
Estrogens and Progestins Cooperatively Shift Breast Cancer Cell Metabolism

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture Methods
2.2. Metabolomics
2.3. RNA-Sequencing (RNA-Seq)
2.4. Seahorse Metabolic Phenotyping
2.5. Transmission Electron Microscopy
2.6. MitoGFP
2.7. Lipid Droplet Staining and Quantitation
2.8. Statistical Analyses
3. Results
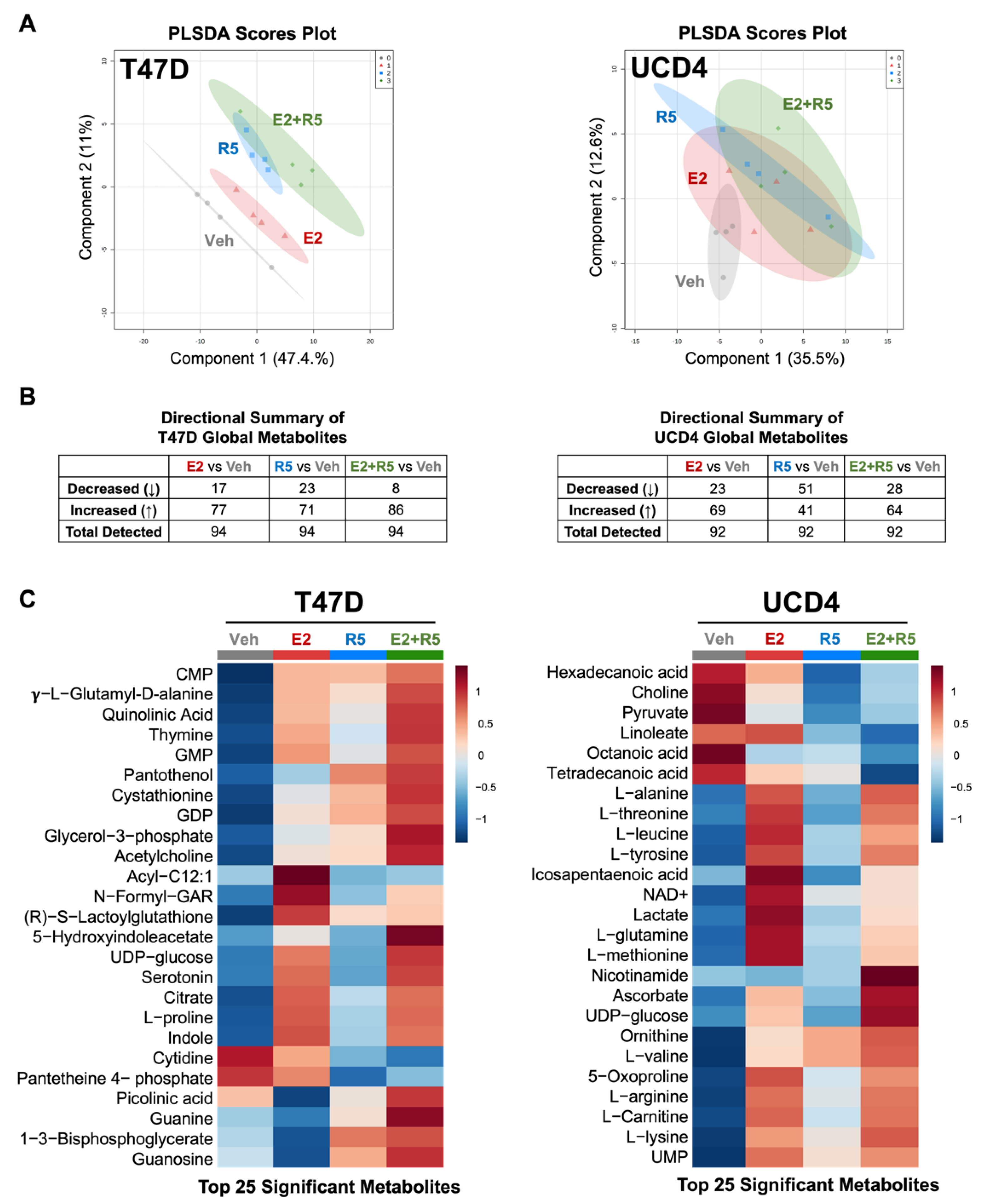
3.1. Estrogens and Progestins Alter Metabolites in Breast Cancer Cells
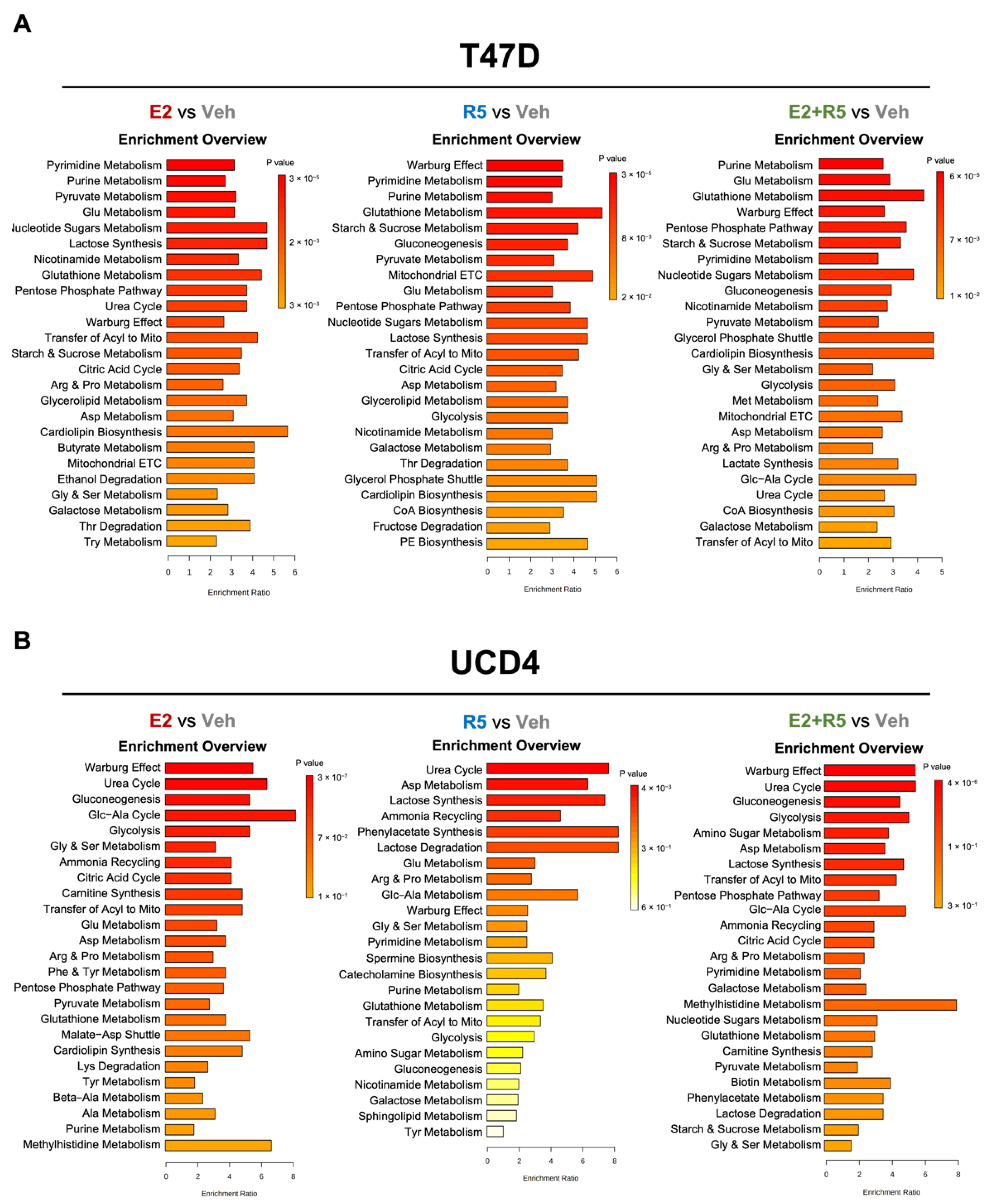
3.2. Estrogen and Progestins Enrich Metabolites Associated with Glucose Metabolism
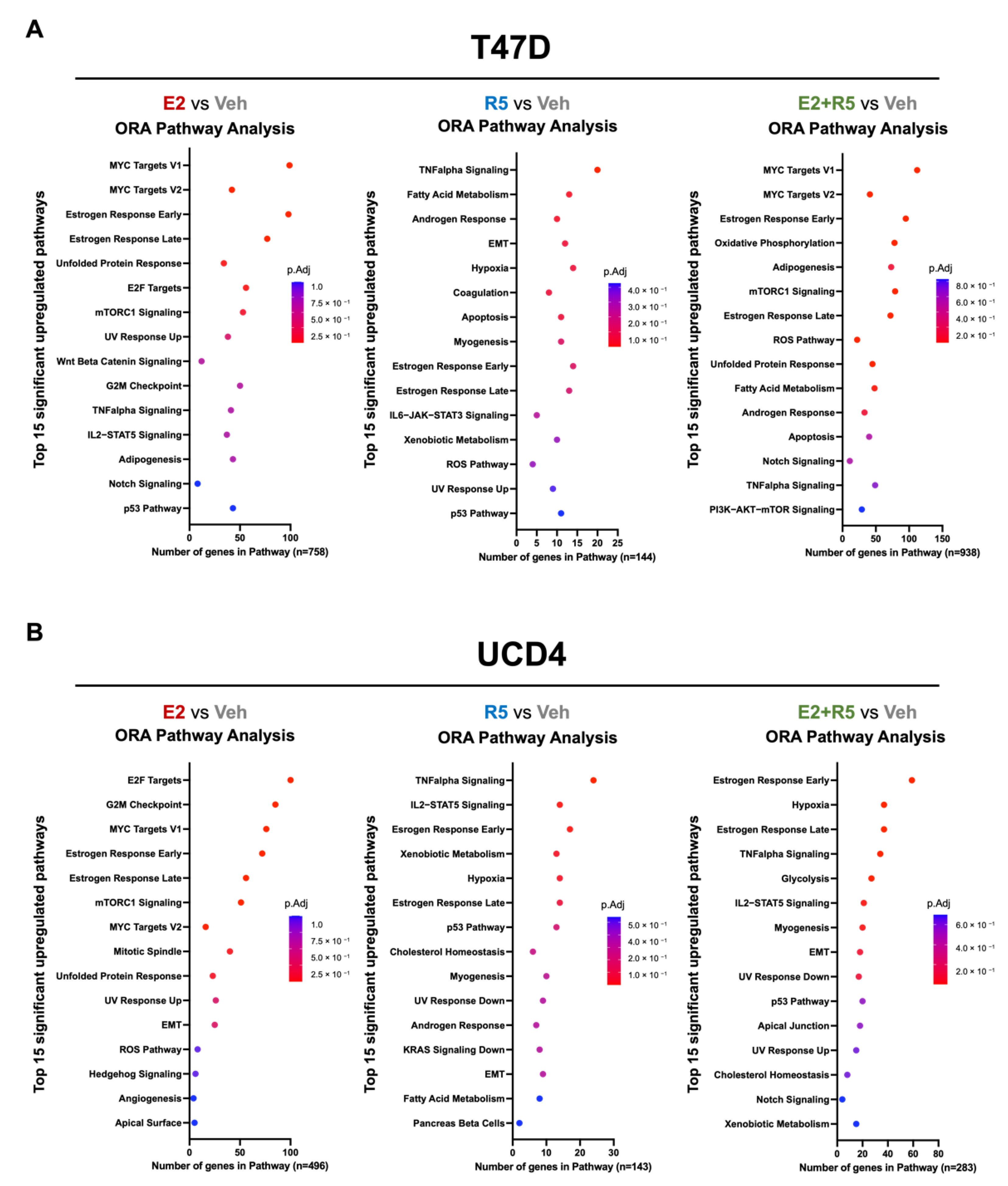
3.3. Estrogen plus Progestins Target Oncogenes That Alter Metabolism and Fatty Acid Metabolism Genes
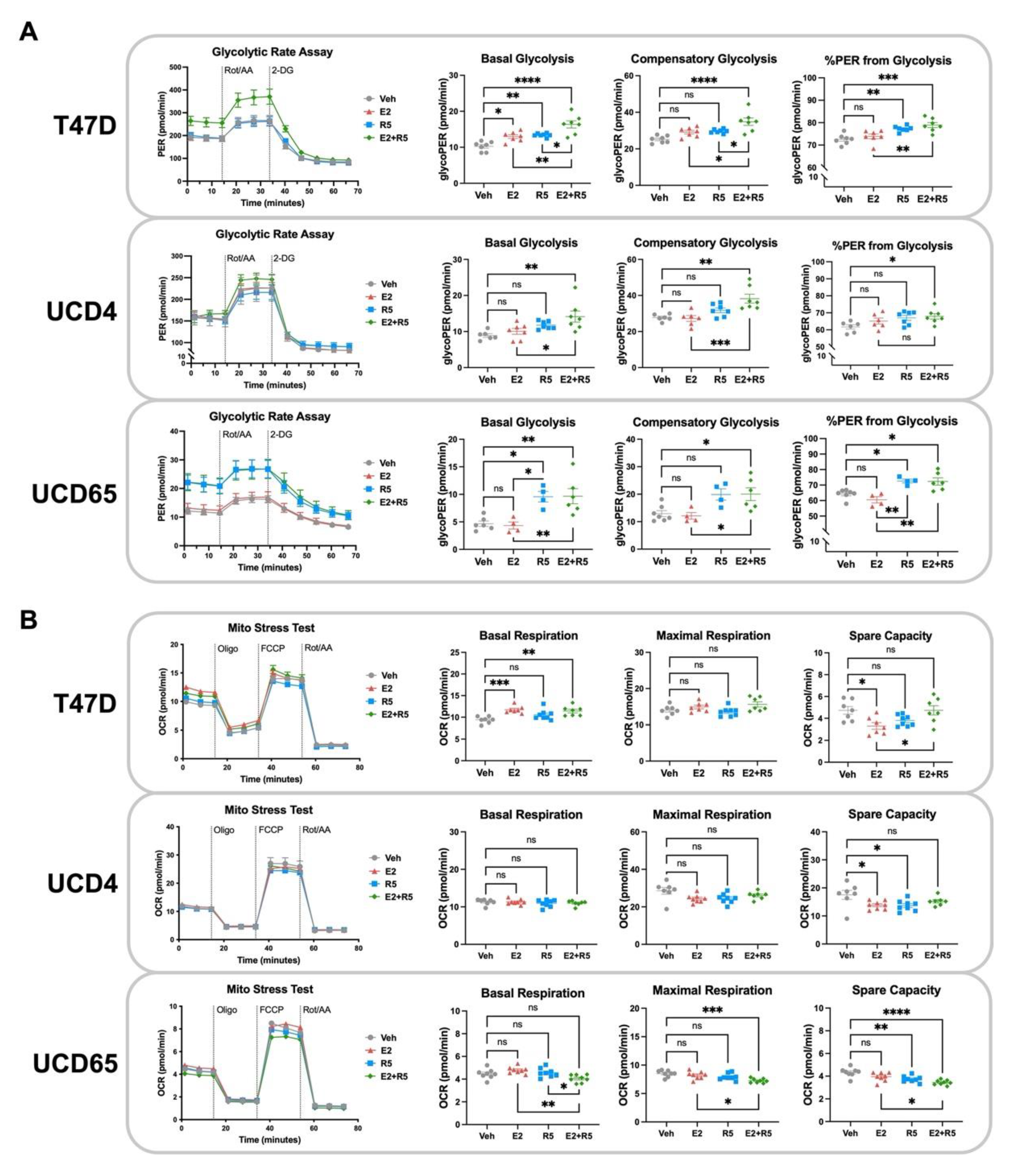
3.4. Glycolysis Is Increased by Estrogen plus Progestin Treatment in Breast Cancer Cells
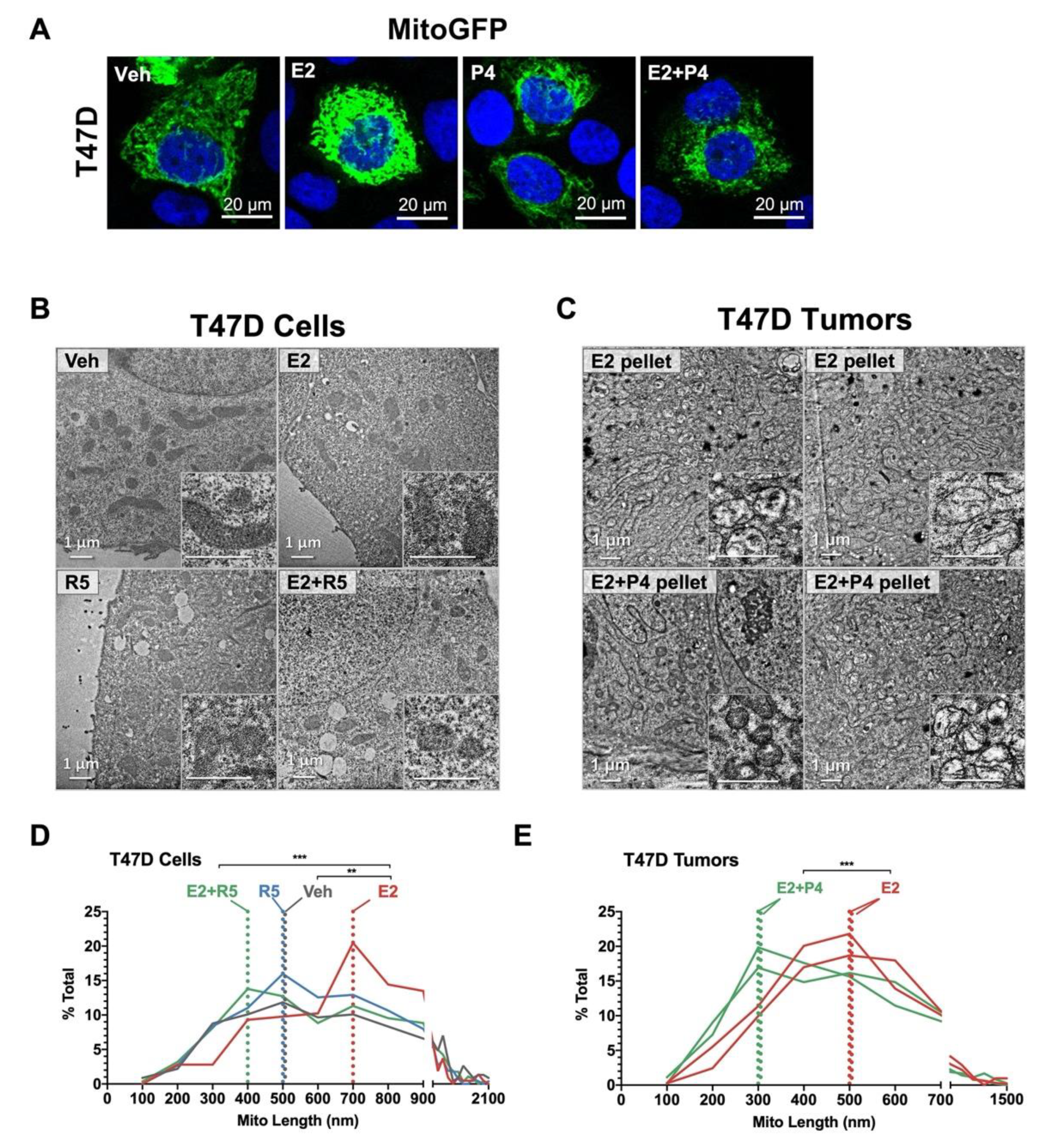
3.5. Progestins Revert Estrogen-Induced Mitochondrial Length in Breast Cancer Cells
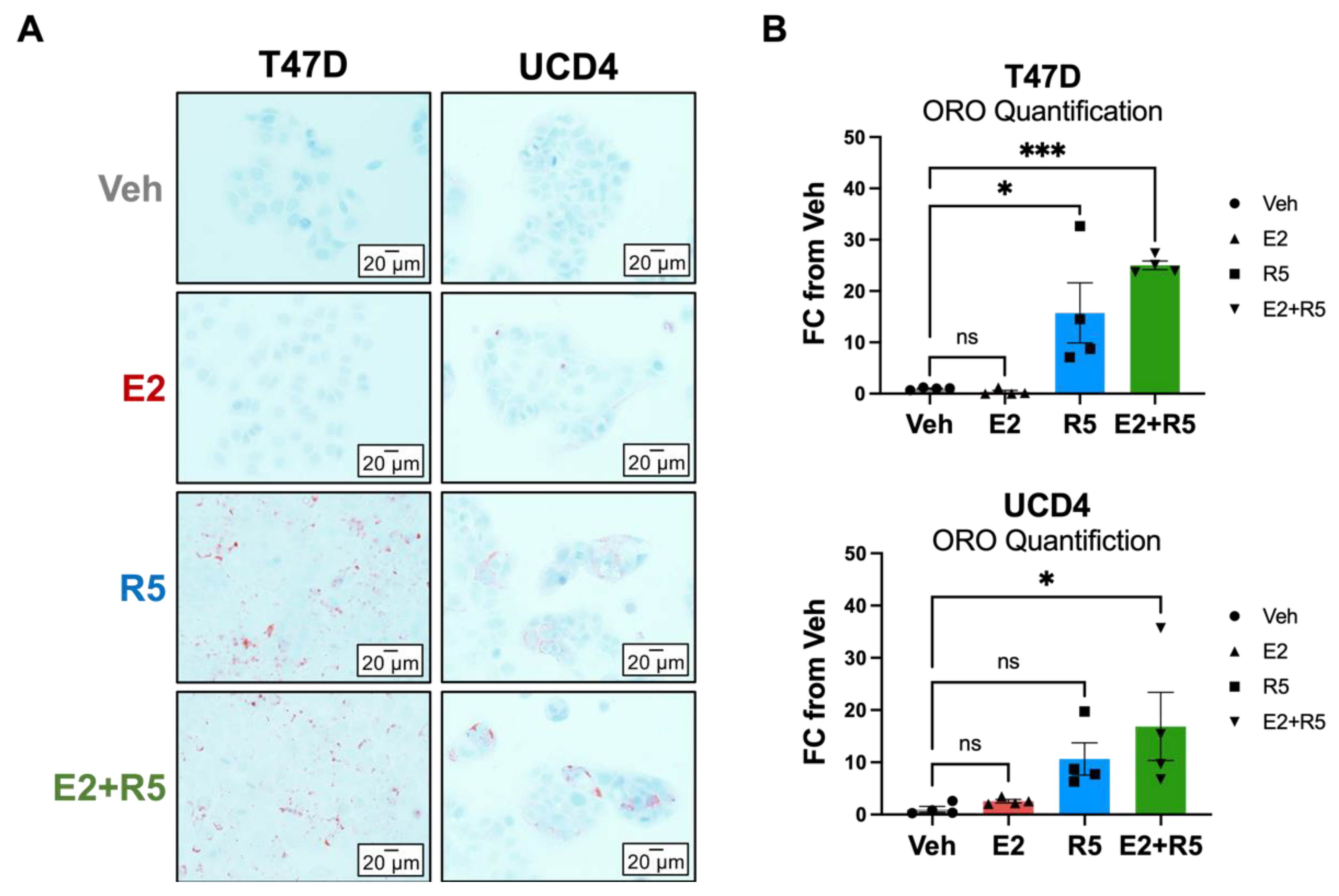
3.6. Glycerolipid Metabolism Is Increased in Breast Cancer Cells Treated with Progestins and Estrogen/Progestin Combination
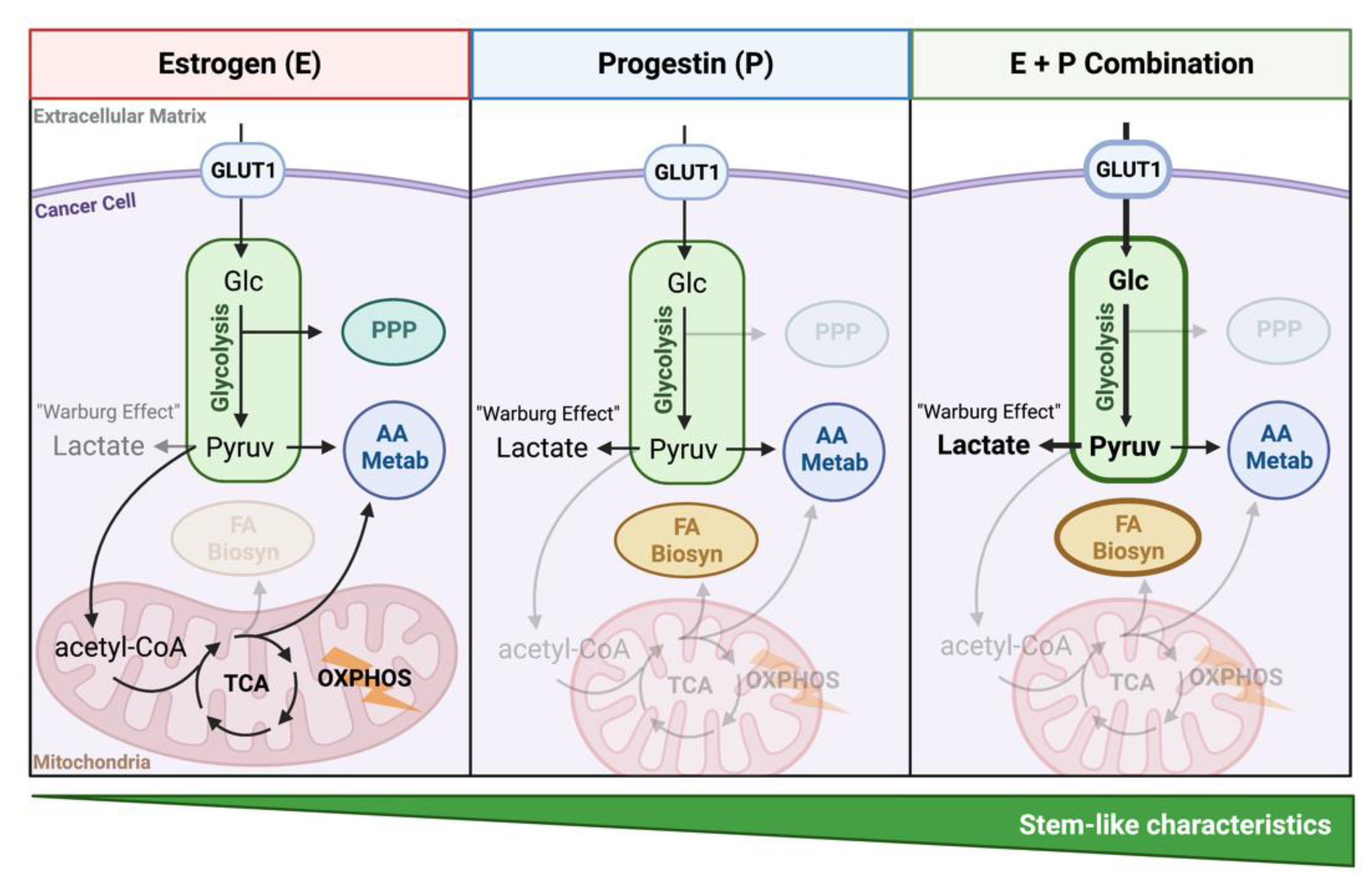
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jatoi, I.; Chen, B.E.; Anderson, W.F.; Rosenberg, P.S. Breast cancer mortality trends in the United States according to estrogen receptor status and age at diagnosis. J. Clin. Oncol. 2007, 25, 1683–1690. [Google Scholar] [CrossRef]
- Siiteri, P.K. Adipose tissue as a source of hormones. Am. J. Clin. Nutr. 1987, 45, 277–282. [Google Scholar] [CrossRef]
- Santoro, N.; Randolph, J.F., Jr. Reproductive hormones and the menopause transition. Obstet. Gynecol. Clin. N. Am. 2011, 38, 455–466. [Google Scholar] [CrossRef]
