
Germline Variants in Cancer Genes from Young Breast Cancer Mexican Patients

 ,
,  , and
, and 
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Ethical Considerations
2.3. Sample and Panel Library Preparation for Sequencing
2.4. Sequencing
2.5. Variant Calling
2.6. Variant Categorization
2.7. Statistical Analysis
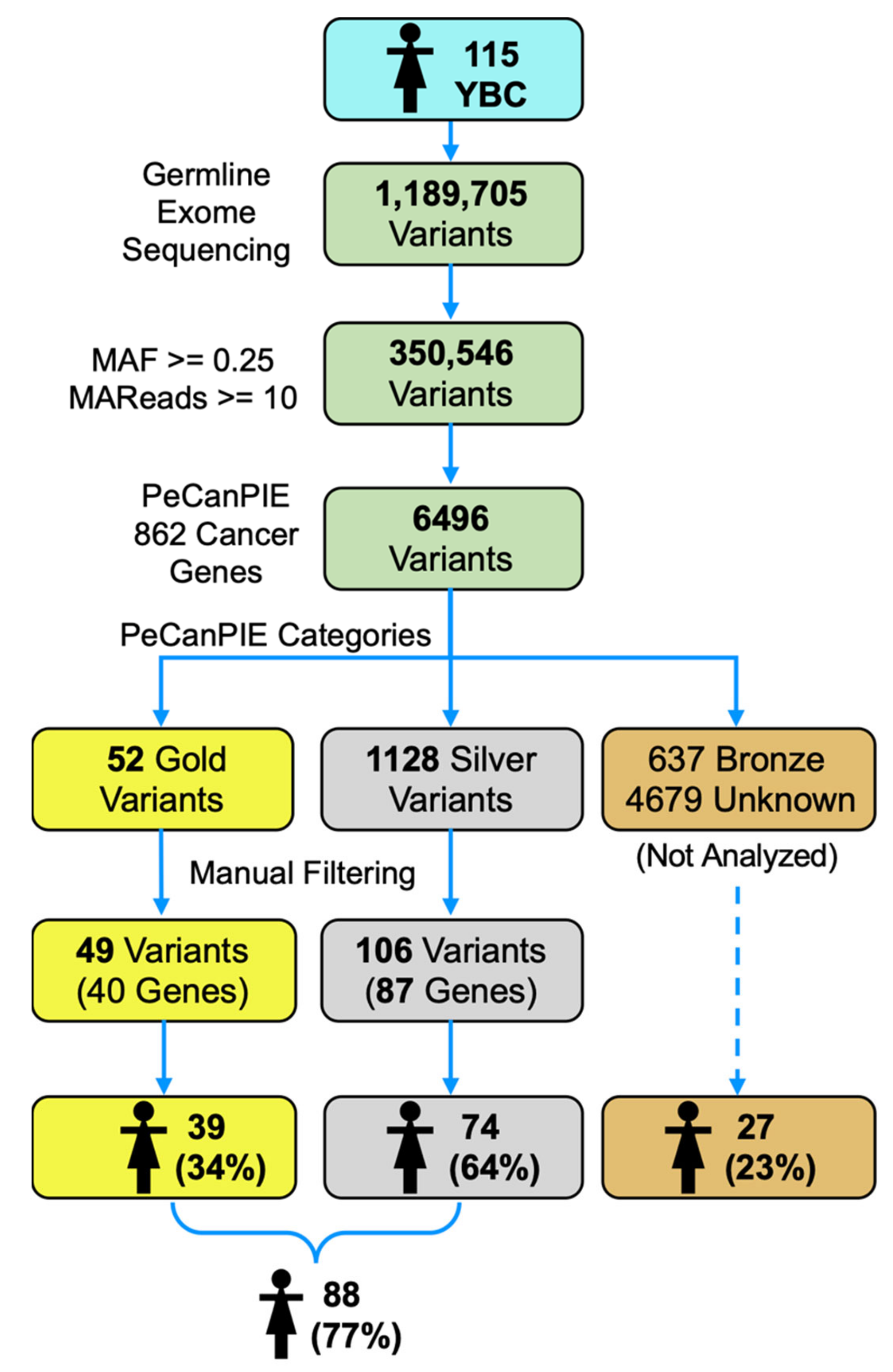
3. Results
3.1. Clinical Population
3.2. Variant Categorization
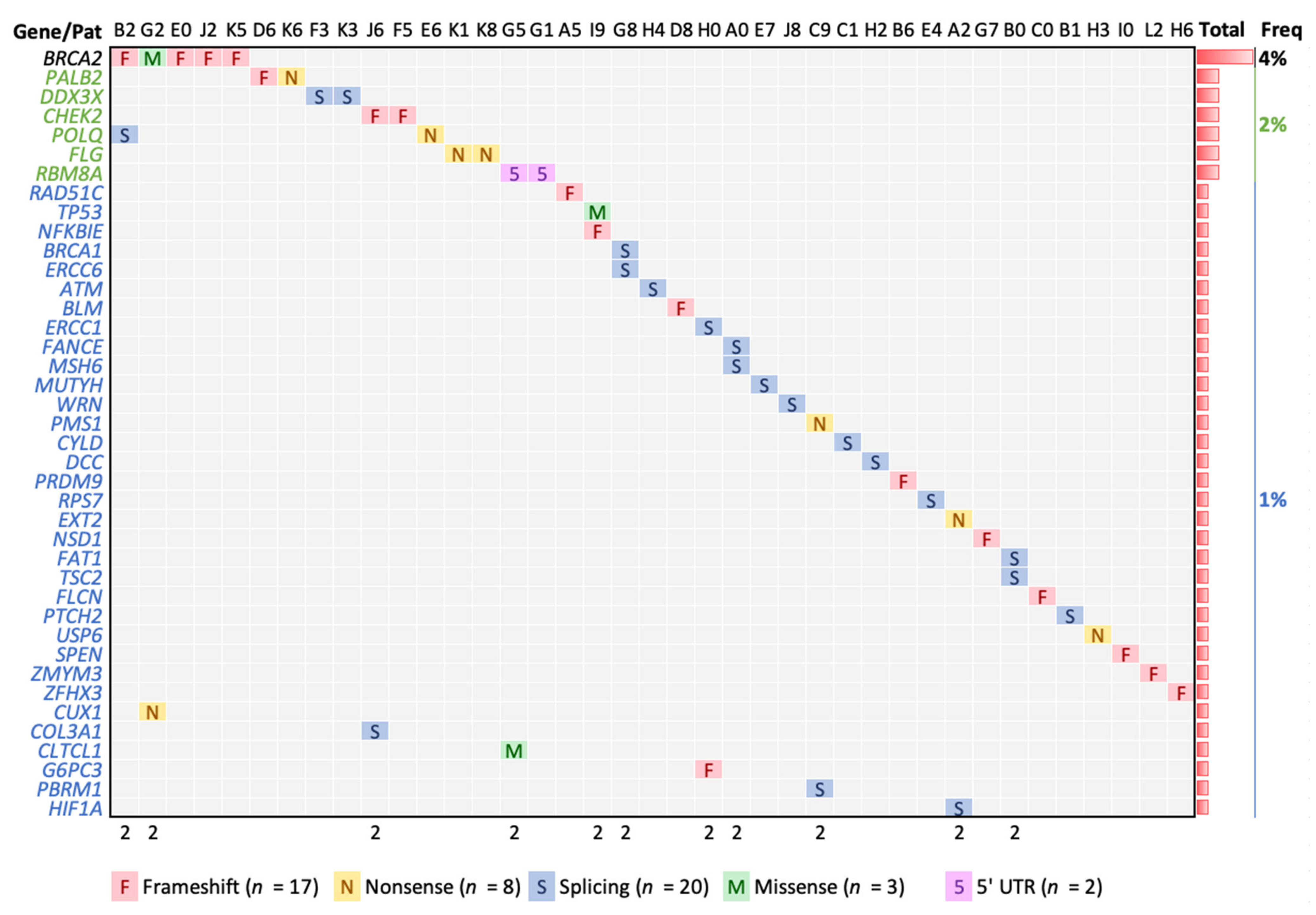
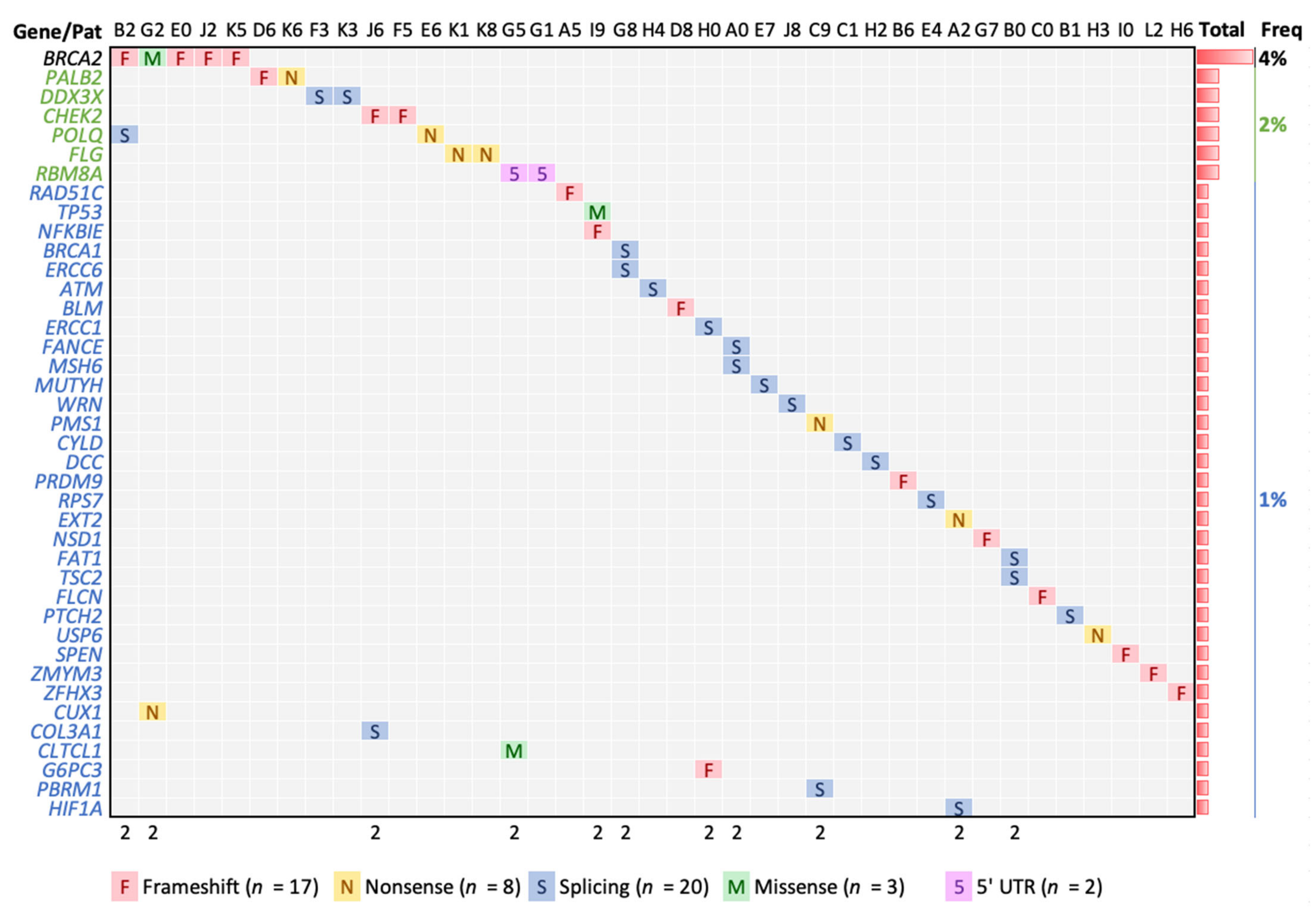
3.3. High Confident Germline Variants in Cancer Genes
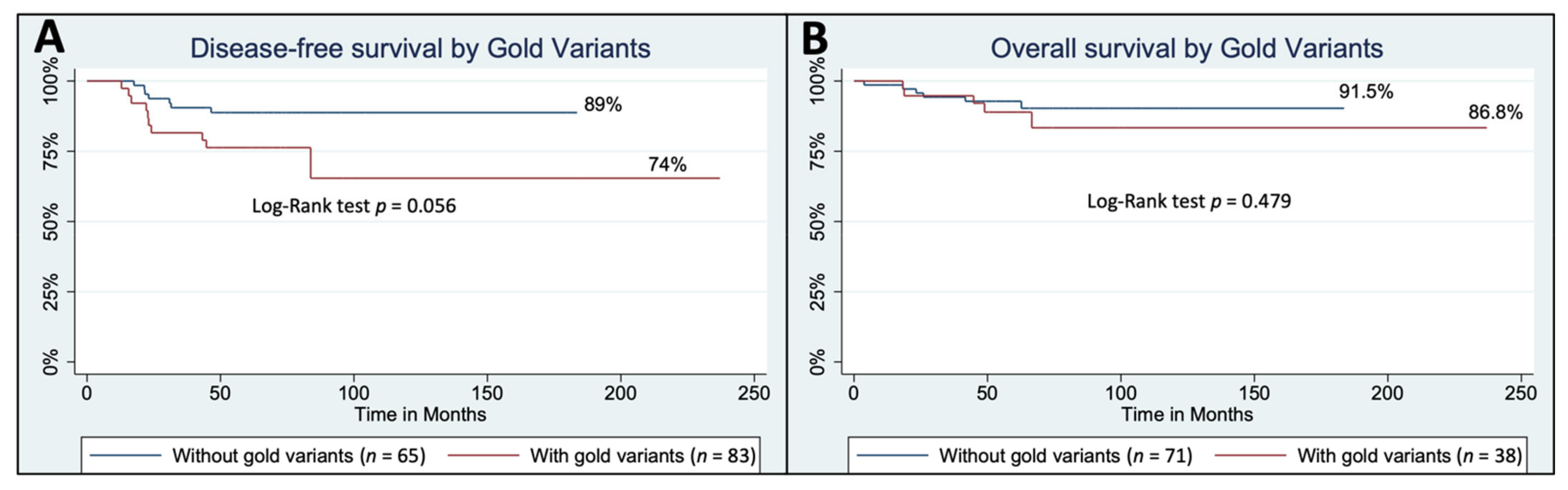
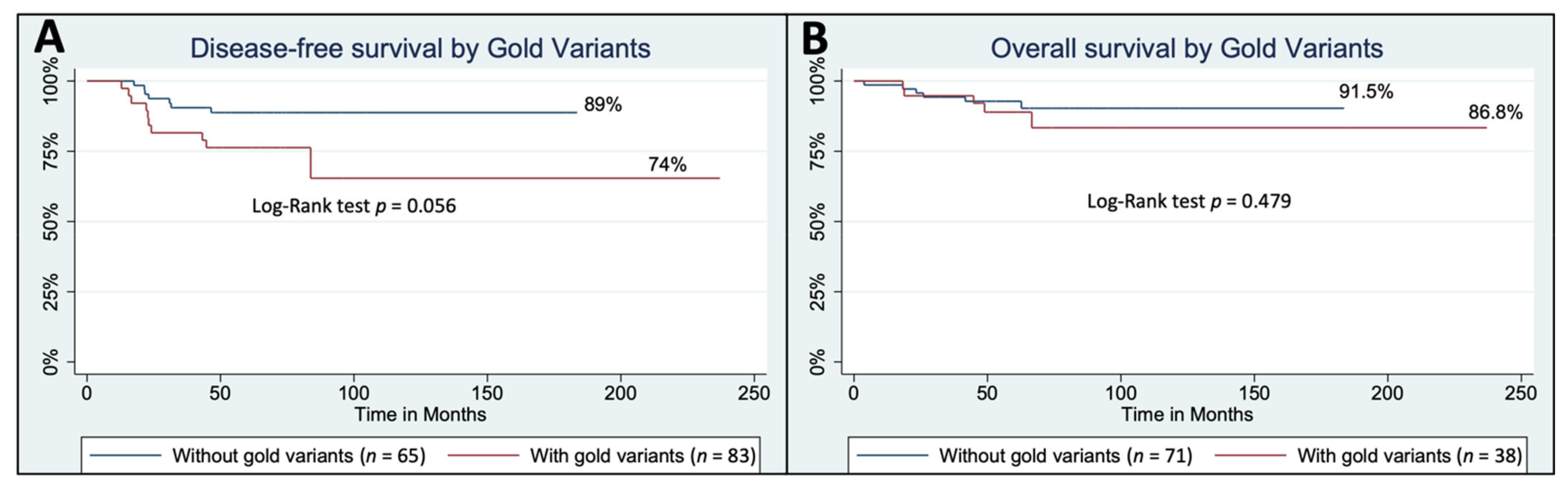
3.4. Modest Confidence Germline Variants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Villarreal-Garza, C.; Platas, A.; Miaja, M.; Fonseca, A.; Mesa-Chavez, F.; Garcia-Garcia, M.; Chapman, J.A.; Lopez-Martinez, E.A.; Pineda, C.; Mohar, A.; et al. Young women with breast cancer in Mexico: Results of the pilot phase of the Joven & Fuerte prospective cohort. J. Glob. Oncol. 2020, 6, 395–406. [Google Scholar] [CrossRef]
- Gómez-Flores-ramos, L.; Castro-Sánchez, A.; Peña-Curiel, O.; Mohar-Betancourt, A. Molecular biology in young women with breast cancer: From tumor gene Expression to DNA mutations. Rev. Investig. Clin. 2017, 69, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudela, E.; Samec, M.; Kubatka, P.; Nachajova, M.; Laucekova, Z.; Liskova, A.; Dokus, K.; Biringer, K.; Simova, D.; Gabonova, E.; et al. Breast cancer in young women: Status quo and advanced disease management by a predictive, preventive, and personalized approach. Cancers 2019, 11, 1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabriel, C.A.; Domchek, S.M. Breast cancer in young women. Breast Cancer Res. 2010, 12, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assi, H.; Khoury, K.; Dbouk, H.; Khalil, L.; Mouhieddine, T.; El Saghir, N. Epidemiology and prognosis of breast cancer in young women. J. Thorac. Dis. 2013, 5 (Suppl. S1), S2–S8. [Google Scholar] [CrossRef] [PubMed]
- Kast, K.; Rhiem, K.; Wappenschmidt, B.; Hahnen, E.; Hauke, J.; Bluemcke, B.; Zarghooni, V.; Herold, N.; Ditsch, N.; Kiechle, M.; et al. Prevalence of BRCA1/2 germline mutations in 21 401 families with breast and ovarian cancer. J. Med. Genet. 2016, 53, 465–471. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, F.C.; Van Overeem Hansen, T.; Sørensen, C.S. Hereditary breast and ovarian cancer: New genes in confined pathways. Nat. Rev. Cancer 2016, 16, 599–612. [Google Scholar] [CrossRef]
- Wendt, C.; Margolin, S. Identifying breast cancer susceptibility genes—A review of the genetic background in familial breast cancer. Acta Oncol. 2019, 58, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Economopoulou, P.; Dimitriadis, G.; Psyrri, A. Beyond BRCA: New hereditary breast cancer susceptibility genes. Cancer Treat. Rev. 2015, 41, 1–8. [Google Scholar] [CrossRef]
- Urbina-Jara, L.K.; Rojas-Martinez, A.; Martinez-Ledesma, E.; Aguilar, D.; Villarreal-Garza, C.; Ortiz-Lopez, R. Landscape of germline mutations in dna repair genes for breast cancer in latin america: Opportunities for parp-like inhibitors and immunotherapy. Genes 2019, 10, 786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urban, R.Q.; Velásquez, C.E.D.; Gitler, R.; Castillo, M.P.R.; Toporek, M.S.; Morales, A.F.; García, O.M.; Esquivel, L.G.; Mejía, G.T.; Dean, M.; et al. Comprehensive Analysis of Germline Variants in Mexican Patients with Hereditary Breast and Ovarian Cancer Susceptibility. Cancers 2018, 10, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fountzilas, C.; Kaklamani, V.G. Multi-gene Panel Testing in Breast Cancer Management. Optim. Breast Cancer Manag. 2018, 173, 121–140. [Google Scholar] [CrossRef]
