
Impact of Cannabinoid Compounds on Skin Cancer

 ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Skin Cancer—Incidence and Current Status of Pharmacotherapy
2. The Endocannabinoid System
2.1. The Endocannabinoid System—A Brief Description of the Components
2.2. The Endocannabinoid System in the Skin
2.3. Endocannabinoid System in Melanoma and Squamous Cell Carcinoma Tissue
3. Inhibition of Tumour Growth of Melanomas and Squamous Cell Carcinomas by Cannabinoids
3.1. Antiproliferative and Proapoptotic Effects of Cannabinoids on Melanoma
3.2. Antiproliferative and Proapoptotic Effects of Cannabinoids on Squamous Cell Carcinomas
3.3. Antiproliferative Effects of Cannabinoids on Kaposi Sarcoma and Cutanous Lymphoma Cells
3.4. Effect of Cannabinoids on Autophagy of Melanoma and Squamous Cell Carcinoma Cells
4. Inhibition of Skin Cancer Angiogenesis by Cannabinoids
5. Inhibition of Melanoma and Squamous Cell Carcinoma Motility, Invasion, and Metastasis by Cannabinoids
5.1. Inhibition of Melanoma Cell Motility, Invasion, and Metastasis by Cannabinoids
5.2. Inhibition of Squamous Cell Carcinoma Motility, Invasion, and Metastasis by Cannabinoids
6. Impact of Cannabinoid Compounds on Skin Tumour Immune Response
7. Cannabinoids and Skin Cancer Prevention
8. Clinical Trials on Cannabinoid Use in Cancer Patients
9. Outlook
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nehal, K.S.; Bichakjian, C.K. Update on Keratinocyte Carcinomas. N. Engl. J. Med. 2018, 379, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Dika, E.; Scarfì, F.; Ferracin, M.; Broseghini, E.; Marcelli, E.; Bortolani, B.; Campione, E.; Riefolo, M.; Ricci, C.; Lambertini, M. Basal Cell Carcinoma: A Comprehensive Review. Int. J. Mol. Sci. 2020, 21, 5572. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Einhorn, L.H.; Meyers, M.L.; Saxman, S.; Destro, A.N.; Panageas, K.S.; Begg, C.B.; Agarwala, S.S.; Schuchter, L.M.; Ernstoff, M.S.; et al. Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. J. Clin. Oncol. 1999, 17, 2745–2751. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Schwartzentruber, D.J.; Hwu, P.; Marincola, F.M.; Topalian, S.L.; Seipp, C.A.; Einhorn, J.H.; White, D.E.; Steinberg, S.M. Prospective randomized trial of the treatment of patients with metastatic melanoma using chemotherapy with cisplatin, dacarbazine, and tamoxifen alone or in combination with interleukin-2 and interferon alfa-2b. J. Clin. Oncol. 1999, 17, 968–975. [Google Scholar] [CrossRef]
- Eton, O.; Legha, S.S.; Bedikian, A.Y.; Lee, J.J.; Buzaid, A.C.; Hodges, C.; Ring, S.E.; Papadopoulos, N.E.; Plager, C.; East, M.J.; et al. Sequential biochemotherapy versus chemotherapy for metastatic melanoma: Results from a phase III randomized trial. J. Clin. Oncol. 2002, 20, 2045–2052. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Minato, N.; Nakano, T.; Honjo, T. Immunological studies on PD-1 deficient mice: Implication of PD-1 as a negative regulator for B cell responses. Int. Immunol. 1998, 10, 1563–1572. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Kraft, S.; Fernandez-Figueras, M.-T.; Richarz, N.A.; Flaherty, K.T.; Hoang, M.P. PDL1 expression in desmoplastic melanoma is associated with tumor aggressiveness and progression. J. Am. Acad. Dermatol. 2017, 77, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Da Pires Silva, I.; Ahmed, T.; Reijers, I.L.M.; Weppler, A.M.; Betof Warner, A.; Patrinely, J.R.; Serra-Bellver, P.; Allayous, C.; Mangana, J.; Nguyen, K.; et al. Ipilimumab alone or ipilimumab plus anti-PD-1 therapy in patients with metastatic melanoma resistant to anti-PD-(L)1 monotherapy: A multicentre, retrospective, cohort study. Lancet Oncol. 2021, 22, 836–847. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [Green Version]
- Hauschild, A.; Grob, J.-J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Vanella, V.; Festino, L.; Trojaniello, C.; Vitale, M.G.; Sorrentino, A.; Paone, M.; Ascierto, P.A. The Role of BRAF-Targeted Therapy for Advanced Melanoma in the Immunotherapy Era. Curr. Oncol. Rep. 2019, 21, 76. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Wunderlich, J.R.; Yang, J.C.; Sherry, R.M.; Topalian, S.L.; Restifo, N.P.; Royal, R.E.; Kammula, U.; White, D.E.; Mavroukakis, S.A.; et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J. Clin. Oncol. 2005, 23, 2346–2357. [Google Scholar] [CrossRef]
- Radvanyi, L.G.; Bernatchez, C.; Zhang, M.; Fox, P.S.; Miller, P.; Chacon, J.; Wu, R.; Lizee, G.; Mahoney, S.; Alvarado, G.; et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin. Cancer Res. 2012, 18, 6758–6770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madan, V.; Lear, J.T.; Szeimies, R.-M. Non-melanoma skin cancer. Lancet 2010, 375, 673–685. [Google Scholar] [CrossRef] [Green Version]
