Anaplastic Large Cell Lymphoma: Molecular Pathogenesis and Treatment


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. ALK-Positive ALCL
2.1. Definition
2.2. History
2.3. Risk Factors
2.4. Clinical Features
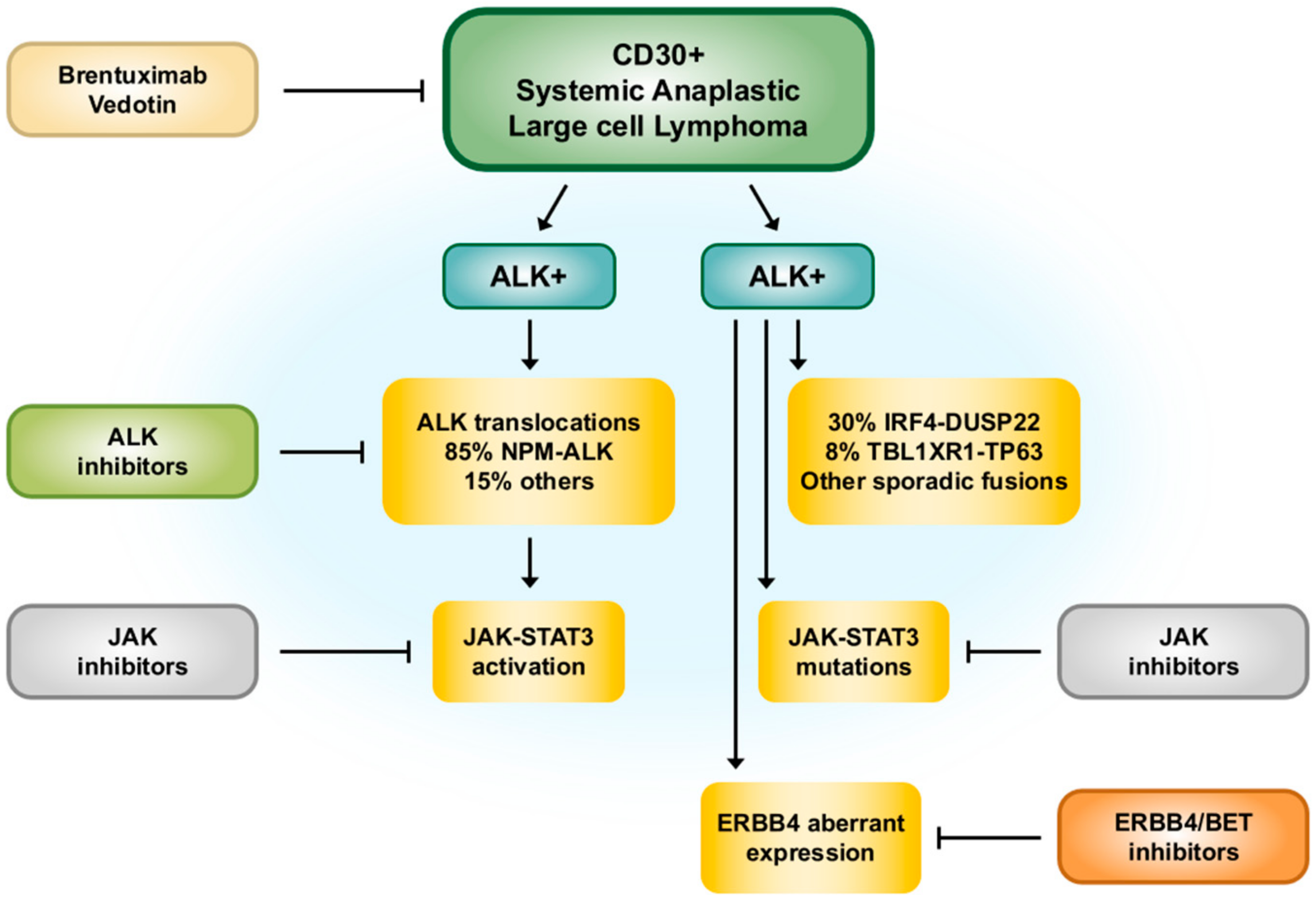
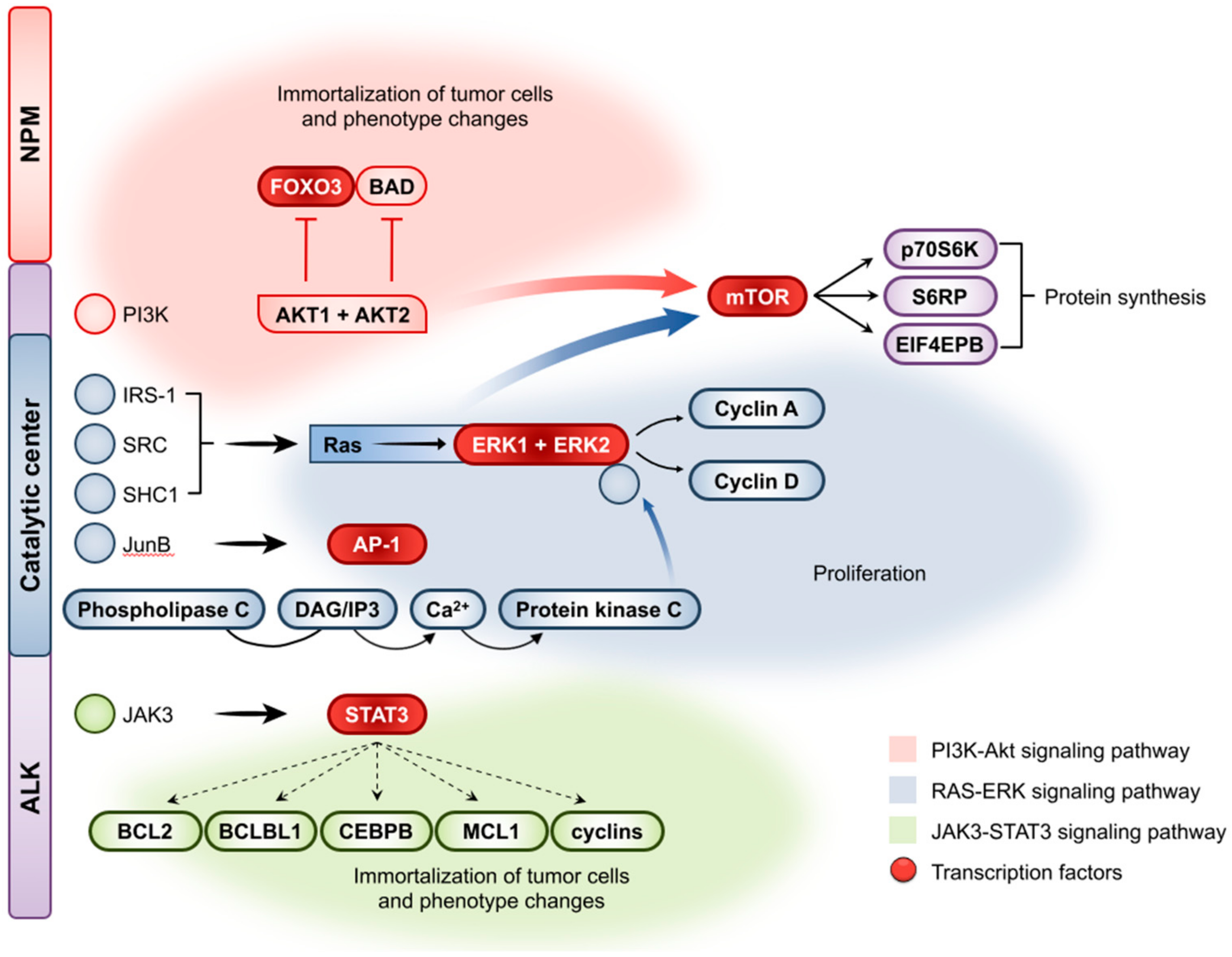
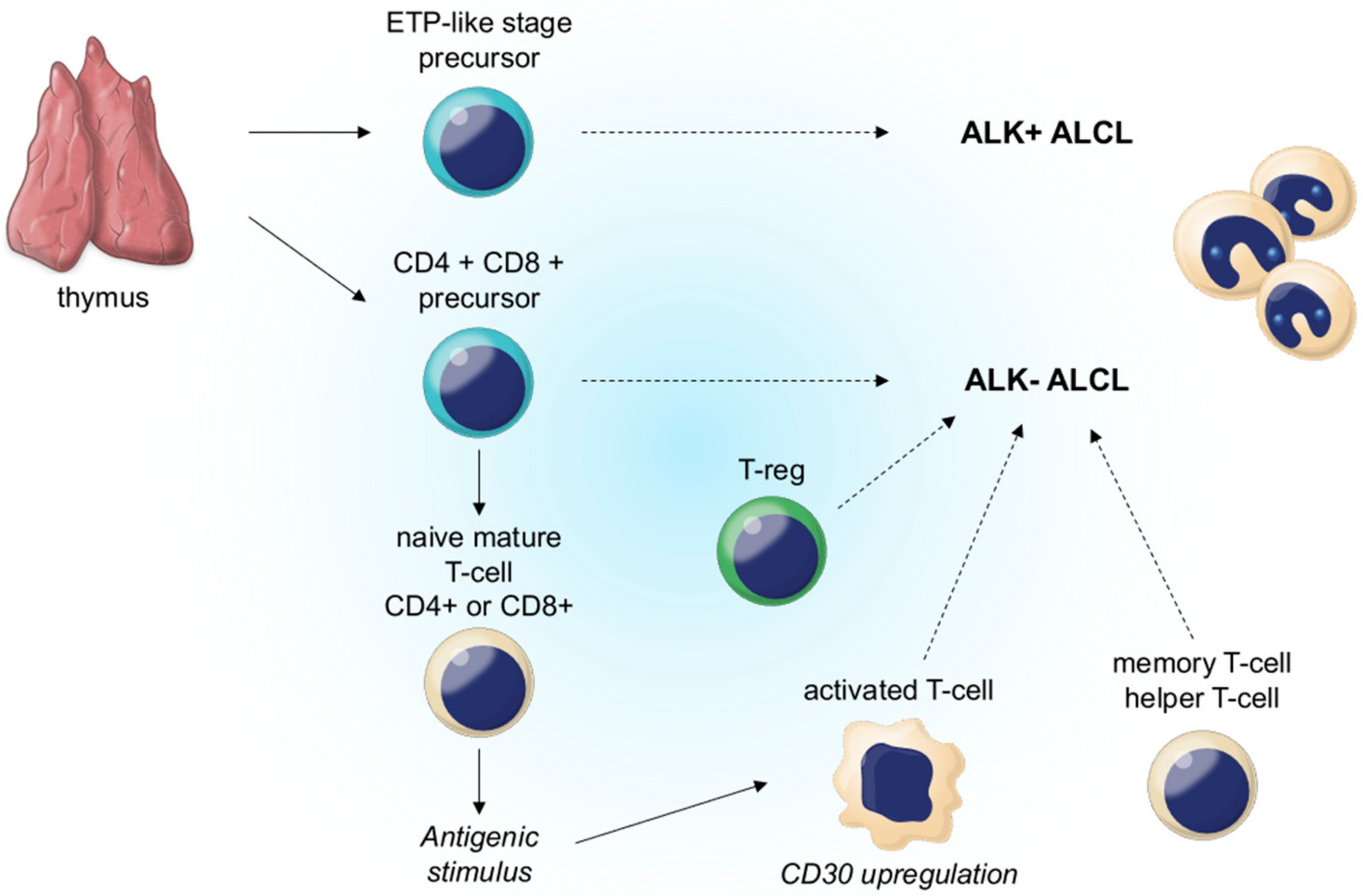