- Spurdle, A.B.; Bowman, M.A.; Shamsani, J.; Kirk, J. Endometrial cancer gene panels: Clinical diagnostic vs research germline DNA testing. Mod. Pathol. 2017, 30, 1048–1068. [Google Scholar] [CrossRef] [PubMed]
- Smolle, M.A.; Kashofer, K.; Riedl, J.M.; Stotz, M.; Gerger, A. Genetic Analysis Using a Gene Panel in 87 Caucasian Patients With Colorectal Cancer: Own Results and Review of Literature. Anticancer Res. 2019, 39, 847–852. [Google Scholar] [CrossRef]
- Yatabe, Y.; Sunami, K.; Goto, K.; Nishio, K.; Aragane, N.; Ikeda, S.; Inoue, A.; Kinoshita, I.; Kimura, H.; Sakamoto, T.; et al. Multiplex gene-panel testing for lung cancer patients. Pathol. Int. 2020, 70, 921–931. [Google Scholar] [CrossRef]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; Flicek, P.; et al. A global reference for human genetic variation. Nature 2015, 68, 526. [Google Scholar]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef]
- Edmonson, M.N.; Patel, A.N.; Hedges, D.J.; Wang, Z.; Rampersaud, E.; Kesserwan, C.A.; Zhou, X.; Liu, Y.; Newman, S.; Rusch, M.C.; et al. Pediatric Cancer Variant Pathogenicity Information Exchange (PeCanPIE): A cloud-based platform for curating and classifying germline variants. Genome Res. 2019, 29, 1555–1565. [Google Scholar] [CrossRef] [Green Version]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.; Parker, M.; Kranenburg, T.A.; Lu, C.; Chen, X.; Ding, L.; Phoenix, T.N.; Hedlund, E.; Wei, L.; Zhu, X.; et al. Novel mutations target distinct subgroups of medulloblastoma. Nature 2012, 488, 43–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winham, S.J.; Larson, N.B.; Armasu, S.M.; Fogarty, Z.C.; Larson, M.C.; McCauley, B.M.; Wang, C.; Lawrenson, K.; Gayther, S.; Cunningham, J.M.; et al. Molecular signatures of X chromosome inactivation and associations with clinical outcomes in epithelial ovarian cancer. Hum. Mol. Genet. 2019, 28, 1331–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scala, M.; Torella, A.; Severino, M.; Morana, G.; Castello, R.; Accogli, A.; Verrico, A.; Vari, M.S.; Cappuccio, G.; Pinelli, M.; et al. Three de novo DDX3X variants associated with distinctive brain developmental abnormalities and brain tumor in intellectually disabled females. Eur. J. Hum. Genet. 2019, 27, 1254–1259. [Google Scholar] [CrossRef] [PubMed]
- Blok, L.S.; Madsen, E.; Juusola, J.; Gilissen, C.; Baralle, D.; Reijnders, M.R.F.; Venselaar, H.; Helsmoortel, C.; Cho, M.T.; Hoischen, A.; et al. Mutations in DDX3X Are a Common Cause of Unexplained Intellectual Disability with Gender-Specific Effects on Wnt Signaling. Am. J. Hum. Genet. 2015, 97, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.-C.; Lee, Y.-H.W.; Tarn, W.-Y. The DEAD-Box RNA Helicase DDX3 Associates with Export Messenger Ribonucleoproteins as well asTip-associated Protein and Participates in Translational Control. Mol. Biol. Cell 2008, 19, 3847–3858. [Google Scholar] [CrossRef] [Green Version]
- Cargill, M.J.; Morales, A.; Ravishankar, S.; Warren, E.H. RNA helicase, DDX3X, is actively recruited to sites of DNA damage in live cells. DNA Repair 2021, 103, 103137. [Google Scholar] [CrossRef]
- Midha, M.K.; Huang, Y.-F.; Yang, H.-H.; Fan, T.-C.; Chang, N.-C.; Chen, T.-H.; Wang, Y.-T.; Kuo, W.-H.; Chang, K.-J.; Shen, C.-Y.; et al. Comprehensive Cohort Analysis of Mutational Spectrum in Early Onset Breast Cancer Patients. Cancers 2020, 12, 2089. [Google Scholar] [CrossRef]