- Boutros, A.; Cecchi, F.; Tanda, E.T.; Croce, E.; Gili, R.; Arecco, L.; Spagnolo, F.; Queirolo, P. Immunotherapy for the Treatment of Cutaneous Squamous Cell Carcinoma. Front. Oncol. 2021, 11, 733917. [Google Scholar] [CrossRef]
- Tóth, K.F.; Ádám, D.; Bíró, T.; Oláh, A. Cannabinoid Signaling in the Skin: Therapeutic Potential of the “C(ut)annabinoid” System. Molecules 2019, 24, 918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinz, B.; Ramer, R. Anti-tumour actions of cannabinoids. Br. J. Pharmacol. 2019, 176, 1384–1394. [Google Scholar] [CrossRef]
- Casanova, M.L.; Blázquez, C.; Martínez-Palacio, J.; Villanueva, C.; Fernández-Aceñero, M.J.; Huffman, J.W.; Jorcano, J.L.; Guzmán, M. Inhibition of skin tumor growth and angiogenesis in vivo by activation of cannabinoid receptors. J. Clin. Investig. 2003, 111, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef]
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar] [CrossRef]
- Bisogno, T.; Melck, D.; Bobrov, M.; Gretskaya, N.M.; Bezuglov, V.V.; de Petrocellis, L.; Di Marzo, V. N-acyl-dopamines: Novel synthetic CB(1) cannabinoid-receptor ligands and inhibitors of anandamide inactivation with cannabimimetic activity in vitro and in vivo. Biochem. J. 2000, 351 Pt 3, 817–824. [Google Scholar] [CrossRef]
- Hanus, L.; Abu-Lafi, S.; Fride, E.; Breuer, A.; Vogel, Z.; Shalev, D.E.; Kustanovich, I.; Mechoulam, R. 2-arachidonyl glyceryl ether, an endogenous agonist of the cannabinoid CB1 receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 3662–3665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, A.C.; Sauer, J.-M.; Knierman, M.D.; Becker, G.W.; Berna, M.J.; Bao, J.; Nomikos, G.G.; Carter, P.; Bymaster, F.P.; Leese, A.B.; et al. Characterization of a novel endocannabinoid, virodhamine, with antagonist activity at the CB1 receptor. J. Pharmacol. Exp. Ther. 2002, 301, 1020–1024. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, R.; Ramer, R.; Hinz, B. Targeting the endocannabinoid system as a potential anticancer approach. Drug Metab. Rev. 2018, 50, 26–53. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A.C. Inhibition of neuroblastoma adenylate cyclase by cannabinoid and nantradol compounds. Life Sci. 1984, 35, 1803–1810. [Google Scholar] [CrossRef]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef]
- Mackie, K.; Devane, W.A.; Hille, B. Anandamide, an endogenous cannabinoid, inhibits calcium currents as a partial agonist in N18 neuroblastoma cells. Mol. Pharmacol. 1993, 44, 498–503. [Google Scholar] [PubMed]
- Sugiura, T.; Kodaka, T.; Nakane, S.; Miyashita, T.; Kondo, S.; Suhara, Y.; Takayama, H.; Waku, K.; Seki, C.; Baba, N.; et al. Evidence that the cannabinoid CB1 receptor is a 2-arachidonoylglycerol receptor. Structure-activity relationship of 2-arachidonoylglycerol, ether-linked analogues, and related compounds. J. Biol. Chem. 1999, 274, 2794–2801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonsiorek, W.; Lunn, C.; Fan, X.; Narula, S.; Lundell, D.; Hipkin, R.W. Endocannabinoid 2-arachidonyl glycerol is a full agonist through human type 2 cannabinoid receptor: Antagonism by anandamide. Mol. Pharmacol. 2000, 57, 1045–1050. [Google Scholar] [PubMed]
- Sugiura, T.; Kondo, S.; Kishimoto, S.; Miyashita, T.; Nakane, S.; Kodaka, T.; Suhara, Y.; Takayama, H.; Waku, K. Evidence that 2-arachidonoylglycerol but not N-palmitoylethanolamine or anandamide is the physiological ligand for the cannabinoid CB2 receptor. Comparison of the agonistic activities of various cannabinoid receptor ligands in HL-60 cells. J. Biol. Chem. 2000, 275, 605–612. [Google Scholar] [CrossRef] [Green Version]
- Savinainen, J.R.; Järvinen, T.; Laine, K.; Laitinen, J.T. Despite substantial degradation, 2-arachidonoylglycerol is a potent full efficacy agonist mediating CB1 receptor-dependent G-protein activation in rat cerebellar membranes. Br. J. Pharmacol. 2001, 134, 664–672. [Google Scholar] [CrossRef] [Green Version]
- Brown, I.; Cascio, M.G.; Wahle, K.W.J.; Smoum, R.; Mechoulam, R.; Ross, R.A.; Pertwee, R.G.; Heys, S.D. Cannabinoid receptor-dependent and -independent anti-proliferative effects of omega-3 ethanolamides in androgen receptor-positive and -negative prostate cancer cell lines. Carcinogenesis 2010, 31, 1584–1591. [Google Scholar] [CrossRef] [Green Version]
- Pertwee, R.G. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br. J. Pharmacol. 2008, 153, 199–215. [Google Scholar] [CrossRef] [Green Version]
- Rosenthaler, S.; Pöhn, B.; Kolmanz, C.; Huu, C.N.; Krewenka, C.; Huber, A.; Kranner, B.; Rausch, W.-D.; Moldzio, R. Differences in receptor binding affinity of several phytocannabinoids do not explain their effects on neural cell cultures. Neurotoxicol. Teratol. 2014, 46, 49–56. [Google Scholar] [CrossRef]
