.B.; Luca, B.A.; Esfahani, M.S.; Azizi, A.; Sworder, B.J.; Nabet, B.Y.; Kurtz, D.M.; Liu, C.L.; Khameneh, F.; Advani, R.H.; et al. The landscape of tumor cell states and ecosystems in diffuse large B cell lymphoma. Cancer Cell 2021, 39, 1422–1437.e10. [Google Scholar] [CrossRef] [PubMed]
- Narrandes, S.; Xu, W. Gene Expression Detection Assay for Cancer Clinical Use. J. Cancer 2018, 9, 2249–2265. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Zhou, X.; Li, R.; Michal, J.J.; Zhang, S.; Dodson, M.V.; Zhang, Z.; Harland, R.M. Whole transcriptome analysis with sequencing: Methods, challenges and potential solutions. Cell. Mol. Life Sci. 2015, 72, 3425–3439. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.N.; Fu, K.; Greiner, T.; Smith, L.; Delabie, J.; Gascoyne, R.; Ott, G.; Rosenwald, A.; Braziel, R.; Campo, E.; et al. The stromal cell marker SPARC predicts for survival in patients with diffuse large B-cell lymphoma treated with rituximab. Am. J. Clin. Pathol. 2011, 135, 54–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keane, C.; Gill, D.; Vari, F.; Cross, D.; Griffiths, L.; Gandhi, M. CD4+ tumor infiltrating lymphocytes are prognostic and independent of R-IPI in patients with DLBCL receiving R-CHOP chemo-immunotherapy. Am. J. Hematol. 2013, 88, 273–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdou, A.G.; Asaad, N.; Kandil, M.; Shabaan, M.; Shams, A. Significance of stromal-1 and stromal-2 signatures and biologic prognostic model in diffuse large B-cell lymphoma. Cancer Biol. Med. 2017, 14, 151–161. [Google Scholar] [CrossRef] [Green Version]
- Hans, C.P.; Weisenburger, D.D.; Greiner, T.C.; Gascoyne, R.D.; Delabie, J.; Ott, G.; Muller-Hermelink, H.K.; Campo, E.; Braziel, R.M.; Jaffe, E.S.; et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004, 103, 275–282. [Google Scholar] [CrossRef]
- Lenz, G.; Nagel, I.; Siebert, R.; Roschke, A.V.; Sanger, W.; Wright, G.W.; Dave, S.S.; Tan, B.; Zhao, H.; Rosenwald, A.; et al. Aberrant immunoglobulin class switch recombination and switch translocations in activated B cell-like diffuse large B cell lymphoma. J. Exp. Med. 2007, 204, 633–643. [Google Scholar] [CrossRef]
- Roider, T.; Seufert, J.; Uvarovskii, A.; Frauhammer, F.; Bordas, M.; Abedpour, N.; Stolarczyk, M.; Mallm, J.P.; Herbst, S.A.; Bruch, P.M.; et al. Dissecting intratumour heterogeneity of nodal B-cell lymphomas at the transcriptional, genetic and drug-response levels. Nat. Cell Biol. 2020, 22, 896–906. [Google Scholar] [CrossRef]
- Plaça, J.R.; Diepstra, A.; Los, T.; Mendeville, M.; Seitz, A.; Lugtenburg, P.J.; Zijlstra, J.; Lam, K.; da Silva, W.A., Jr.; Ylstra, B.; et al. Reproducibility of Gene Expression Signatures in Diffuse Large B-Cell Lymphoma. Cancers 2022, 14, 1346. [Google Scholar] [CrossRef]