- Kunadirek, P.; Chuaypen, N.; Jenjaroenpun, P.; Wongsurawat, T.; Pinjaroen, N.; Sirichindakul, P.; Nookaew, I.; Tangkijvanich, P. Cell-Free DNA Analysis by Whole-Exome Sequencing for Hepatocellular Carcinoma: A Pilot Study in Thailand. Cancers 2021, 13, 2229. [Google Scholar] [CrossRef]
- Gellert, P.; Segal, C.V.; Gao, Q.; López-Knowles, E.; Martin, L.A.; Dodson, A.; Li, T.; Miller, C.A.; Lu, C.; Mardis, E.R.; et al. Impact of mutational profiles on response of primary oestrogen receptor-positive breast cancers to oestrogen deprivation. Nat. Commun. 2016, 7, 13294. [Google Scholar] [CrossRef] [Green Version]
- Albers, C.A.; Paul, D.S.; Schulze, H.; Freson, K.; Stephens, J.C.; Smethurst, P.A.; Jolley, J.D.; Cvejic, A.; Kostadima, M.; Bertone, P.; et al. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat. Genet. 2012, 44, 435–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, N.; Chen, H.; Zhao, N.; Yi, Y.; Li, C. A Comprehensive Pan-Cancer Analysis of RBM8A Based on Data Mining. J. Oncol. 2021, 2021, 9983354. [Google Scholar] [CrossRef] [PubMed]
- Encinas, G.; Sabelnykova, V.Y.; De Lyra, E.C.; Katayama, M.L.H.; Maistro, S.; Valle, P.W.M.D.V.; Pereira, G.F.D.L.; Rodrigues, L.M.; Serio, P.A.D.M.P.; De Gouvêa, A.C.R.C.; et al. Somatic mutations in early onset luminal breast cancer. Oncotarget 2018, 9, 22460–22479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalkh, N.; Chouery, E.; Haidar, Z.; Khater, C.; Atallah, D.; Ali, H.; Marafie, M.J.; Al-Mulla, M.R.; Al-Mulla, F.; Megarbane, A. Next-generation sequencing in familial breast cancer patients from Lebanon. BMC Med. Genom. 2017, 10, 8. [Google Scholar] [CrossRef] [Green Version]
- Sokolenko, A.P.; Preobrazhenskaya, E.V.; Aleksakhina, S.N.; Iyevleva, A.G.; Mitiushkina, N.V.; Zaitseva, O.A.; Yatsuk, O.S.; Tiurin, V.I.; Strelkova, T.N.; Togo, A.V.; et al. Candidate gene analysis of BRCA1/2 mutation-negative high-risk Russian breast cancer patients. Cancer Lett. 2015, 359, 259–261. [Google Scholar] [CrossRef]
- Ellingson, M.S.; Hart, S.N.; Kalari, K.R.; Suman, V.; Schahl, K.A.; Dockter, T.J.; Felten, S.J.; Sinnwell, J.P.; Thompson, K.J.; Tang, X.; et al. Exome sequencing reveals frequent deleterious germline variants in cancer susceptibility genes in women with invasive breast cancer undergoing neoadjuvant chemotherapy. Breast Cancer Res. Treat. 2015, 153, 435–443. [Google Scholar] [CrossRef] [Green Version]
- Tedaldi, G.; Tebaldi, M.; Zampiga, V.; Danesi, R.; Arcangeli, V.; Ravegnani, M.; Cangini, I.; Pirini, F.; Petracci, E.; Rocca, A.; et al. Multiple-gene panel analysis in a case series of 255 women with hereditary breast and ovarian cancer. Oncotarget 2017, 8, 47064–47075. [Google Scholar] [CrossRef]
- Thompson, E.R.; Doyle, M.A.; Ryland, G.L.; Rowley, S.M.; Choong, D.Y.H.; Tothill, R.W.; Thorne, H.; Barnes, D.R.; Li, J.; Ellul, J.; et al. Exome Sequencing Identifies Rare Deleterious Mutations in DNA Repair Genes FANCC and BLM as Potential Breast Cancer Susceptibility Alleles. PLoS Genet. 2012, 8, e1002894. [Google Scholar] [CrossRef] [Green Version]
- Sokolenko, A.P.; Bogdanova, N.; Kluzniak, W.; Preobrazhenskaya, E.V.; Kuligina, E.S.; Iyevleva, A.G.; Aleksakhina, S.N.; Mitiushkina, N.V.; Gorodnova, T.V.; Bessonov, A.A.; et al. Double heterozygotes among breast cancer patients analyzed for BRCA1, CHEK2, ATM, NBN/NBS1, and BLM germ-line mutations. Breast Cancer Res. Treat. 2014, 145, 553–562. [Google Scholar] [CrossRef]