2.5. Molecular Pathogenesis
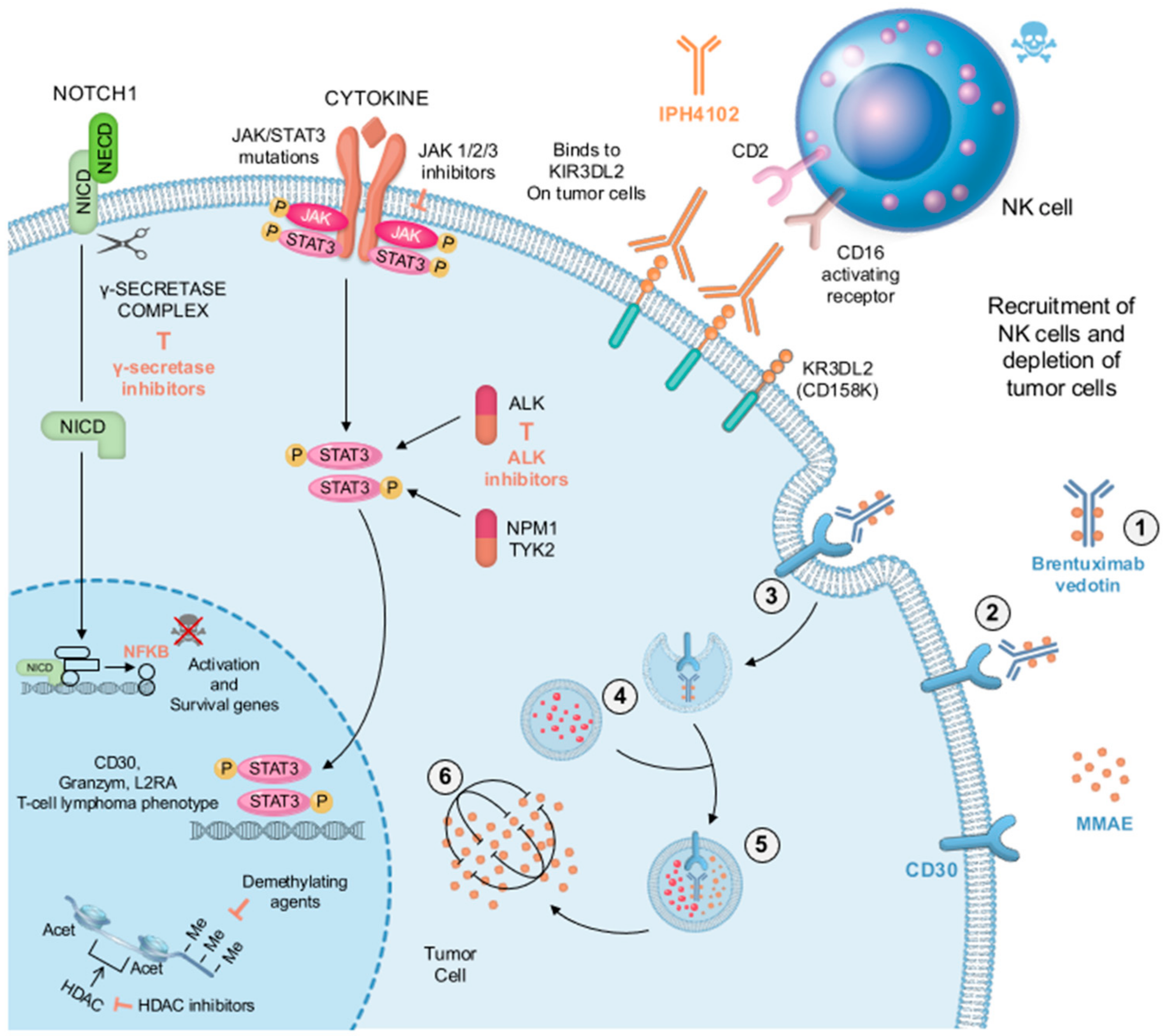
2.6. Management
3. ALK-Negative ALCL