- Chamuleau, M.E.D.; Burggraaff, C.N.; Nijland, M.; Bakunina, K.; Mous, R.; Lugtenburg, P.J.; Dierickx, D.; van Imhoff, G.W.; Vermaat, J.S.P.; Marijt, E.A.F.; et al. Treatment of patients with MYC rearrangement positive large B-cell lymphoma with R-CHOP plus lenalidomide: Results of a multicenter HOVON phase II trial. Haematologica 2020, 105, 2805–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Küppers, R.; Rajewsky, K.; Hansmann, M.L. Diffuse large cell lymphomas are derived from mature B cells carrying V region genes with a high load of somatic mutation and evidence of selection for antibody expression. Eur. J. Immunol. 1997, 27, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trabucco, S.E.; Sokol, E.S.; Maund, S.L.; Moore, J.A.; Frampton, G.M.; Albacker, L.A.; Oestergaard, M.Z.; Venstrom, J.; Sehn, L.H.; Bolen, C.R. Prediction and characterization of diffuse large B-cell lymphoma cell-of-origin subtypes using targeted sequencing. Future Oncol. 2021, 17, 4171–4183. [Google Scholar] [CrossRef]
- Karube, K.; Enjuanes, A.; Dlouhy, I.; Jares, P.; Martin-Garcia, D.; Nadeu, F.; Ordonez, G.R.; Rovira, J.; Clot, G.; Royo, C.; et al. Integrating genomic alterations in diffuse large B-cell lymphoma identifies new relevant pathways and potential therapeutic targets. Leukemia 2018, 32, 675–684. [Google Scholar] [CrossRef]
- Wright, G.W.; Huang, D.W.; Phelan, J.D.; Coulibaly, Z.A.; Roulland, S.; Young, R.M.; Wang, J.Q.; Schmitz, R.; Morin, R.D.; Tang, J.; et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 2020, 37, 551–568.e14. [Google Scholar] [CrossRef]
- Lacy, S.E.; Barrans, S.L.; Beer, P.A.; Painter, D.; Smith, A.G.; Roman, E.; Cooke, S.L.; Ruiz, C.; Glover, P.; Van Hoppe, S.J.L.; et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: A Haematological Malignancy Research Network report. Blood 2020, 135, 1759–1771. [Google Scholar] [CrossRef]
- Weber, T.; Schmitz, R. Molecular Subgroups of Diffuse Large B Cell Lymphoma: Biology and Implications for Clinical Practice. Curr. Oncol. Rep. 2022, 24, 13–21. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, L.; Guan, Y.Q.; Shen, K.F.; Zhang, M.L.; Cai, H.D.; Wang, J.C.; Wang, Y.; Huang, L.; Cao, Y.; et al. Novel bioinformatic classification system for genetic signatures identification in diffuse large B-cell lymphoma. BMC Cancer 2020, 20, 714. [Google Scholar] [CrossRef]
- Shouval, R.; Alarcon Tomas, A.; Fein, J.A.; Flynn, J.R.; Markovits, E.; Mayer, S.; Olaide Afuye, A.; Alperovich, A.; Anagnostou, T.; Besser, M.J.; et al. Impact of TP53 Genomic Alterations in Large B-Cell Lymphoma Treated With CD19-Chimeric Antigen Receptor T-Cell Therapy. J. Clin. Oncol. 2021, 40, jco2102143. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcelis, L.; Antoranz, A.; Delsupehe, A.M.; Biesemans, P.; Ferreiro, J.F.; Debackere, K.; Vandenberghe, P.; Verhoef, G.; Gheysens, O.; Cattoretti, G.; et al. In-depth characterization of the tumor microenvironment in central nervous system lymphoma reveals implications for immune-checkpoint therapy. Cancer Immunol. Immunother. 2020, 69, 1751–1766. [Google Scholar] [CrossRef] [PubMed]
- Keane, C.; Vari, F.; Hertzberg, M.; Cao, K.A.; Green, M.R.; Han, E.; Seymour, J.F.; Hicks, R.J.; Gill, D.; Crooks, P.; et al. Ratios of T-cell immune effectors and checkpoint molecules as prognostic biomarkers in diffuse large B-cell lymphoma: A population-based study. Lancet Haematol. 2015, 2, e445–e455. [Google Scholar] [CrossRef] [Green Version]