- Torrezan, G.T.; de Almeida, F.G.d.S.R.; Figueiredo, M.C.P.; Barros, B.D.d.F.; de Paula, C.A.A.; Valieris, R.; de Souza, J.E.S.; Ramalho, R.F.; da Silva, F.C.C.; Ferreira, E.N.; et al. Complex Landscape of Germline Variants in Brazilian Patients With Hereditary and Early Onset Breast Cancer. Front. Genet. 2018, 9, 161. [Google Scholar] [CrossRef] [Green Version]
- Scarpitta, R.; Zanna, I.; Aretini, P.; Gambino, G.; Scatena, C.; Mei, B.; Ghilli, M.; Rossetti, E.; Roncella, M.; Congregati, C.; et al. Germline investigation in male breast cancer of DNA repair genes by next-generation sequencing. Breast Cancer Res. Treat. 2019, 178, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Oliver, J.; Quezada Urban, R.; Franco Cortés, C.A.; Díaz Velásquez, C.E.; Montealegre Paez, A.L.; Pacheco-Orozco, R.A.; Castro Rojas, C.; García-Robles, R.; López Rivera, J.J.; Gaitán Chaparro, S.; et al. Latin American Study of Hereditary Breast and Ovarian Cancer LACAM: A Genomic Epidemiology Approach. Front. Oncol. 2019, 9, 1429. [Google Scholar] [CrossRef] [PubMed]
- Verhoeft, K.R.; Ngan, H.L.; Lui, V.W.Y. The cylindromatosis (CYLD) gene and head and neck tumorigenesis. Cancers Head Neck 2016, 1, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heddar, A.; Fermey, P.; Coutant, S.; Angot, E.; Sabourin, J.-C.; Michelin, P.; Parodi, N.; Charbonnier, F.; Vezain, M.; Bougeard, G.; et al. Familial solitary chondrosarcoma resulting from germline EXT2 mutation. Genes Chromosom. Cancer 2017, 56, 128–134. [Google Scholar] [CrossRef]
- Santos, S.C.L.; Rizzo, I.M.P.O.; Takata, R.I.; Speck-Martins, C.E.; Brum, J.M.; Sollaci, C. Analysis of mutations in EXT1 and EXT2 in Brazilian patients with multiple osteochondromas. Mol. Genet. Genom. Med. 2018, 6, 382–392. [Google Scholar] [CrossRef]
- Afshar, A.R.; Pekmezci, M.; Bloomer, M.M.; Cadenas, N.J.; Stevers, M.; Banerjee, A.; Roy, R.; Olshen, A.B.; Van Ziffle, J.; Onodera, C.; et al. Next-Generation Sequencing of Retinoblastoma Identifies Pathogenic Alterations beyond RB1 Inactivation That Correlate with Aggressive Histopathologic Features. Ophthalmology 2020, 127, 804–813. [Google Scholar] [CrossRef]
- Park, S.; Supek, F.; Lehner, B. Systematic discovery of germline cancer predisposition genes through the identification of somatic second hits. Nat. Commun. 2018, 9, 2601. [Google Scholar] [CrossRef] [Green Version]
- Benusiglio, P.R.; Couvé, S.; Gilbert-Dussardier, B.; Deveaux, S.; Le Jeune, H.; Da Costa, M.; Fromont, G.; Memeteau, F.; Yacoub, M.; Coupier, I.; et al. A germline mutation in PBRM1 predisposes to renal cell carcinoma. J. Med. Genet. 2015, 52, 426–430. [Google Scholar] [CrossRef]
- Spinella, J.-F.; Healy, J.; Saillour, V.; Richer, C.; Cassart, P.; Ouimet, M.; Sinnett, D. Whole-exome sequencing of a rare case of familial childhood acute lymphoblastic leukemia reveals putative predisposing mutations in Fanconi anemia genes. BMC Cancer 2015, 15, 539. [Google Scholar] [CrossRef] [Green Version]
- Taeubner, J.; Brozou, T.; Qin, N.; Bartl, J.; Ginzel, S.; Schaper, J.; Felsberg, J.; Fulda, S.; Vokuhl, C.; Borkhardt, A.; et al. Congenital embryonal rhabdomyosarcoma caused by heterozygous concomitant PTCH1 and PTCH2 germline mutations. Eur. J. Hum. Genet. 2018, 26, 137–142. [Google Scholar] [CrossRef]
- Li, N.; McInerny, S.; Zethoven, M.; Cheasley, D.; Lim, B.W.X.; Rowley, S.M.; Devereux, L.; Grewal, N.; Ahmadloo, S.; Byrne, D.; et al. Combined Tumor Sequencing and Case-Control Analyses of RAD51C in Breast Cancer. J. Natl. Cancer Inst. 2019, 111, 1332–1338. [Google Scholar] [CrossRef] [PubMed]