- Thomas, A.; Baillie, G.L.; Phillips, A.M.; Razdan, R.K.; Ross, R.A.; Pertwee, R.G. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br. J. Pharmacol. 2007, 150, 613–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakowiecki, J.; Abel, R.; Orzeł, U.; Pasznik, P.; Preissner, R.; Filipek, S. Allosteric Modulation of the CB1 Cannabinoid Receptor by Cannabidiol-A Molecular Modeling Study of the N-Terminal Domain and the Allosteric-Orthosteric Coupling. Molecules 2021, 26, 2456. [Google Scholar] [CrossRef]
- Russo, E.; Guy, G.W. A tale of two cannabinoids: The therapeutic rationale for combining tetrahydrocannabinol and cannabidiol. Med. Hypotheses 2006, 66, 234–246. [Google Scholar] [CrossRef]
- Zygmunt, P.M.; Petersson, J.; Andersson, D.A.; Chuang, H.; Sørgård, M.; Di Marzo, V.; Julius, D.; Högestätt, E.D. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 1999, 400, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Hanus, L.; de Petrocellis, L.; Tchilibon, S.; Ponde, D.E.; Brandi, I.; Moriello, A.S.; Davis, J.B.; Mechoulam, R.; Di Marzo, V. Molecular targets for cannabidiol and its synthetic analogues: Effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br. J. Pharmacol. 2001, 134, 845–852. [Google Scholar] [CrossRef]
- Huang, S.M.; Bisogno, T.; Trevisani, M.; Al-Hayani, A.; de Petrocellis, L.; Fezza, F.; Tognetto, M.; Petros, T.J.; Krey, J.F.; Chu, C.J.; et al. An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc. Natl. Acad. Sci. USA 2002, 99, 8400–8405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, M.; Millns, P.; Smart, D.; Wright, J.E.; Kendall, D.A.; Ralevic, V. Noladin ether, a putative endocannabinoid, attenuates sensory neurotransmission in the rat isolated mesenteric arterial bed via a non-CB1/CB2 Gi/o linked receptor. Br. J. Pharmacol. 2004, 142, 509–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, N.; Neeper, M.P.; Liu, Y.; Hutchinson, T.L.; Lubin, M.L.; Flores, C.M. TRPV2 is activated by cannabidiol and mediates CGRP release in cultured rat dorsal root ganglion neurons. J. Neurosci. 2008, 28, 6231–6238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.-O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Lauckner, J.E.; Jensen, J.B.; Chen, H.-Y.; Lu, H.-C.; Hille, B.; Mackie, K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc. Natl. Acad. Sci. USA 2008, 105, 2699–2704. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Gaetani, S.; Oveisi, F.; Lo Verme, J.; Serrano, A.; Rodríguez de Fonseca, F.; Rosengarth, A.; Luecke, H.; Di Giacomo, B.; Tarzia, G.; et al. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-α. Nature 2003, 425, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Lo Verme, J.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15–19. [Google Scholar] [CrossRef]
- Artmann, A.; Petersen, G.; Hellgren, L.I.; Boberg, J.; Skonberg, C.; Nellemann, C.; Hansen, S.H.; Hansen, H.S. Influence of dietary fatty acids on endocannabinoid and N-acylethanolamine levels in rat brain, liver and small intestine. Biochim. Biophys. Acta 2008, 1781, 200–212. [Google Scholar] [CrossRef]
- Liu, J.; Li, H.; Burstein, S.H.; Zurier, R.B.; Chen, J.D. Activation and binding of peroxisome proliferator-activated receptor γ by synthetic cannabinoid ajulemic acid. Mol. Pharmacol. 2003, 63, 983–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, S.E.; Tarling, E.J.; Bennett, A.J.; Kendall, D.A.; Randall, M.D. Novel time-dependent vascular actions of Δ9-tetrahydrocannabinol mediated by peroxisome proliferator-activated receptor gamma. Biochem. Biophys. Res. Commun. 2005, 337, 824–831. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, S.E.; Sun, Y.; Bennett, A.J.; Randall, M.D.; Kendall, D.A. Time-dependent vascular actions of cannabidiol in the rat aorta. Eur. J. Pharmacol. 2009, 612, 61–68. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, S.E. Cannabinoids go nuclear: Evidence for activation of peroxisome proliferator-activated receptors. Br. J. Pharmacol. 2007, 152, 576–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
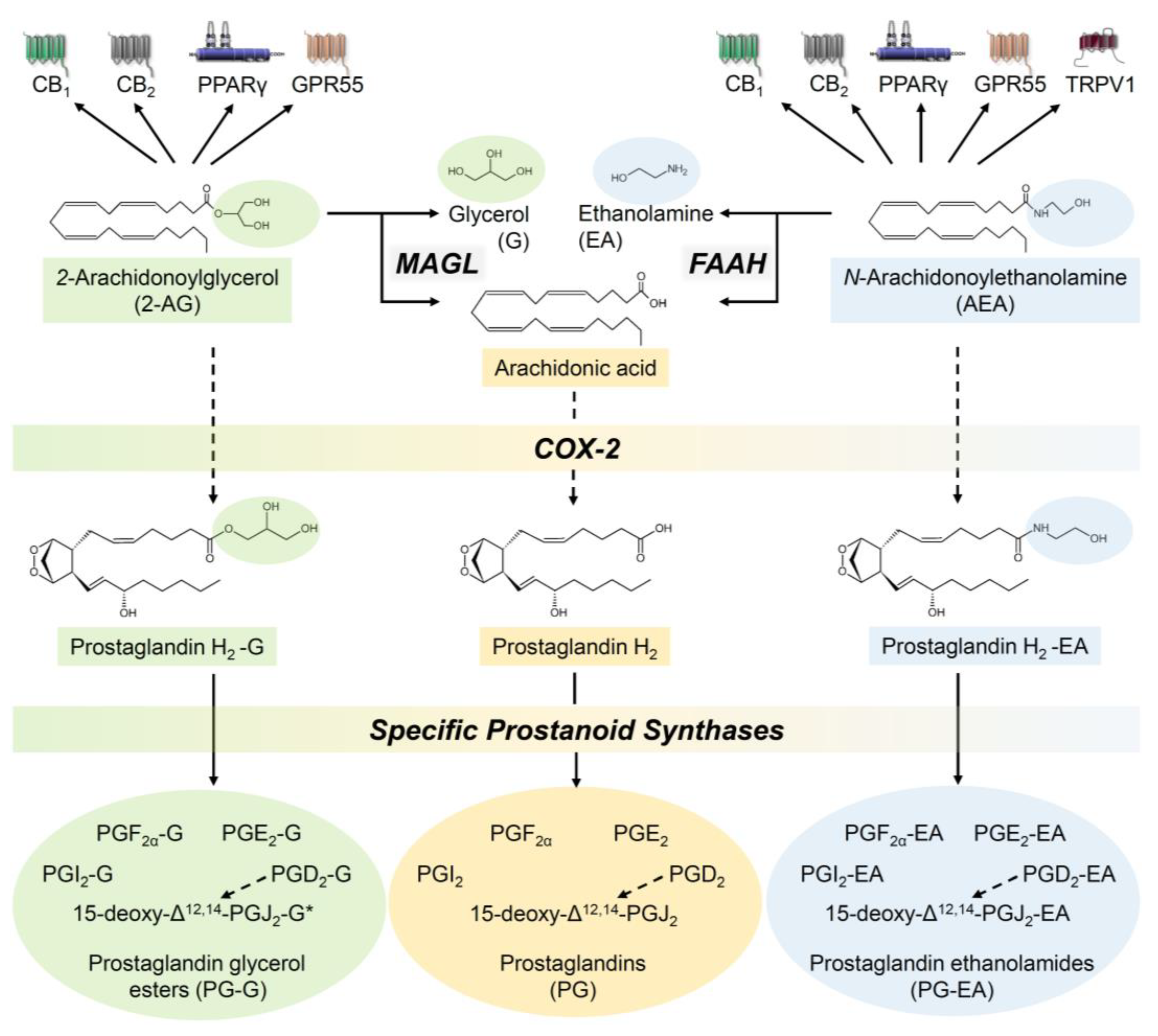
- Di Marzo, V. The endocannabinoid system: Its general strategy of action, tools for its pharmacological manipulation and potential therapeutic exploitation. Pharmacol. Res. 2009, 60, 77–84. [Google Scholar] [CrossRef]
- Deutsch, D.G.; Chin, S.A. Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem. Pharmacol. 1993, 46, 791–796. [Google Scholar] [CrossRef]