- Kuo, H.P.; Ezell, S.A.; Schweighofer, K.J.; Cheung, L.W.K.; Hsieh, S.; Apatira, M.; Sirisawad, M.; Eckert, K.; Hsu, S.J.; Chen, C.T.; et al. Combination of Ibrutinib and ABT-199 in Diffuse Large B-Cell Lymphoma and Follicular Lymphoma. Mol. Cancer Ther. 2017, 16, 1246–1256. [Google Scholar] [CrossRef] [Green Version]
- Nowakowski, G.S.; Chiappella, A.; Gascoyne, R.D.; Scott, D.W.; Zhang, Q.; Jurczak, W.; Özcan, M.; Hong, X.; Zhu, J.; Jin, J.; et al. ROBUST: A Phase III Study of Lenalidomide Plus R-CHOP Versus Placebo Plus R-CHOP in Previously Untreated Patients With ABC-Type Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2021, 39, 1317–1328. [Google Scholar] [CrossRef]
- Younes, A.; Sehn, L.H.; Johnson, P.; Zinzani, P.L.; Hong, X.; Zhu, J.; Patti, C.; Belada, D.; Samoilova, O.; Suh, C.; et al. Randomized Phase III Trial of Ibrutinib and Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone in Non-Germinal Center B-Cell Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2019, 37, 1285–1295. [Google Scholar] [CrossRef]
- Richardson, P.G.; Sonneveld, P.; Schuster, M.W.; Irwin, D.; Stadtmauer, E.A.; Facon, T.; Harousseau, J.L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H.; et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2005, 352, 2487–2498. [Google Scholar] [CrossRef] [Green Version]
- Robak, T.; Huang, H.; Jin, J.; Zhu, J.; Liu, T.; Samoilova, O.; Pylypenko, H.; Verhoef, G.; Siritanaratkul, N.; Osmanov, E.; et al. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N. Engl. J. Med. 2015, 372, 944–953. [Google Scholar] [CrossRef] [Green Version]
- Wilson, W.H.; Wright, G.W.; Huang, D.W.; Hodkinson, B.; Balasubramanian, S.; Fan, Y.; Vermeulen, J.; Shreeve, M.; Staudt, L.M. Effect of ibrutinib with R-CHOP chemotherapy in genetic subtypes of DLBCL. Cancer Cell 2021, 39, 1643–1653.e3. [Google Scholar] [CrossRef]
- Hartert, K.T.; Wenzl, K.; Krull, J.E.; Manske, M.; Sarangi, V.; Asmann, Y.; Larson, M.C.; Maurer, M.J.; Slager, S.; Macon, W.R.; et al. Targeting of inflammatory pathways with R2CHOP in high-risk DLBCL. Leukemia 2021, 35, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Morschhauser, F.; Feugier, P.; Flinn, I.W.; Gasiorowski, R.; Greil, R.; Illés, Á.; Johnson, N.A.; Larouche, J.F.; Lugtenburg, P.J.; Patti, C.; et al. A phase 2 study of venetoclax plus R-CHOP as first-line treatment for patients with diffuse large B-cell lymphoma. Blood 2021, 137, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Coccaro, N.; Anelli, L.; Zagaria, A.; Perrone, T.; Specchia, G.; Albano, F. Molecular Complexity of Diffuse Large B-Cell Lymphoma: Can It Be a Roadmap for Precision Medicine? Cancers 2020, 12, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, R.D.; Arthur, S.E.; Hodson, D.J. Molecular profiling in diffuse large B-cell lymphoma: Why so many types of subtypes? Br. J. Haematol. 2021, 196, 814–829. [Google Scholar] [CrossRef]
- Scarfò, I.; Maus, M.V. Current approaches to increase CAR T cell potency in solid tumors: Targeting the tumor microenvironment. J. Immunother. Cancer 2017, 5, 28. [Google Scholar] [CrossRef] [Green Version]