- Collaborative Group on Hormonal Factors in Breast Cancer. Type and timing of menopausal hormone therapy and breast cancer risk: Individual participant meta-analysis of the worldwide epidemiological evidence. Lancet 2019, 394, 1159–1168. [Google Scholar] [CrossRef]
- Carroll, J.S.; Hickey, T.E.; Tarulli, G.A.; Williams, M.; Tilley, W.D. Deciphering the divergent roles of progestogens in breast cancer. Nat. Rev. Cancer 2017, 17, 54–64. [Google Scholar] [CrossRef]
- Boonyaratanakornkit, V.; Pateetin, P. The role of ovarian sex steroids in metabolic homeostasis, obesity, and postmenopausal breast cancer: Molecular mechanisms and therapeutic implications. BioMed Res. Int. 2015, 2015, 140196. [Google Scholar] [CrossRef]
- Hussein, S.; Khanna, P.; Yunus, N.; Gatza, M.L. Nuclear Receptor-Mediated Metabolic Reprogramming and the Impact on HR+ Breast Cancer. Cancers 2021, 13, 4808. [Google Scholar] [CrossRef]
- Frasor, J.; Danes, J.M.; Komm, B.; Chang, K.C.; Lyttle, C.R.; Katzenellenbogen, B.S. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: Insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology 2003, 144, 4562–4574. [Google Scholar] [CrossRef]
- Richer, J.K.; Jacobsen, B.M.; Manning, N.G.; Abel, M.G.; Wolf, D.M.; Horwitz, K.B. Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J. Biol. Chem. 2002, 277, 5209–5218. [Google Scholar] [CrossRef] [PubMed]
- Boonyaratanakornkit, V.; McGowan, E.; Sherman, L.; Mancini, M.A.; Cheskis, B.J.; Edwards, D.P. The role of extranuclear signaling actions of progesterone receptor in mediating progesterone regulation of gene expression and the cell cycle. Mol. Endocrinol. 2007, 21, 359–375. [Google Scholar] [CrossRef] [PubMed]
- Diep, C.H.; Daniel, A.R.; Mauro, L.J.; Knutson, T.P.; Lange, C.A. Progesterone action in breast, uterine, and ovarian cancers. J. Mol. Endocrinol. 2015, 54, R31–R53. [Google Scholar] [CrossRef]