- Blankman, J.L.; Simon, G.M.; Cravatt, B.F. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem. Biol. 2007, 14, 1347–1356. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Ives, D.; Ramesha, C.S. Synthesis of prostaglandin E2 ethanolamide from anandamide by cyclooxygenase-2. J. Biol. Chem. 1997, 272, 21181–21186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozak, K.R.; Rowlinson, S.W.; Marnett, L.J. Oxygenation of the endocannabinoid, 2-arachidonylglycerol, to glyceryl prostaglandins by cyclooxygenase-2. J. Biol. Chem. 2000, 275, 33744–33749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozak, K.R.; Crews, B.C.; Morrow, J.D.; Wang, L.-H.; Ma, Y.H.; Weinander, R.; Jakobsson, P.-J.; Marnett, L.J. Metabolism of the endocannabinoids, 2-arachidonylglycerol and anandamide, into prostaglandin, thromboxane, and prostacyclin glycerol esters and ethanolamides. J. Biol. Chem. 2002, 277, 44877–44885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felder, C.C.; Nielsen, A.; Briley, E.M.; Palkovits, M.; Priller, J.; Axelrod, J.; Nguyen, D.N.; Richardson, J.M.; Riggin, R.M.; Koppel, G.A.; et al. Isolation and measurement of the endogenous cannabinoid receptor agonist, anandamide, in brain and peripheral tissues of human and rat. FEBS Lett. 1996, 393, 231–235. [Google Scholar] [CrossRef] [Green Version]
- Calignano, A.; La Rana, G.; Giuffrida, A.; Piomelli, D. Control of pain initiation by endogenous cannabinoids. Nature 1998, 394, 277–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ständer, S.; Schmelz, M.; Metze, D.; Luger, T.; Rukwied, R. Distribution of cannabinoid receptor 1 (CB1) and 2 (CB2) on sensory nerve fibers and adnexal structures in human skin. J. Dermatol. Sci. 2005, 38, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Del Río, C.; Millán, E.; García, V.; Appendino, G.; DeMesa, J.; Muñoz, E. The endocannabinoid system of the skin. A potential approach for the treatment of skin disorders. Biochem. Pharmacol. 2018, 157, 122–133. [Google Scholar]
- Rosenfield, R.L.; Deplewski, D.; Kentsis, A.; Ciletti, N. Mechanisms of androgen induction of sebocyte differentiation. Dermatology 1998, 196, 43–46. [Google Scholar] [CrossRef]
- Yin, K.; Smith, A.G. Nuclear receptor function in skin health and disease: Therapeutic opportunities in the orphan and adopted receptor classes. Cell. Mol. Life Sci. 2016, 73, 3789–3800. [Google Scholar] [CrossRef]
- Karsak, M.; Gaffal, E.; Date, R.; Wang-Eckhardt, L.; Rehnelt, J.; Petrosino, S.; Starowicz, K.; Steuder, R.; Schlicker, E.; Cravatt, B.; et al. Attenuation of allergic contact dermatitis through the endocannabinoid system. Science 2007, 316, 1494–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka-Takaichi, M.; Sugawara, K.; Sumitomo, R.; Tsuruta, D. The Mast Cell-SCF-CB1 Interaction Is a Key Player in Seborrheic Keratosis. J. Histochem. Cytochem. 2020, 68, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Magina, S.; Esteves-Pinto, C.; Moura, E.; Serrão, M.P.; Moura, D.; Petrosino, S.; Di Marzo, V.; Vieira-Coelho, M.A. Inhibition of basal and ultraviolet B-induced melanogenesis by cannabinoid CB1 receptors: A keratinocyte-dependent effect. Arch. Dermatol. Res. 2011, 303, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Siracusa, R.; Impellizzeri, D.; Cordaro, M.; Gugliandolo, E.; Peritore, A.F.; Di Paola, R.; Cuzzocrea, S. Topical Application of Adelmidrol + Trans-Traumatic Acid Enhances Skin Wound Healing in a Streptozotocin-Induced Diabetic Mouse Model. Front. Pharmacol. 2018, 9, 871. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.-L.; Yu, T.-S.; Li, X.-N.; Fan, Y.-Y.; Ma, W.-X.; Du, Y.; Zhao, R.; Guan, D.-W. Cannabinoid receptor type 2 is time-dependently expressed during skin wound healing in mice. Int. J. Legal Med. 2012, 126, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, K.; Bíró, T.; Tsuruta, D.; Tóth, B.I.; Kromminga, A.; Zákány, N.; Zimmer, A.; Funk, W.; Gibbs, B.F.; Zimmer, A.; et al. Endocannabinoids limit excessive mast cell maturation and activation in human skin. J. Allergy Clin. Immunol. 2012, 129, 726–738.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Yang, J.; Zhao, H.; Fang, X.; Li, H. Cannabinoid receptor 2 is upregulated in melanoma. J. Cancer Res. Ther. 2012, 8, 549–554. [Google Scholar] [CrossRef]
- Hijiya, N.; Shibata, T.; Daa, T.; Hamanaka, R.; Uchida, T.; Matsuura, K.; Tsukamoto, Y.; Nakada, C.; Iha, H.; Inomata, M.; et al. Overexpression of cannabinoid receptor 1 in esophageal squamous cell carcinoma is correlated with metastasis to lymph nodes and distant organs, and poor prognosis. Pathol. Int. 2017, 67, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Klein Nulent, T.J.W.; van Diest, P.J.; van der Groep, P.; Leusink, F.K.J.; Kruitwagen, C.L.J.J.; Koole, R.; van Cann, E.M. Cannabinoid receptor-2 immunoreactivity is associated with survival in squamous cell carcinoma of the head and neck. Br. J. Oral Maxillofac. Surg. 2013, 51, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, S.; Giaginis, C.; Alexandrou, P.; Rodriguez, J.; Tasoulas, J.; Danas, E.; Patsouris, E.; Klijanienko, J. Evaluation of cannabinoid CB1 and CB2 receptors expression in mobile tongue squamous cell carcinoma: Associations with clinicopathological parameters and patients’ survival. Tumour Biol. 2016, 37, 3647–3656. [Google Scholar] [CrossRef] [PubMed]