- Van de Donk, N.; Themeli, M.; Usmani, S.Z. Determinants of response and mechanisms of resistance of CAR T-cell therapy in multiple myeloma. Blood Cancer Discov. 2021, 2, 302–318. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Viardot, A.; Goebeler, M.E.; Hess, G.; Neumann, S.; Pfreundschuh, M.; Adrian, N.; Zettl, F.; Libicher, M.; Sayehli, C.; Stieglmaier, J.; et al. Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood 2016, 127, 1410–1416. [Google Scholar] [CrossRef]
- Hutchings, M.; Morschhauser, F.; Iacoboni, G.; Carlo-Stella, C.; Offner, F.C.; Sureda, A.; Salles, G.; Martínez-Lopez, J.; Crump, M.; Thomas, D.N.; et al. Glofitamab, a Novel, Bivalent CD20-Targeting T-Cell-Engaging Bispecific Antibody, Induces Durable Complete Remissions in Relapsed or Refractory B-Cell Lymphoma: A Phase I Trial. J. Clin. Oncol. 2021, 39, 1959–1970. [Google Scholar] [CrossRef]
- Kahle, X.U.; Montes de Jesus, F.M.; Glaudemans, A.; Lub-de Hooge, M.N.; Jorritsma-Smit, A.; Plattel, W.J.; van Meerten, T.; Diepstra, A.; van den Berg, A.; Kwee, T.C.; et al. Molecular imaging in lymphoma beyond (18)F-FDG-PET: Understanding the biology and its implications for diagnostics and therapy. Lancet Haematol. 2020, 7, e479–e489. [Google Scholar] [CrossRef]
- Ijsselsteijn, M.E.; van der Breggen, R.; Farina Sarasqueta, A.; Koning, F.; de Miranda, N. A 40-Marker Panel for High Dimensional Characterization of Cancer Immune Microenvironments by Imaging Mass Cytometry. Front. Immunol. 2019, 10, 2534. [Google Scholar] [CrossRef] [PubMed]
- Newell, E.W.; Cheng, Y. Mass cytometry: Blessed with the curse of dimensionality. Nat. Immunol. 2016, 17, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Bandura, D.R.; Baranov, V.I.; Ornatsky, O.I.; Antonov, A.; Kinach, R.; Lou, X.; Pavlov, S.; Vorobiev, S.; Dick, J.E.; Tanner, S.D. Mass cytometry: Technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal. Chem. 2009, 81, 6813–6822. [Google Scholar] [CrossRef] [PubMed]
- Nijland, M.; Seitz, A.; Terpstra, M.; van Imhoff, G.W.; Kluin, P.M.; van Meerten, T.; Atayar, Ç.; van Kempen, L.C.; Diepstra, A.; Kok, K.; et al. Mutational Evolution in Relapsed Diffuse Large B-Cell Lymphoma. Cancers 2018, 10, 459. [Google Scholar] [CrossRef] [Green Version]
- Albitar, M.; Zhang, H.; Goy, A.; Xu-Monette, Z.Y.; Bhagat, G.; Visco, C.; Tzankov, A.; Fang, X.; Zhu, F.; Dybkaer, K.; et al. Determining clinical course of diffuse large B-cell lymphoma using targeted transcriptome and machine learning algorithms. Blood Cancer J. 2022, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, M.K.; Hoang, T.; Law, S.C.; Brosda, S.; O’Rourke, K.; Tobin, J.W.D.; Vari, F.; Murigneux, V.; Fink, L.; Gunawardana, J.; et al. EBV-associated primary CNS lymphoma occurring after immunosuppression is a distinct immunobiological entity. Blood 2021, 137, 1468–1477. [Google Scholar] [CrossRef] [PubMed]
- De Groen, R.A.; van Eijk, R.; Boehringer, S.; van Wezel, T.; Raghoo, R.; Ruano, D.; Jansen, P.M.; Briaire-de Bruijn, I.; de Groot, F.A.; Kleiverda, K.; et al. Frequent mutated B2M, EZH2, IRF8, and TNFRSF14 in primary bone diffuse large B-cell lymphoma reflect a GCB phenotype. Blood Adv. 2021, 5, 3760–3776. [Google Scholar] [CrossRef]