- Chen, J.Q.; Delannoy, M.; Cooke, C.; Yager, J.D. Mitochondrial localization of ERalpha and ERbeta in human MCF7 cells. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E1011–E1022. [Google Scholar] [CrossRef]
- Pedram, A.; Razandi, M.; Wallace, D.C.; Levin, E.R. Functional estrogen receptors in the mitochondria of breast cancer cells. Mol. Biol. Cell 2006, 17, 2125–2137. [Google Scholar] [CrossRef] [PubMed]
- Behera, M.A.; Dai, Q.; Garde, R.; Saner, C.; Jungheim, E.; Price, T.M. Progesterone stimulates mitochondrial activity with subsequent inhibition of apoptosis in MCF-10A benign breast epithelial cells. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1089–E1096. [Google Scholar] [CrossRef][Green Version]
- Dai, Q.; Shah, A.A.; Garde, R.V.; Yonish, B.A.; Zhang, L.; Medvitz, N.A.; Miller, S.E.; Hansen, E.L.; Dunn, C.N.; Price, T.M. A truncated progesterone receptor (PR-M) localizes to the mitochondrion and controls cellular respiration. Mol. Endocrinol. 2013, 27, 741–753. [Google Scholar] [CrossRef] [PubMed]
- Tidwell, T.R.; Soreide, K.; Hagland, H.R. Aging, Metabolism, and Cancer Development: From Peto’s Paradox to the Warburg Effect. Aging Dis. 2017, 8, 662–676. [Google Scholar] [CrossRef]
- Forbes, N.S.; Meadows, A.L.; Clark, D.S.; Blanch, H.W. Estradiol stimulates the biosynthetic pathways of breast cancer cells: Detection by metabolic flux analysis. Metab. Eng. 2006, 8, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Hitz, C.A.; Gijon, M.A.; Bergman, B.C.; Eckel, R.H.; Jacobsen, B.M. Progestin modulates the lipid profile and sensitivity of breast cancer cells to docetaxel. Mol. Cell. Endocrinol. 2012, 363, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Medina, R.A.; Meneses, A.M.; Vera, J.C.; Guzman, C.; Nualart, F.; Astuya, A.; Garcia, M.A.; Kato, S.; Carvajal, A.; Pinto, M.; et al. Estrogen and progesterone up-regulate glucose transporter expression in ZR-75-1 human breast cancer cells. Endocrinology 2003, 144, 4527–4535. [Google Scholar] [CrossRef]