- Baba, Y.; Funakoshi, T.; Mori, M.; Emoto, K.; Masugi, Y.; Ekmekcioglu, S.; Amagai, M.; Tanese, K. Expression of monoacylglycerol lipase as a marker of tumour invasion and progression in malignant melanoma. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 2038–2045. [Google Scholar] [CrossRef]
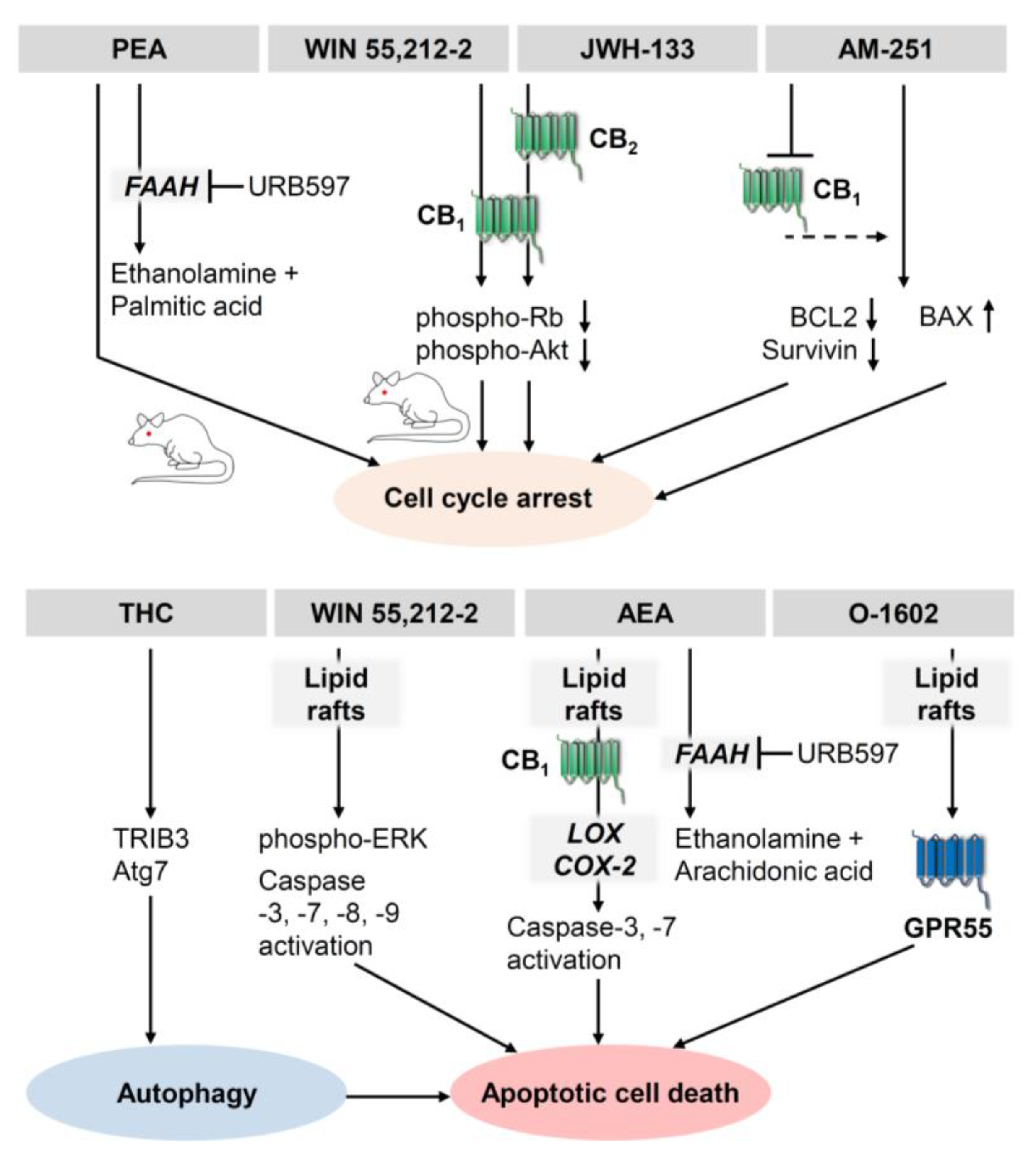
- Blázquez, C.; Carracedo, A.; Barrado, L.; Real, P.J.; Fernández-Luna, J.L.; Velasco, G.; Malumbres, M.; Guzmán, M. Cannabinoid receptors as novel targets for the treatment of melanoma. FASEB J. 2006, 20, 2633–2635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scuderi, M.R.; Cantarella, G.; Scollo, M.; Lempereur, L.; Palumbo, M.; Saccani-Jotti, G.; Bernardini, R. The antimitogenic effect of the cannabinoid receptor agonist WIN55212-2 on human melanoma cells is mediated by the membrane lipid raft. Cancer Lett. 2011, 310, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Simmerman, E.; Qin, X.; Yu, J.C.; Baban, B. Cannabinoids as a Potential New and Novel Treatment for Melanoma: A Pilot Study in a Murine Model. J. Surg. Res. 2019, 235, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Burch, R.; Mortuza, A.; Blumenthal, E.; Mustafa, A. Effects of cannabidiol (CBD) on the inhibition of melanoma cells in vitro. J. Immunoass. Immunochem. 2021, 42, 285–291. [Google Scholar] [CrossRef]
- Naderi, J.; Dana, N.; Javanmard, S.H.; Amooheidari, A.; Yahay, M.; Vaseghi, G. Effects of standardized Cannabis sativa extract and ionizing radiation in melanoma cells in vitro. J. Cancer Res. Ther. 2020, 16, 1495–1499. [Google Scholar]
- Adinolfi, B.; Romanini, A.; Vanni, A.; Martinotti, E.; Chicca, A.; Fogli, S.; Nieri, P. Anticancer activity of anandamide in human cutaneous melanoma cells. Eur. J. Pharmacol. 2013, 718, 154–159. [Google Scholar] [CrossRef]
- Hamtiaux, L.; Masquelier, J.; Muccioli, G.G.; Bouzin, C.; Feron, O.; Gallez, B.; Lambert, D.M. The association of N-palmitoylethanolamine with the FAAH inhibitor URB597 impairs melanoma growth through a supra-additive action. BMC Cancer 2012, 12, 92. [Google Scholar] [CrossRef] [Green Version]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Ng, S.-W.; Cravatt, B.F. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010, 140, 49–61. [Google Scholar] [CrossRef] [Green Version]
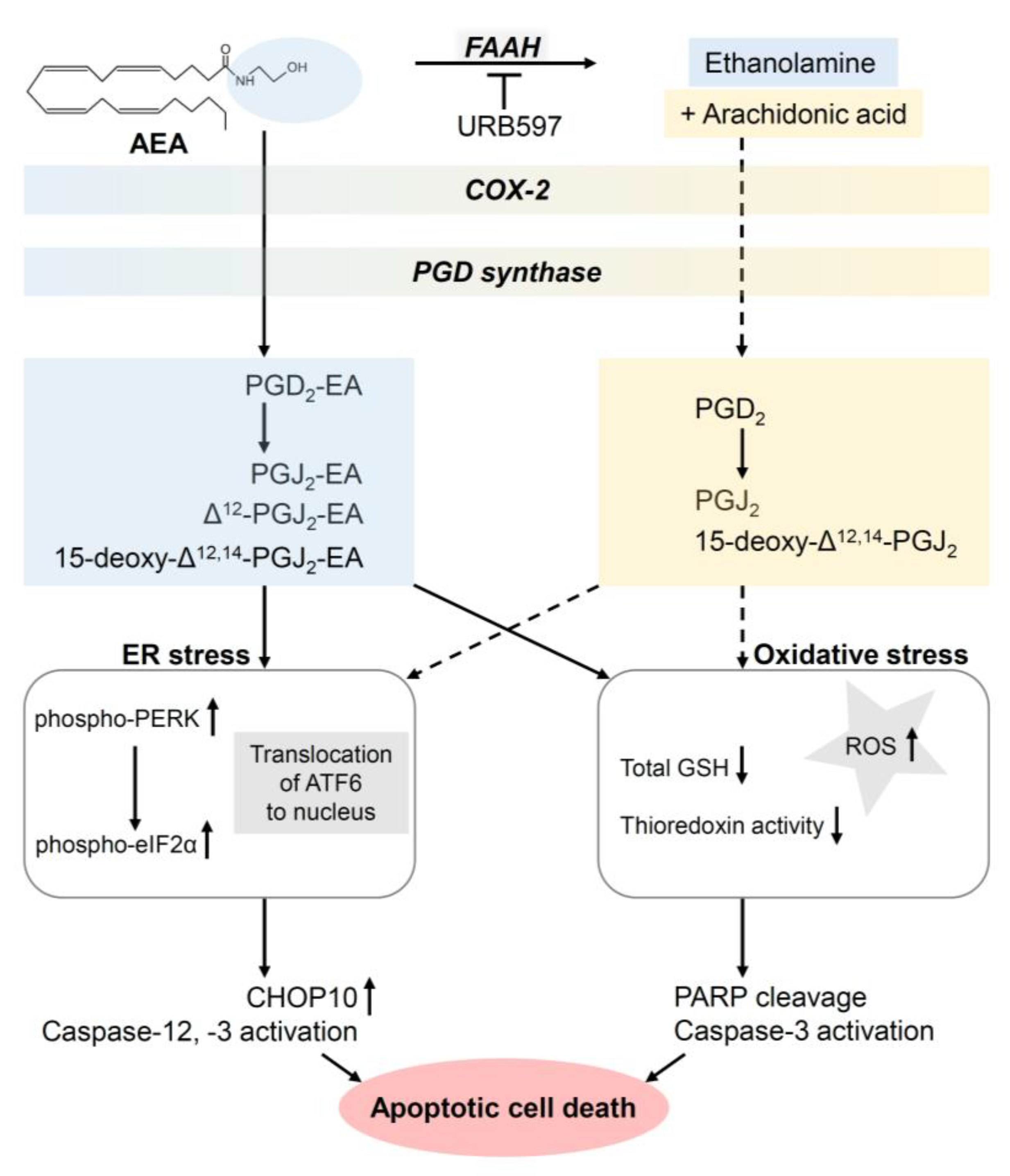
- Elhassanny, A.E.M.; Ladin, D.A.; Soliman, E.; Albassam, H.; Morris, A.; Kobet, R.; Thayne, K.; Burns, C.; Danell, A.S.; van Dross, R. Prostaglandin D2-ethanolamide induces skin cancer apoptosis by suppressing the activity of cellular antioxidants. Prostaglandins Other Lipid Mediat. 2019, 142, 9–23. [Google Scholar] [CrossRef]
- Ladin, D.A.; Soliman, E.; Escobedo, R.; Fitzgerald, T.L.; Yang, L.V.; Burns, C.; van Dross, R. Synthesis and Evaluation of the Novel Prostamide, 15-Deoxy, Δ12,14-Prostamide J2, as a Selective Antitumor Therapeutic. Mol. Cancer Ther. 2017, 16, 838–849. [Google Scholar] [CrossRef] [Green Version]
- Carpi, S.; Fogli, S.; Romanini, A.; Pellegrino, M.; Adinolfi, B.; Podestà, A.; Costa, B.; Da Pozzo, E.; Martini, C.; Breschi, M.C.; et al. AM251 induces apoptosis and G2/M cell cycle arrest in A375 human melanoma cells. Anticancer Drugs 2015, 26, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Carpi, S.; Fogli, S.; Polini, B.; Montagnani, V.; Podestà, A.; Breschi, M.C.; Romanini, A.; Stecca, B.; Nieri, P. Tumor-promoting effects of cannabinoid receptor type 1 in human melanoma cells. Toxicol. In Vitro 2017, 40, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Kenessey, I.; Bánki, B.; Márk, A.; Varga, N.; Tóvári, J.; Ladányi, A.; Rásó, E.; Tímár, J. Revisiting CB1 receptor as drug target in human melanoma. Pathol. Oncol. Res. 2012, 18, 857–866. [Google Scholar] [CrossRef]
- Pucci, M.; Pasquariello, N.; Battista, N.; Di Tommaso, M.; Rapino, C.; Fezza, F.; Zuccolo, M.; Jourdain, R.; Finazzi Agrò, A.; Breton, L.; et al. Endocannabinoids stimulate human melanogenesis via type-1 cannabinoid receptor. J. Biol. Chem. 2012, 287, 15466–15478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dross, R.T. Metabolism of anandamide by COX-2 is necessary for endocannabinoid-induced cell death in tumorigenic keratinocytes. Mol. Carcinog. 2009, 48, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Kuc, C.; Jenkins, A.; van Dross, R.T. Arachidonoyl ethanolamide (AEA)-induced apoptosis is mediated by J-series prostaglandins and is enhanced by fatty acid amide hydrolase (FAAH) blockade. Mol. Carcinog. 2012, 51, 139–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soliman, E.; Henderson, K.L.; Danell, A.S.; van Dross, R. Arachidonoyl-ethanolamide activates endoplasmic reticulum stress-apoptosis in tumorigenic keratinocytes: Role of cyclooxygenase-2 and novel J-series prostamides. Mol. Carcinog. 2016, 55, 117–130. [Google Scholar] [CrossRef]