- Blombery, P.; Fox, L.C.; Ryland, G.L.; Thompson, E.R.; Lickiss, J.; McBean, M.; Yerneni, S.; Hughes, D.; Greenway, A.; Mechinaud, F.; et al. Utility of clinical comprehensive genomic characterization for diagnostic categorization in patients presenting with hypocellular bone marrow failure syndromes. Haematologica 2021, 106, 64–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Song, B.; Guo, S.; Li, G.; Jin, G. Identification of germline and somatic mutations in pancreatic adenosquamous carcinoma using whole exome sequencing. Cancer Biomark. 2020, 27, 389–397. [Google Scholar] [CrossRef]
- Sigurdson, A.J.; Brenner, A.V.; Roach, J.A.; Goudeva, L.; Müller, J.A.; Nerlich, K.; Reiners, C.; Schwab, R.; Pfeiffer, L.; Waldenberger, M.; et al. Selected single-nucleotide polymorphisms in FOXE1, SERPINA5, FTO, EVPL, TICAM1 and SCARB1 are associated with papillary and follicular thyroid cancer risk: Replication study in a German population. Carcinogenesis 2016, 37, 677–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, C.J.; Miranda, M.A.; O’Hern, M.J.; McElroy, J.P.; Coombes, K.R.; Bundschuh, R.; Cohn, D.E.; Mutch, D.G.; Goodfellow, P.J. Patterns of CTCF and ZFHX3 Mutation and Associated Outcomes in Endometrial Cancer. J. Natl. Cancer Inst. 2015, 107, 11. [Google Scholar] [CrossRef] [Green Version]
- Rai, R.; Sharma, K.L.; Tiwari, S.; Misra, S.; Kumar, A.; Mittal, B. DCC (deleted in colorectal carcinoma) gene variants confer increased susceptibility to gallbladder cancer (Ref. No.: Gene-D-12-01446). Gene 2013, 518, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Lubinski, J.; Huzarski, T.; Møller, P.; Armel, S.; Karlan, B.Y.; Senter, L.; Eisen, A.; Foulkes, W.D.; Singer, C.F.; et al. Weight Gain and the Risk of Ovarian Cancer in BRCA1 and BRCA2 Mutation Carriers. Cancer Epidemiol. Biomark. Prev. 2021, 30, 2038–2043. [Google Scholar] [CrossRef]
- Qian, F.; Rookus, M.A.; Leslie, G.; Risch, H.A.; Greene, M.H.; Aalfs, C.M.; Adank, M.A.; Adlard, J.; Agnarsson, B.A.; Ahmed, M.; et al. Mendelian randomisation study of height and body mass index as modifiers of ovarian cancer risk in 22,588 BRCA1 and BRCA2 mutation carriers. Br. J. Cancer 2019, 121, 180–192. [Google Scholar] [CrossRef] [Green Version]
- Qian, F.; Wang, S.; Mitchell, J.; McGuffog, L.; Barrowdale, D.; Leslie, G.; Oosterwijk, J.C.; Chung, W.K.; Evans, D.G.; Engel, C.; et al. Height and Body Mass Index as Modifiers of Breast Cancer Risk in BRCA1/2 Mutation Carriers: A Mendelian Randomization Study. J. Natl. Cancer Inst. 2019, 111, 350–364. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.A.; Jian, J.-W.; Hung, C.-F.; Peng, H.-P.; Yang, C.-F.; Cheng, H.-C.S.; Yang, A.-S. Germline breast cancer susceptibility gene mutations and breast cancer outcomes. BMC Cancer 2018, 18, 315. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Wu, J.; Zhang, C.; Sun, S.; Zhang, J.; Liu, W.; Zhang, Z.; Huang, J. BRCA mutations and survival in breast cancer: An updated systematic review and meta-analysis. Oncotarget 2016, 7, 70113–70127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhoog, L.C.; Brekelmans, C.T.M.; Seynaeve, C.; Dahmen, G.; Van Geel, A.N.; Bartels, C.C.M.; Tilanus-Linthorst, M.M.A.; Wagner, A.; Devilee, P.; Halley, D.J.J.; et al. Survival in Hereditary Breast Cancer Associated with Germline Mutations of BRCA2. J. Clin. Oncol. 1999, 17, 3396–3402. [Google Scholar] [CrossRef] [PubMed]