- Finlay-Schultz, J.; Jacobsen, B.M.; Riley, D.; Paul, K.V.; Turner, S.; Ferreira-Gonzalez, A.; Harrell, J.C.; Kabos, P.; Sartorius, C.A. New generation breast cancer cell lines developed from patient-derived xenografts. Breast Cancer Res. 2020, 22, 68. [Google Scholar] [CrossRef]
- D’Alessandro, A.; Moore, H.B.; Moore, E.E.; Wither, M.; Nemkov, T.; Gonzalez, E.; Slaughter, A.; Fragoso, M.; Hansen, K.C.; Silliman, C.C.; et al. Early hemorrhage triggers metabolic responses that build up during prolonged shock. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 308, R1034–R1044. [Google Scholar] [CrossRef]
- D’Alessandro, A.; Nemkov, T.; Kelher, M.; West, F.B.; Schwindt, R.K.; Banerjee, A.; Moore, E.E.; Silliman, C.C.; Hansen, K.C. Routine storage of red blood cell (RBC) units in additive solution-3: A comprehensive investigation of the RBC metabolome. Transfusion 2015, 55, 1155–1168. [Google Scholar] [CrossRef]
- Gehrke, S.; Rice, S.; Stefanoni, D.; Wilkerson, R.B.; Nemkov, T.; Reisz, J.A.; Hansen, K.C.; Lucas, A.; Cabrales, P.; Drew, K.; et al. Red Blood Cell Metabolic Responses to Torpor and Arousal in the Hibernator Arctic Ground Squirrel. J. Proteome Res. 2019, 18, 1827–1841. [Google Scholar] [CrossRef]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’Donovan, C. MetaboLights: A resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 2020, 48, D440–D444. [Google Scholar] [CrossRef]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wishart, D.S. Metabolomic data processing, analysis, and interpretation using MetaboAnalyst. Curr. Protoc. Bioinform. 2011, 34, 14.10.1–14.10.48. [Google Scholar] [CrossRef]