- Park, S.-W.; Kim, J.-E.; Oh, S.-M.; Cha, W.-J.; Hah, J.-H.; Sung, M.-W. Anticancer effects of anandamide on head and neck squamous cell carcinoma cells via the production of receptor-independent reactive oxygen species. Head Neck 2015, 37, 1187–1192. [Google Scholar] [CrossRef]
- Park, S.-W.; Hah, J.H.; Oh, S.-M.; Jeong, W.-J.; Sung, M.-W. 5-lipoxygenase mediates docosahexaenoyl ethanolamide and N-arachidonoyl-L-alanine-induced reactive oxygen species production and inhibition of proliferation of head and neck squamous cell carcinoma cells. BMC Cancer 2016, 16, 458. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-C.; Tung, T.-H.; Huang, C.-C.; Lin, S.-Y.; Chao, S.-C.; Chiu, S.-P.; Lee, S.-P.; Lo, C.-M. Electrochemical Assessment of Anticancer Compounds on the Human Tongue Squamous Carcinoma Cells. Sensors 2020, 20, 2632. [Google Scholar] [CrossRef]
- Go, Y.Y.; Kim, S.R.; Kim, D.Y.; Chae, S.-W.; Song, J.-J. Cannabidiol enhances cytotoxicity of anti-cancer drugs in human head and neck squamous cell carcinoma. Sci. Rep. 2020, 10, 20622. [Google Scholar] [CrossRef] [PubMed]
- Önay, Ö.; Köse, S.; Süslü, N.; Korkusuz, P.; Nemutlu, E.; Aydın, C.; Hoşal, Ş. Human laryngeal squamous cell carcinoma cell line release of endogenous anandamide and 2-arachidonoylglycerol, and their antiproliferative effect via exogenous supplementation: An in vitro study. Cell Tissue Bank. 2022, 23, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Gómez, E.; Andradas, C.; Flores, J.M.; Quintanilla, M.; Paramio, J.M.; Guzmán, M.; Sánchez, C. The orphan receptor GPR55 drives skin carcinogenesis and is upregulated in human squamous cell carcinomas. Oncogene 2013, 32, 2534–2542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Sadat, S.H.; Ebisumoto, K.; Sakai, A.; Panuganti, B.A.; Ren, S.; Goto, Y.; Haft, S.; Fukusumi, T.; Ando, M.; et al. Cannabinoids Promote Progression of HPV-Positive Head and Neck Squamous Cell Carcinoma via p38 MAPK Activation. Clin. Cancer Res. 2020, 26, 2693–2703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maor, Y.; Yu, J.; Kuzontkoski, P.M.; Dezube, B.J.; Zhang, X.; Groopman, J.E. Cannabidiol inhibits growth and induces programmed cell death in kaposi sarcoma-associated herpesvirus-infected endothelium. Genes Cancer 2012, 3, 512–520. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, J.F.; Kunos, G.; Groopman, J.E. Cannabinoid modulation of Kaposi’s sarcoma-associated herpesvirus infection and transformation. Cancer Res. 2007, 67, 7230–7237. [Google Scholar] [CrossRef] [Green Version]
- Luca, T.; Di Benedetto, G.; Scuderi, M.R.; Palumbo, M.; Clementi, S.; Bernardini, R.; Cantarella, G. The CB1/CB2 receptor agonist WIN-55,212-2 reduces viability of human Kaposi’s sarcoma cells in vitro. Eur. J. Pharmacol. 2009, 616, 16–21. [Google Scholar] [CrossRef]
- Mazuz, M.; Tiroler, A.; Moyal, L.; Hodak, E.; Nadarajan, S.; Vinayaka, A.C.; Gorovitz-Haris, B.; Lubin, I.; Drori, A.; Drori, G.; et al. Synergistic cytotoxic activity of cannabinoids from cannabis sativa against cutaneous T-cell lymphoma (CTCL) in-vitro and ex-vivo. Oncotarget 2020, 11, 1141–1156. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, J.L.; Hill, D.S.; McKee, C.S.; Hernandez-Tiedra, S.; Lorente, M.; Lopez-Valero, I.; Eleni Anagnostou, M.; Babatunde, F.; Corazzari, M.; Redfern, C.P.F.; et al. Exploiting cannabinoid-induced cytotoxic autophagy to drive melanoma cell death. J. Investig. Dermatol. 2015, 135, 1629–1637. [Google Scholar] [CrossRef] [Green Version]
- Capes-Davis, A.; Theodosopoulos, G.; Atkin, I.; Drexler, H.G.; Kohara, A.; MacLeod, R.A.F.; Masters, J.R.; Nakamura, Y.; Reid, Y.A.; Reddel, R.R.; et al. Check your cultures! A list of cross-contaminated or misidentified cell lines. Int. J. Cancer 2010, 127, 1–8. [Google Scholar] [CrossRef]
- Haskó, J.; Fazakas, C.; Molnár, J.; Nyúl-Tóth, Á.; Herman, H.; Hermenean, A.; Wilhelm, I.; Persidsky, Y.; Krizbai, I.A. CB2 receptor activation inhibits melanoma cell transmigration through the blood-brain barrier. Int. J. Mol. Sci. 2014, 15, 8063–8074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaseghi, G.; Taki, M.J.; Javanmard, S.H. Standardized Cannabis sativa extract attenuates tau and stathmin gene expression in the melanoma cell line. Iran. J. Basic Med. Sci. 2017, 20, 1178–1181. [Google Scholar] [PubMed]