- Vermaat, J.S.; Somers, S.F.; de Wreede, L.C.; Kraan, W.; de Groen, R.A.L.; Schrader, A.M.R.; Kerver, E.D.; Scheepstra, C.G.; Berenschot, H.; Deenik, W.; et al. MYD88 mutations identify a molecular subgroup of diffuse large B-cell lymphoma with an unfavorable prognosis. Haematologica 2020, 105, 424–434. [Google Scholar] [CrossRef] [Green Version]
- Schrader, A.M.R.; Jansen, P.M.; Willemze, R.; Vermeer, M.H.; Cleton-Jansen, A.M.; Somers, S.F.; Veelken, H.; van Eijk, R.; Kraan, W.; Kersten, M.J.; et al. High prevalence of MYD88 and CD79B mutations in intravascular large B-cell lymphoma. Blood 2018, 131, 2086–2089. [Google Scholar] [CrossRef] [Green Version]
- Kersten, M.J.; Kraan, W.; Doorduijn, J.; Bromberg, J.; Lam, K.; Kluin, P.M.; van der Holt, B.J.; Spaargaren, M.; Pals, S.T. Diffuse large B cell lymphomas relapsing in the CNS lack oncogenic MYD88 and CD79B mutations. Blood Cancer J. 2014, 4, e266. [Google Scholar] [CrossRef] [Green Version]
- Kraan, W.; van Keimpema, M.; Horlings, H.M.; Schilder-Tol, E.J.; Oud, M.E.; Noorduyn, L.A.; Kluin, P.M.; Kersten, M.J.; Spaargaren, M.; Pals, S.T. High prevalence of oncogenic MYD88 and CD79B mutations in primary testicular diffuse large B-cell lymphoma. Leukemia 2014, 28, 719–720. [Google Scholar] [CrossRef] [PubMed]
- Schrader, A.M.R.; de Groen, R.A.L.; Willemze, R.; Jansen, P.M.; Quint, K.D.; van Wezel, T.; van Eijk, R.; Ruano, D.; Tensen, C.P.; Hauben, E.; et al. Cell-of-origin classification using the Hans and Lymph2Cx algorithms in primary cutaneous large B-cell lymphomas. Virchows Arch. 2022. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Xu, P.P.; Wang, N.; Yi, H.M.; Dong, L.; Fu, D.; Huang, J.Y.; Huang, H.Y.; Janin, A.; Cheng, S.; et al. Influence of oncogenic mutations and tumor microenvironment alterations on extranodal invasion in diffuse large B-cell lymphoma. Clin. Transl. Med. 2020, 10, e221. [Google Scholar] [CrossRef] [PubMed]
- Candelaria, M.; Oñate-Ocaña, L.F.; Corona-Herrera, J.; Barrera-Carmona, C.; Ponce-Martínez, M.; Gutiérrez-Hernández, O.; Avilés-Salas, A.; Cacho-Díaz, B. Clinical characteristics of primary extranodal versus nodal diffuse large b-cell lymphoma: A retrospective cohort study in a cancer center. Rev. Investig. Clin. 2019, 71, 349–358. [Google Scholar]
- Mottok, A.; Wright, G.; Rosenwald, A.; Ott, G.; Ramsower, C.; Campo, E.; Braziel, R.M.; Delabie, J.; Weisenburger, D.D.; Song, J.Y.; et al. Molecular classification of primary mediastinal large B-cell lymphoma using routinely available tissue specimens. Blood 2018, 132, 2401–2405. [Google Scholar] [CrossRef] [Green Version]
- Klapper, W.; Kreuz, M.; Kohler, C.W.; Burkhardt, B.; Szczepanowski, M.; Salaverria, I.; Hummel, M.; Loeffler, M.; Pellissery, S.; Woessmann, W.; et al. Patient age at diagnosis is associated with the molecular characteristics of diffuse large B-cell lymphoma. Blood 2012, 119, 1882–1887. [Google Scholar] [CrossRef]
- Mogilenko, D.A.; Shchukina, I.; Artyomov, M.N. Immune ageing at single-cell resolution. Nat. Rev. Immunol. 2021, 1–15. [Google Scholar] [CrossRef]