- Fettig, L.M.; McGinn, O.; Finlay-Schultz, J.; LaBarbera, D.V.; Nordeen, S.K.; Sartorius, C.A. Cross talk between progesterone receptors and retinoic acid receptors in regulation of cytokeratin 5-positive breast cancer cells. Oncogene 2017, 36, 6074–6084. [Google Scholar] [CrossRef][Green Version]
- Finlay-Schultz, J.; Gillen, A.E.; Brechbuhl, H.M.; Ivie, J.J.; Matthews, S.B.; Jacobsen, B.M.; Bentley, D.L.; Kabos, P.; Sartorius, C.A. Breast Cancer Suppression by Progesterone Receptors Is Mediated by Their Modulation of Estrogen Receptors and RNA Polymerase III. Cancer Res. 2017, 77, 4934–4946. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Ge, T.; Yang, J.; Zhou, S.; Wang, Y.; Li, Y.; Tong, X. The Role of the Pentose Phosphate Pathway in Diabetes and Cancer. Front. Endocrinol. 2020, 11, 365. [Google Scholar] [CrossRef]
- Mahendralingam, M.J.; Kim, H.; McCloskey, C.W.; Aliar, K.; Casey, A.E.; Tharmapalan, P.; Pellacani, D.; Ignatchenko, V.; Garcia-Valero, M.; Palomero, L.; et al. Mammary epithelial cells have lineage-rooted metabolic identities. Nat. Metab. 2021, 3, 665–681. [Google Scholar] [CrossRef]
- Klinge, C.M. Estrogenic control of mitochondrial function and biogenesis. J. Cell. Biochem. 2008, 105, 1342–1351. [Google Scholar] [CrossRef]
- Avagliano, A.; Ruocco, M.R.; Aliotta, F.; Belviso, I.; Accurso, A.; Masone, S.; Montagnani, S.; Arcucci, A. Mitochondrial Flexibility of Breast Cancers: A Growth Advantage and a Therapeutic Opportunity. Cells 2019, 8, 401. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef]
- Axlund, S.D.; Sartorius, C.A. Progesterone regulation of stem and progenitor cells in normal and malignant breast. Mol. Cell. Endocrinol. 2012, 357, 71–79. [Google Scholar] [CrossRef]