- Sailler, S.; Schmitz, K.; Jäger, E.; Ferreiros, N.; Wicker, S.; Zschiebsch, K.; Pickert, G.; Geisslinger, G.; Walter, C.; Tegeder, I.; et al. Regulation of circulating endocannabinoids associated with cancer and metastases in mice and humans. Oncoscience 2014, 1, 272–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakami, Y.; Klein, T.W.; Newton, C.; Djeu, J.Y.; Dennert, G.; Specter, S.; Friedman, H. Suppression by cannabinoids of a cloned cell line with natural killer cell activity. Proc. Soc. Exp. Biol. Med. 1988, 187, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.W.; Kawakami, Y.; Newton, C.; Friedman, H. Marijuana components suppress induction and cytolytic function of murine cytotoxic T cells in vitro and in vivo. J. Toxicol. Environ. Health 1991, 32, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.; Niggemann, B.; Zaenker, K.S.; Entschladen, F. Anandamide is an endogenous inhibitor for the migration of tumor cells and T lymphocytes. Cancer Immunol. Immunother. 2004, 53, 723–728. [Google Scholar] [CrossRef]
- Börner, C.; Smida, M.; Höllt, V.; Schraven, B.; Kraus, J. Cannabinoid receptor type 1- and 2-mediated increase in cyclic AMP inhibits T cell receptor-triggered signaling. J. Biol. Chem. 2009, 284, 35450–35460. [Google Scholar] [CrossRef] [Green Version]
- Haustein, M.; Ramer, R.; Linnebacher, M.; Manda, K.; Hinz, B. Cannabinoids increase lung cancer cell lysis by lymphokine-activated killer cells via upregulation of ICAM-1. Biochem. Pharmacol. 2014, 92, 312–325. [Google Scholar] [CrossRef]
- Glodde, N.; Jakobs, M.; Bald, T.; Tüting, T.; Gaffal, E. Differential role of cannabinoids in the pathogenesis of skin cancer. Life Sci. 2015, 138, 35–40. [Google Scholar] [CrossRef]
- Oláh, A.; Markovics, A.; Szabó-Papp, J.; Szabó, P.T.; Stott, C.; Zouboulis, C.C.; Bíró, T. Differential effectiveness of selected non-psychotropic phytocannabinoids on human sebocyte functions implicates their introduction in dry/seborrhoeic skin and acne treatment. Exp. Dermatol. 2016, 25, 701–707. [Google Scholar] [CrossRef] [Green Version]
- Odan, M.; Ishizuka, N.; Hiramatsu, Y.; Inagaki, M.; Hashizume, H.; Fujii, Y.; Mitsumori, S.; Morioka, Y.; Soga, M.; Deguchi, M.; et al. Discovery of S-777469: An orally available CB2 agonist as an antipruritic agent. Bioorg. Med. Chem. Lett. 2012, 22, 2803–2806. [Google Scholar] [CrossRef] [PubMed]
- Haruna, T.; Soga, M.; Morioka, Y.; Imura, K.; Furue, Y.; Yamamoto, M.; Hayakawa, J.; Deguchi, M.; Arimura, A.; Yasui, K. The Inhibitory Effect of S-777469, a Cannabinoid Type 2 Receptor Agonist, on Skin Inflammation in Mice. Pharmacology 2017, 99, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Burstein, S. Molecular Mechanisms for the Inflammation-Resolving Actions of Lenabasum. Mol. Pharmacol. 2021, 99, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, J.; Nakae, D.; Yasukawa, K. Structure-dependent inhibitory effects of synthetic cannabinoids against 12-O-tetradecanoylphorbol-13-acetate-induced inflammation and skin tumour promotion in mice. J. Pharm. Pharmacol. 2013, 65, 1223–1230. [Google Scholar] [CrossRef]
- Zheng, D.; Bode, A.M.; Zhao, Q.; Cho, Y.-Y.; Zhu, F.; Ma, W.-Y.; Dong, Z. The cannabinoid receptors are required for ultraviolet-induced inflammation and skin cancer development. Cancer Res. 2008, 68, 3992–3998. [Google Scholar] [CrossRef] [Green Version]
- Jastrząb, A.; Gęgotek, A.; Skrzydlewska, E. Cannabidiol Regulates the Expression of Keratinocyte Proteins Involved in the Inflammation Process through Transcriptional Regulation. Cells 2019, 8, 827. [Google Scholar] [CrossRef] [Green Version]
- Gęgotek, A.; Atalay, S.; Domingues, P.; Skrzydlewska, E. The Differences in the Proteome Profile of Cannabidiol-Treated Skin Fibroblasts following UVA or UVB Irradiation in 2D and 3D Cell Cultures. Cells 2019, 8, 995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Łuczaj, W.; Jastrząb, A.; do Rosário Domingues, M.; Domingues, P.; Skrzydlewska, E. Changes in Phospholipid/Ceramide Profiles and Eicosanoid Levels in the Plasma of Rats Irradiated with UV Rays and Treated Topically with Cannabidiol. Int. J. Mol. Sci. 2021, 22, 8700. [Google Scholar] [CrossRef]
- Guzmán, M.; Duarte, M.J.; Blázquez, C.; Ravina, J.; Rosa, M.C.; Galve-Roperh, I.; Sánchez, C.; Velasco, G.; González-Feria, L. A pilot clinical study of Δ9-tetrahydrocannabinol in patients with recurrent glioblastoma multiforme. Br. J. Cancer 2006, 95, 197–203. [Google Scholar] [CrossRef]
- Twelves, C.; Sabel, M.; Checketts, D.; Miller, S.; Tayo, B.; Jove, M.; Brazil, L.; Short, S.C. A phase 1b randomised, placebo-controlled trial of nabiximols cannabinoid oromucosal spray with temozolomide in patients with recurrent glioblastoma. Br. J. Cancer 2021, 124, 1379–1387. [Google Scholar] [CrossRef]