- Downing, J.R.; Wilson, R.K.; Zhang, J.; Mardis, E.R.; Pui, C.H.; Ding, L.; Ley, T.J.; Evans, W.E. The pediatric cancer genome project. Nat. Genet. 2012, 44, 619–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bose, R.; Kavuri, S.M.; Searleman, A.C.; Shen, W.; Shen, D.; Koboldt, D.C.; Monsey, J.; Goel, N.; Aronson, A.B.; Li, S.; et al. Activating HER2 Mutations in HER2 Gene Amplification Negative Breast Cancer. Cancer Discov. 2013, 3, 224–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Costa E Silva Carvalho, S.; Cury, N.M.; Brotto, D.B.; De Araujo, L.F.; Rosa, R.C.A.; Texeira, L.A.; Plaça, J.R.; Marques, A.A.; Peronni, K.C.; Ruy, P.D.C.; et al. Germline variants in DNA repair genes associated with hereditary breast and ovarian cancer syndrome: Analysis of a 21 gene panel in the Brazilian population. BMC Med. Genom. 2020, 13, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couch, F.J.; Shimelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations Between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol. 2017, 3, 1190–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fazel, A.; Hasanpour-Heidari, S.; Salamat, F.; Rajaie, S.; Kazeminezhad, V.; Naeimi-Tabiei, M.; Jahangirrad, A.; Sedaghat, S.M.; Hosseinpoor, R.; Ghasemi-Kebria, F.; et al. Marked increase in breast cancer incidence in young women: A 10-year study from Northern Iran, 2004–2013. Cancer Epidemiol. 2019, 62, 101573. [Google Scholar] [CrossRef]
- Dimitrova, N.; Znaor, A.; Agius, D.; Eser, S.; Sekerija, M.; Ryzhov, A.; Primic-Žakelj, M.; Coebergh, J.W.; Agius, D.; Coza, D.; et al. Breast cancer in South-Eastern European countries since 2000: Rising incidence and decreasing mortality at young and middle ages. Eur. J. Cancer 2017, 83, 43–55. [Google Scholar] [CrossRef]
- Rocha-Brischiliari, S.C.; De Oliveira, R.R.; Andrade, L.; Brischiliari, A.; Gravena, A.A.F.; Carvalho, M.D.D.B.; Pelloso, S.M. The Rise in Mortality from Breast Cancer in Young Women: Trend Analysis in Brazil. PLoS ONE 2017, 12, e0168950. [Google Scholar] [CrossRef]
- Colonna, M.; Delafosse, P.; Uhry, Z.; Poncet, F.; Arveux, P.; Molinie, F.; Cherie-Challine, L.; Grosclaude, P. Is breast cancer incidence increasing among young women? An analysis of the trend in France for the period 1983–2002. Breast 2008, 17, 289–292. [Google Scholar] [CrossRef]
- Friebel, T.M.; Domchek, S.M.; Rebbeck, T.R. Modifiers of Cancer Risk in BRCA1 and BRCA2 Mutation Carriers: Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2014, 106, dju091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lek, M.; Karczewski, K.; Minikel, E.; Samocha, K.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.; Ware, J.; Hill, A.; Cummings, B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. bioRxiv 2015, 536, 030338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamath-Loeb, A.S.; Welcsh, P.; Waite, M.; Adman, E.T.; Loeb, L.A. The Enzymatic Activities of the Werner Syndrome Protein Are Disabled by the Amino Acid Polymorphism R834C. J. Biol. Chem. 2004, 279, 55499–55505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamath-Loeb, A.S.; Zavala-van Rankin, D.G.; Flores-Morales, J.; Emond, M.J.; Sidorova, J.M.; Carnevale, A.; Del Carmen Cárdenas-Cortés, M.; Norwood, T.H.; Monnat, R.J.; Loeb, L.A.; et al. Homozygosity for the WRN Helicase-Inactivating Variant, R834C, does not confer a Werner syndrome clinical phenotype. Sci. Rep. 2017, 7, srep44081. [Google Scholar] [CrossRef] [Green Version]
- Dorling, L.; Carvalho, S.; Allen, J.; González-Neira, A.; Luccarini, C.; Wahlström, C.; Pooley, K.A.; Parsons, M.T.; Fortuno, C. Breast Cancer Risk Genes—Association Analysis in More than 113,000 Women. N. Engl. J. Med. 2021, 384, 428–439. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Data | n | Frequency (%) | |