- Simoes, B.M.; Alferez, D.G.; Howell, S.J.; Clarke, R.B. The role of steroid hormones in breast cancer stem cells. Endocr.-Relat. Cancer 2015, 22, T177–T186. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Horwitz, K.B.; Sartorius, C.A. 90 YEARS OF PROGESTERONE: Progesterone and progesterone receptors in breast cancer: Past, present, future. J. Mol. Endocrinol. 2020, 65, T49–T63. [Google Scholar] [CrossRef]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Shang, L.; Brooks, M.D.; Jiagge, E.; Zhu, Y.; Buschhaus, J.M.; Conley, S.; Fath, M.A.; Davis, A.; Gheordunescu, E.; et al. Targeting Breast Cancer Stem Cell State Equilibrium through Modulation of Redox Signaling. Cell Metab. 2018, 28, 69–86.e66. [Google Scholar] [CrossRef] [PubMed]
- Mancini, R.; Noto, A.; Pisanu, M.E.; De Vitis, C.; Maugeri-Sacca, M.; Ciliberto, G. Metabolic features of cancer stem cells: The emerging role of lipid metabolism. Oncogene 2018, 37, 2367–2378. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, M.; Sanchez-Alvarez, R.; Sotgia, F.; Lisanti, M.P. The ER-alpha mutation Y537S confers Tamoxifen-resistance via enhanced mitochondrial metabolism, glycolysis and Rho-GDI/PTEN signaling: Implicating TIGAR in somatic resistance to endocrine therapy. Aging 2018, 10, 4000–4023. [Google Scholar] [CrossRef] [PubMed]
- Zinger, L.; Merenbakh-Lamin, K.; Klein, A.; Elazar, A.; Journo, S.; Boldes, T.; Pasmanik-Chor, M.; Spitzer, A.; Rubinek, T.; Wolf, I. Ligand-binding Domain-activating Mutations of ESR1 Rewire Cellular Metabolism of Breast Cancer Cells. Clin. Cancer Res. 2019, 25, 2900–2914. [Google Scholar] [CrossRef] [PubMed]
- Cruceriu, D.; Baldasici, O.; Balacescu, O.; Berindan-Neagoe, I. The dual role of tumor necrosis factor-alpha (TNF-alpha) in breast cancer: Molecular insights and therapeutic approaches. Cell. Oncol. 2020, 43, 1–18. [Google Scholar] [CrossRef]