- Vermaat, J.S.; Pals, S.T.; Younes, A.; Dreyling, M.; Federico, M.; Aurer, I.; Radford, J.; Kersten, M.J.; Eha Lymphoma Group. Precision medicine in diffuse large B-cell lymphoma: Hitting the target. Haematologica 2015, 100, 989–993. [Google Scholar]
- You, H.; Xu-Monette, Z.Y.; Wei, L.; Nunns, H.; Nagy, M.L.; Bhagat, G.; Fang, X.; Zhu, F.; Visco, C.; Tzankov, A.; et al. Genomic complexity is associated with epigenetic regulator mutations and poor prognosis in diffuse large B-cell lymphoma. Oncoimmunology 2021, 10, 1928365. [Google Scholar] [CrossRef]
- Abdulla, M.; Alexsson, A.; Sundstrom, C.; Ladenvall, C.; Mansouri, L.; Lindskog, C.; Berglund, M.; Cavelier, L.; Enblad, G.; Hollander, P.; et al. PD-L1 and IDO1 are potential targets for treatment in patients with primary diffuse large B-cell lymphoma of the CNS. Acta Oncol. 2021, 60, 531–538. [Google Scholar] [CrossRef]
- Cheng, J.; Zhao, L.; Zhang, Y.; Qin, Y.; Guan, Y.; Zhang, T.; Liu, C.; Zhou, J. Understanding the Mechanisms of Resistance to CAR T-Cell Therapy in Malignancies. Front. Oncol. 2019, 9, 1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croci, G.A.; Au-Yeung, R.K.H.; Reinke, S.; Staiger, A.M.; Koch, K.; Oschlies, I.; Richter, J.; Poeschel, V.; Held, G.; Loeffler, M.; et al. SPARC-positive macrophages are the superior prognostic factor in the microenvironment of diffuse large B-cell lymphoma and independent of MYC rearrangement and double-/triple-hit status. Ann. Oncol. 2021, 32, 1400–1409. [Google Scholar] [CrossRef] [PubMed]
- Durek, P.; Nordstrom, K.; Gasparoni, G.; Salhab, A.; Kressler, C.; de Almeida, M.; Bassler, K.; Ulas, T.; Schmidt, F.; Xiong, J.; et al. Epigenomic Profiling of Human CD4(+) T Cells Supports a Linear Differentiation Model and Highlights Molecular Regulators of Memory Development. Immunity 2016, 45, 1148–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gennert, D.G.; Lynn, R.C.; Granja, J.M.; Weber, E.W.; Mumbach, M.R.; Zhao, Y.; Duren, Z.; Sotillo, E.; Greenleaf, W.J.; Wong, W.H.; et al. Dynamic chromatin regulatory landscape of human CAR T cell exhaustion. Proc. Natl. Acad. Sci. USA 2021, 118, e2104758118. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, J.; Ruella, M.; Houot, R. Born to survive: How cancer cells resist CAR T cell therapy. J. Hematol. Oncol. 2021, 14, 199. [Google Scholar] [CrossRef] [PubMed]
- Leveille, E.; Johnson, N.A. Genetic Events Inhibiting Apoptosis in Diffuse Large B Cell Lymphoma. Cancers 2021, 13, 2167. [Google Scholar] [CrossRef] [PubMed]
- Manfroi, B.; De Grandis, M.; Moreaux, J.; Tabruyn, S.; Mayol, J.F.; Quintero, M.; Righini, C.; Sturm, N.; Aurrand-Lions, M.; Huard, B. The microenvironment of DLBCL is characterized by noncanonical macrophages recruited by tumor-derived CCL5. Blood Adv. 2021, 5, 4338–4351. [Google Scholar] [CrossRef]
- Xu, T.; Chai, J.; Wang, K.; Jia, Q.; Liu, Y.; Wang, Y.; Xu, J.; Yu, K.; Zhao, D.; Ma, J.; et al. Tumor Immune Microenvironment Components and Checkpoint Molecules in Anaplastic Variant of Diffuse Large B-Cell Lymphoma. Front. Oncol. 2021, 11, 638154. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).