|---|---|---|---|
| Patients | n | 115 | |
| Age | Mean | 33.9 | |
| Interquartile range | 31–38 | ||
| Age at menarche | Mean | 12.34 | |
| Interquartile range | 11–13 | ||
| Parity | Nulliparous | 33 | 28.7 |
| 1 child | 27 | 23.2 | |
| 2 children | 32 | 23.5 | |
| >3 children | 23 | 20.0 | |
| Breastfeeding | No | 37 | 32.2 |
| Yes | 78 | 67.8 | |
| Luminal B (Her2-positive) | 19 | 13.2 | |
| Luminal B (Her2-negative) | 31 | 28.3 | |
| Her2-positive (non-luminal) | 8 | 6.6 | |
| Triple Negative | 30 | 28.3 | |
| Histology | Ductal | 98 | 85.2 |
| Lobular | 6 | 5.2 | |
| Mixed | 9 | 7.8 | |
| Others | 2 | 1.7 | |
| Clinical Stage | I | 15 | 13.0 |
| II | 45 | 39.1 | |
| III | 49 | 42.6 | |
| IV | 6 | 5.2 | |
| Consumption of Hormonal contraceptives | No | 50 | 43.5 |
| Yes | 65 | 56.55 | |
| First-grade family history of cancer | No | 90 | 78.3 |
| Yes | 25 | 21.7 | |
| Second-grade family history of cancer | No | 78 | 67.8 |
| Yes | 37 | 32.2 | |
| Body Mass Index | Normal (BMI < 25) | 44 | 38.3 |
| Overweight | 45 | 39.1 | |
| Obesity (BMI > 30) | 26 | 22.6 |
| Chr | Position | Ref * | Alt * | Depths | Gene | LA + | Type | AA Chg | Pat | pLI | AF Lat § | ProtSize |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 11 | 108175579 | G | A | 21,19 | ATM | Splice | E1892_E37 | H4 | 0 | 1 × 10−4 | 3056 | |
| 15 | 91341429 | A | AG | 15,25 | BLM | Frameshift | T1074fs | D8 | 0 | - | 1417 | |
| 17 | 41258472 | C | A | 54,35 | BRCA1 | Splice | R71_E4 | G8 | 0 | 3 × 10−5 | 1863 | |
| 13 | 32914033 | CA | C | 18,20 | BRCA2 | Frameshift | S1848fs | E0 | 0 | 3 × 10−5 | 3418 | |
| 13 | 32914122 | AC | A | 52,30 | BRCA2 | Frameshift | N1877fs | J2 | 0 | 3 × 10−5 | 3418 | |
| 13 | 32930653 | C | CA | 15,17 | BRCA2 | Frameshift | S2509fs | B2 | 0 | - | 3418 | |
| 13 | 32937507 | A | G | 44,30 | BRCA2 | Missense | D2723G | G2 | 0 | A:C 0 | 3418 | |
| 13 | 32954260 | CG | C | 23,17 | BRCA2 | Frameshift | V3079fs | K5 | 0 | 2 × 10−4 | 3418 | |
| 22 | 29091718 | TA | T | 29,23 | CHEK2 | Frameshift | L413fs | J6 | 0 | - | 543 | |
| 22 | 29115401 | A | ATGAT | 28,16 | CHEK2 | Frameshift | M222fs | F5 | 0 | 1 × 10−4 | 543 | |
| 22 | 19222211 | C | T | 33,36 | CLTCL1 | No | Missense | E330K | G5 | 0 | 9 × 10−4 | 1640 |
| 2 | 189868190 | T | A | 35,26 | COL3A1 | No | Splice | P869P | J6 | 1 | 9 × 10−5 | 1466 |
| 7 | 101926377 | C | T | 57,41 | CUX1 | No | Nonsense | Q678 * | G2 | 1 | 3 × 10−3 | 678 |
| 16 | 50818362 | T | A | 30,39 | CYLD | No | Splice | I650_E13 | C1 | 1 | 3 × 10−5 | 956 |
| 18 | 50734187 | G | A | 28,23 | DCC | No | Splice | V621_E11 | H2 | 0.99 | 2 × 10−4 | 1447 |
| X | 41206109 | T | C | 46,39 | DDX3X | No | Splice | G539_E15 | K3 | 1 | 4 × 10−5 | 662 |
| X | 41206562 | T | C | 44,41 | DDX3X | No | Splice | S590_E16 | F3 | 1 | 2 × 10−3 | 662 |
| 19 | 45917294 | T | C | 21,22 | ERCC1 | Splice | V235_E7 | H0 | 0 | 6 × 10−5 | 297 | |
| 10 | 50680422 | C | T | 16,25 | ERCC6 | No | Splice | R975_E16 | G8 | 0 | C:A 3 × 10−5 | 1493 |
| 11 | 44228353 | G | A | 33,34 | EXT2 | No | Nonsense | W535 * | A2 | 0 | 1 × 10−4 | 718 |
| 6 | 35425330 | C | T | 25,34 | FANCE | No | Splice | D286_E3 | A0 | 0 | 3 × 10−4 | 536 |
| 4 | 187530955 | C | G | 34,21 | FAT1 | No | Splice | T3356T | B0 | 0 | 0 | 4588 |
| 17 | 17117000 | CG | C | 45,40 | FLCN | No | Frameshift | R570fs | C0 | 0.79 | C:T 3 × 10−5 | 579 |
| 1 | 152285000 | G | A | 46,43 | FLG | No | Nonsense | R788 * | K8 | 0 | 6 × 10−4 | 4061 |