- Benz, C.C. Impact of aging on the biology of breast cancer. Crit. Rev. Oncol. Hematol. 2008, 66, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Sahin, S.; Erdem, G.U.; Karatas, F.; Aytekin, A.; Sever, A.R.; Ozisik, Y.; Altundag, K. The association between body mass index and immunohistochemical subtypes in breast cancer. Breast 2017, 32, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, R.; Picon-Ruiz, M.; Aurrekoetxea-Rodriguez, I.; Nunes de Paiva, V.; D’Amico, M.; Yoon, H.; Radhakrishnan, R.; Morata-Tarifa, C.; Ince, T.; Lippman, M.E.; et al. The Major Pre- and Postmenopausal Estrogens Play Opposing Roles in Obesity-Driven Mammary Inflammation and Breast Cancer Development. Cell Metab. 2020, 31, 1154–1172.e9. [Google Scholar] [CrossRef] [PubMed]
- Giles, E.D.; Wellberg, E.A.; Astling, D.P.; Anderson, S.M.; Thor, A.D.; Jindal, S.; Tan, A.C.; Schedin, P.S.; Maclean, P.S. Obesity and overfeeding affecting both tumor and systemic metabolism activates the progesterone receptor to contribute to postmenopausal breast cancer. Cancer Res. 2012, 72, 6490–6501. [Google Scholar] [CrossRef]
- Chlebowski, R.T.; Anderson, G.L.; Gass, M.; Lane, D.S.; Aragaki, A.K.; Kuller, L.H.; Manson, J.E.; Stefanick, M.L.; Ockene, J.; Sarto, G.E.; et al. Estrogen plus progestin and breast cancer incidence and mortality in postmenopausal women. JAMA 2010, 304, 1684–1692. [Google Scholar] [CrossRef] [PubMed]
- Chlebowski, R.T.; Anderson, G.L.; Aragaki, A.K.; Manson, J.E.; Stefanick, M.L.; Pan, K.; Barrington, W.; Kuller, L.H.; Simon, M.S.; Lane, D.; et al. Association of Menopausal Hormone Therapy With Breast Cancer Incidence and Mortality During Long-term Follow-up of the Women’s Health Initiative Randomized Clinical Trials. JAMA 2020, 324, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Santen, R.J. Risk of breast cancer with progestins: Critical assessment of current data. Steroids 2003, 68, 953–964. [Google Scholar] [CrossRef]
- Horwitz, K.B.; Sartorius, C.A. Progestins in hormone replacement therapies reactivate cancer stem cells in women with preexisting breast cancers: A hypothesis. J. Clin. Endocrinol. Metab. 2008, 93, 3295–3298. [Google Scholar] [CrossRef]