- Ramer, R.; Wittig, F.; Hinz, B. The Endocannabinoid System as a Pharmacological Target for New Cancer Therapies. Cancers 2021, 13, 5701. [Google Scholar] [CrossRef] [PubMed]
- Whiting, P.F.; Wolff, R.F.; Deshpande, S.; Di Nisio, M.; Duffy, S.; Hernandez, A.V.; Keurentjes, J.C.; Lang, S.; Misso, K.; Ryder, S.; et al. Cannabinoids for Medical Use: A Systematic Review and Meta-analysis. JAMA 2015, 313, 2456–2473. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hong, P.J.; May, C.; Rehman, Y.; Oparin, Y.; Hong, C.J.; Hong, B.Y.; AminiLari, M.; Gallo, L.; Kaushal, A.; et al. Medical cannabis or cannabinoids for chronic non-cancer and cancer related pain: A systematic review and meta-analysis of randomised clinical trials. BMJ 2021, 374, n1034. [Google Scholar] [CrossRef]
- Simon, L.; Baldwin, C.; Kalea, A.Z.; Slee, A. Cannabinoid interventions for improving cachexia outcomes in cancer: A systematic review and meta-analysis. J. Cachexia Sarcopenia Muscle 2022, 13, 23–41. [Google Scholar] [CrossRef]
- Elliott, D.A.; Nabavizadeh, N.; Romer, J.L.; Chen, Y.; Holland, J.M. Medical marijuana use in head and neck squamous cell carcinoma patients treated with radiotherapy. Support. Care Cancer 2016, 24, 3517–3524. [Google Scholar] [CrossRef]
- Zutt, M.; Hänssle, H.; Emmert, S.; Neumann, C.; Kretschmer, L. Dronabinol zur supportiven Therapie metastasierter maligner Melanome mit Lebermetastasen. Hautarzt 2006, 57, 423–427. [Google Scholar] [CrossRef]
- Taha, T.; Meiri, D.; Talhamy, S.; Wollner, M.; Peer, A.; Bar-Sela, G. Cannabis Impacts Tumor Response Rate to Nivolumab in Patients with Advanced Malignancies. Oncologist 2019, 24, 549–554. [Google Scholar] [CrossRef] [Green Version]
- Bar-Sela, G.; Cohen, I.; Campisi-Pinto, S.; Lewitus, G.M.; Oz-Ari, L.; Jehassi, A.; Peer, A.; Turgeman, I.; Vernicova, O.; Berman, P.; et al. Cannabis Consumption Used by Cancer Patients during Immunotherapy Correlates with Poor Clinical Outcome. Cancers 2020, 12, 2447. [Google Scholar] [CrossRef]
- Liang, C.; McClean, M.D.; Marsit, C.; Christensen, B.; Peters, E.; Nelson, H.H.; Kelsey, K.T. A population-based case-control study of marijuana use and head and neck squamous cell carcinoma. Cancer Prev. Res. 2009, 2, 759–768. [Google Scholar] [CrossRef] [Green Version]
- Rosenblatt, K.A.; Daling, J.R.; Chen, C.; Sherman, K.J.; Schwartz, S.M. Marijuana use and risk of oral squamous cell carcinoma. Cancer Res. 2004, 64, 4049–4054. [Google Scholar] [CrossRef] [Green Version]
- Ghasemiesfe, M.; Barrow, B.; Leonard, S.; Keyhani, S.; Korenstein, D. Association Between Marijuana Use and Risk of Cancer: A Systematic Review and Meta-analysis. JAMA Netw. Open 2019, 2, e1916318. [Google Scholar] [CrossRef] [PubMed]
- Clark, T.M. Scoping Review and Meta-Analysis Suggests that Cannabis Use May Reduce Cancer Risk in the United States. Cannabis Cannabinoid Res. 2021, 6, 413–434. [Google Scholar] [CrossRef] [PubMed]
- Adler, B.L.; DeLeo, V.A. Allergenic ingredients in commercial topical cannabinoid preparations. J. Am. Acad. Dermatol. 2019, 81, 847–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, A.P.; Green, B.J.; Sussman, G.; Berlin, N.; Lata, H.; Chandra, S.; ElSohly, M.A.; Hettick, J.M.; Beezhold, D.H. Characterization of Cannabis sativa allergens. Ann. Allergy Asthma Immunol. 2013, 111, 32–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, M.; Kirchhof, M.G. Dermatology-Related Uses of Medical Cannabis Promoted by Dispensaries in Canada, Europe, and the United States. J. Cutan. Med. Surg. 2019, 23, 178–184. [Google Scholar] [CrossRef]
- Shi, S.; Brant, A.R.; Sabolch, A.; Pollom, E. False News of a Cannabis Cancer Cure. Cureus 2019, 11, e3918. [Google Scholar] [CrossRef] [Green Version]
- Zenone, M.; Snyder, J.; Caulfield, T. Crowdfunding Cannabidiol (CBD) for Cancer: Hype and Misinformation on GoFundMe. Am. J. Public Health 2020, 110, S294–S299. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramer, R.; Wendt, F.; Wittig, F.; Schäfer, M.; Boeckmann, L.; Emmert, S.; Hinz, B. Impact of Cannabinoid Compounds on Skin Cancer. Cancers 2022, 14, 1769. https://doi.org/10.3390/cancers14071769
Ramer R, Wendt F, Wittig F, Schäfer M, Boeckmann L, Emmert S, Hinz B. Impact of Cannabinoid Compounds on Skin Cancer. Cancers. 2022; 14(7):1769. https://doi.org/10.3390/cancers14071769
Chicago/Turabian StyleRamer, Robert, Franziska Wendt, Felix Wittig, Mirijam Schäfer, Lars Boeckmann, Steffen Emmert, and Burkhard Hinz. 2022. "Impact of Cannabinoid Compounds on Skin Cancer" Cancers 14, no. 7: 1769. https://doi.org/10.3390/cancers14071769
APA StyleRamer, R., Wendt, F., Wittig, F., Schäfer, M., Boeckmann, L., Emmert, S., & Hinz, B. (2022). Impact of Cannabinoid Compounds on Skin Cancer. Cancers, 14(7), 1769. https://doi.org/10.3390/cancers14071769