| 1 | 152285861 | G | A | 45,34 | FLG | No | Nonsense | R501 * | K1 | 0 | 4 × 10−3 | 4061 |
| 17 | 42148542 | TC | T | 11,14 | G6PC3 | No | Frameshift | I70fs | H0 | 0 | 8 × 10−4 | 346 |
| 14 | 62203827 | G | A | 20,20 | HIF1A | No | Splice | D417_E9 | A2 | 0 | 2 × 10−4 | 826 |
| 2 | 48033791 | GT(26) | G | 15,12 | MSH6 | Splice | R1334_E9 | A0 | 0 | 1 × 10−4 & | 1360 | |
| 1 | 45797228 | C | T | 25,23 | MUTYH | Splice | G396_E13 | E7 | 0 | 3 × 10−3 | 546 | |
| 6 | 44233331 | G | GC | 21,12 | NFKBIE | No | Frameshift | A57fs | I9 | 0.77 | 3 × 10−4 | 500 |
| 5 | 176722446 | TC(6) | T | 27,25 | NSD1 | No | Frameshift | S2424fs | G7 | 1 | - | 2696 |
| 16 | 23641062 | CAG | C | 25,39 | PALB2 | Frameshift | S804fs | D6 | 0 | 1 × 10−4 | 1186 | |
| 16 | 23641139 | G | C | 24,36 | PALB2 | Nonsense | S779 * | K6 | 0 | 9 × 10−5 | 1186 | |
| 3 | 52620706 | TG | T | 15,12 | PBRM1 | No | Splice | E1017_E21 | C9 | 1 | 0 & | 1689 |
| 2 | 190728500 | C | T | 26,29 | PMS1 | No | Nonsense | R630 * | C9 | 0 | 1 × 10−4 | 932 |
| 3 | 121168273 | T | C | 19,22 | POLQ | Splice | I2385V | B2 | 0 | - | 2590 | |
| 3 | 121207489 | A | T | 20,15 | POLQ | Nonsense | L1430 * | E6 | 0 | 9 × 10−5 | 2590 | |
| 5 | 23527845 | CA | C | 31,42 | PRDM9 | No | Frameshift | T883fs | B6 | 0 | 6 × 10−5 | 894 |
| 1 | 45294985 | C | T | 14,13 | PTCH2 | No | Splice | L406_E10 | B1 | 0 | 3 × 10−5 | 1203 |
| 17 | 56774167 | C | CT | 47,59 | RAD51C | Frameshift | A173fs | A5 | 0 | - | 376 | |
| 1 | 145507646 | G | A | 15,27 | RBM8A | No | UTR_5 | E1_UTR_5 | G1 | 0.57 | 1 × 10−2 | 174 |
| 1 | 145507646 | G | A | 18,24 | RBM8A | No | UTR_5 | E1_UTR_5 | G5 | 0.57 | 1 × 10−2 | 174 |
| 2 | 3623181 | G | A | 51,58 | RPS7 | No | Splice | E4 | 0.95 | - | 194 | |
| 1 | 16262459 | G | GC(27) | 27,17 | SPEN | No | Frameshift | A3242fs | I0 | 1 | 9 × 10−4 & | 3664 |
| 17 | 7578406 | C | T | 22,29 | TP53 | Missense | R175H | I9 | 0.53 | 0 | 393 | |
| 16 | 2124201 | C | T | 44,31 | TSC2 | No | Splice | R786C | B0 | 1 | 0 | 1807 |
| 17 | 5074956 | T | A | 83,62 | USP6 | No | Nonsense | Y1343 * | H3 | 0 | 9 × 10−5 | 1406 |
| 8 | 31014882 | A | G | 13,13 | WRN | Splice | K1274_E33 | J8 | 0 | 1 × 10−4 | 1432 | |
| 16 | 72991713 | C | CC(9) | 20,14 | ZFHX3 | No | Frameshift | A778fs | H6 | 1 | 0 | 3703 |
| X | 70466308 | GTGGT | G | 28,11 | ZMYM3 | No | Frameshift | P821fs | L2 | 1 | - | 1370 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Flores-Ramos, L.; Barraza-Arellano, A.L.; Mohar, A.; Trujillo-Martínez, M.; Grimaldo, L.; Ortiz-Lopez, R.; Treviño, V. Germline Variants in Cancer Genes from Young Breast Cancer Mexican Patients. Cancers 2022, 14, 1647. https://doi.org/10.3390/cancers14071647
Gómez-Flores-Ramos L, Barraza-Arellano AL, Mohar A, Trujillo-Martínez M, Grimaldo L, Ortiz-Lopez R, Treviño V. Germline Variants in Cancer Genes from Young Breast Cancer Mexican Patients. Cancers. 2022; 14(7):1647. https://doi.org/10.3390/cancers14071647
Chicago/Turabian StyleGómez-Flores-Ramos, Liliana, Angélica Leticia Barraza-Arellano, Alejandro Mohar, Miguel Trujillo-Martínez, Lizbeth Grimaldo, Rocío Ortiz-Lopez, and Víctor Treviño. 2022. "Germline Variants in Cancer Genes from Young Breast Cancer Mexican Patients" Cancers 14, no. 7: 1647. https://doi.org/10.3390/cancers14071647
APA StyleGómez-Flores-Ramos, L., Barraza-Arellano, A. L., Mohar, A., Trujillo-Martínez, M., Grimaldo, L., Ortiz-Lopez, R., & Treviño, V. (2022). Germline Variants in Cancer Genes from Young Breast Cancer Mexican Patients. Cancers, 14(7), 1647. https://doi.org/10.3390/cancers14071647