- Yu, M.; Chen, S.; Hong, W.; Gu, Y.; Huang, B.; Lin, Y.; Zhou, Y.; Jin, H.; Deng, Y.; Tu, L.; et al. Prognostic role of glycolysis for cancer outcome: Evidence from 86 studies. J. Cancer Res. Clin. Oncol. 2019, 145, 967–999. [Google Scholar] [CrossRef]
- Kamaraju, S.; Fowler, A.M.; Weil, E.; Wisinski, K.B.; Truong, T.H.; Lehr, M.; Chaudhary, L.N.; Cheng, Y.C.; Chitambar, C.R.; Rui, H.; et al. Leveraging Antiprogestins in the Treatment of Metastatic Breast Cancer. Endocrinology 2021, 162, bqab060. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ward, A.V.; Matthews, S.B.; Fettig, L.M.; Riley, D.; Finlay-Schultz, J.; Paul, K.V.; Jackman, M.; Kabos, P.; MacLean, P.S.; Sartorius, C.A. Estrogens and Progestins Cooperatively Shift Breast Cancer Cell Metabolism. Cancers 2022, 14, 1776. https://doi.org/10.3390/cancers14071776
Ward AV, Matthews SB, Fettig LM, Riley D, Finlay-Schultz J, Paul KV, Jackman M, Kabos P, MacLean PS, Sartorius CA. Estrogens and Progestins Cooperatively Shift Breast Cancer Cell Metabolism. Cancers. 2022; 14(7):1776. https://doi.org/10.3390/cancers14071776
Chicago/Turabian StyleWard, Ashley V., Shawna B. Matthews, Lynsey M. Fettig, Duncan Riley, Jessica Finlay-Schultz, Kiran V. Paul, Matthew Jackman, Peter Kabos, Paul S. MacLean, and Carol A. Sartorius. 2022. "Estrogens and Progestins Cooperatively Shift Breast Cancer Cell Metabolism" Cancers 14, no. 7: 1776. https://doi.org/10.3390/cancers14071776
APA StyleWard, A. V., Matthews, S. B., Fettig, L. M., Riley, D., Finlay-Schultz, J., Paul, K. V., Jackman, M., Kabos, P., MacLean, P. S., & Sartorius, C. A. (2022). Estrogens and Progestins Cooperatively Shift Breast Cancer Cell Metabolism. Cancers, 14(7), 1776. https://doi.org/10.3390/cancers14071776