3.1. Definition
3.2. History
3.3. Risk Factors
3.4. Clinical Features
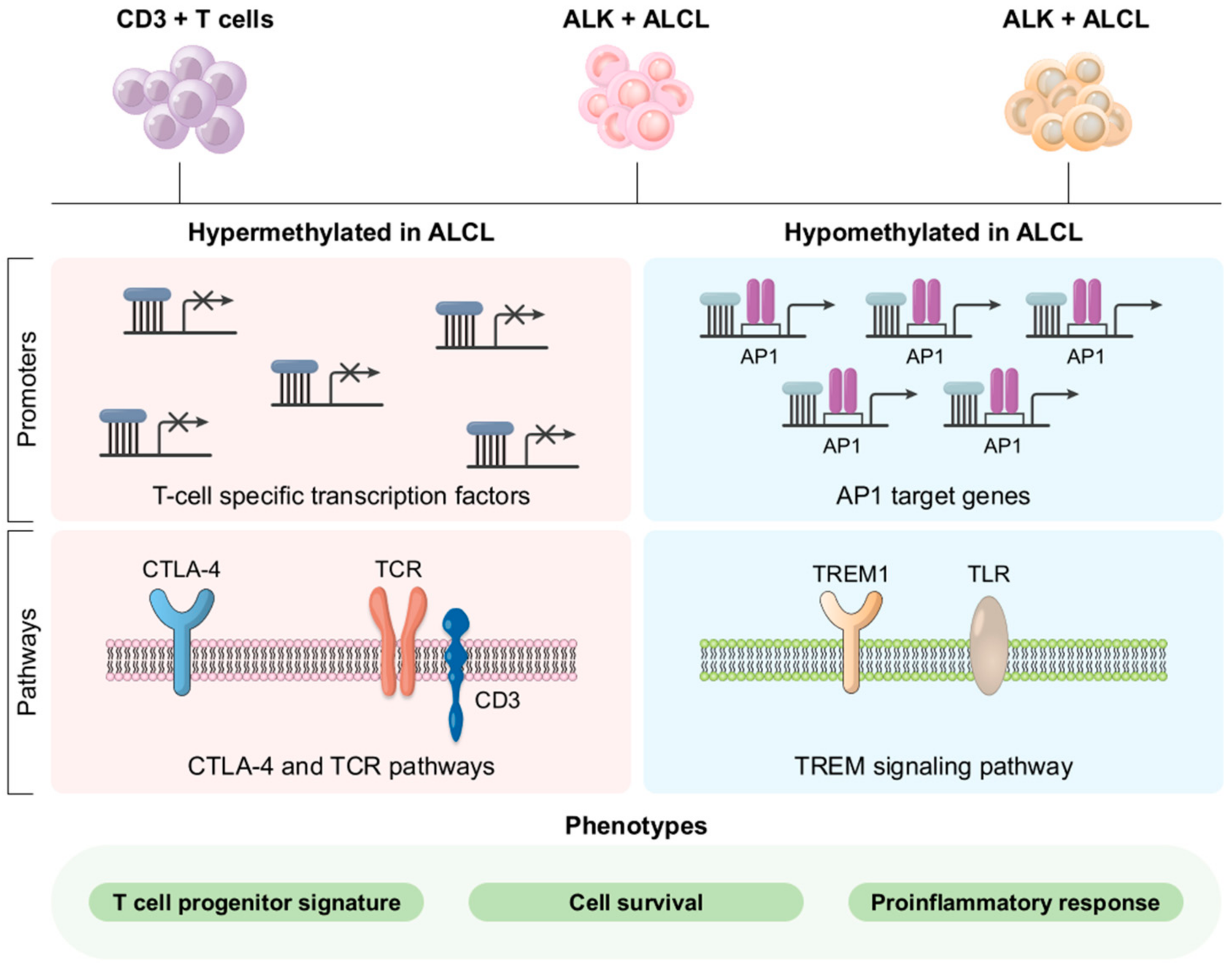
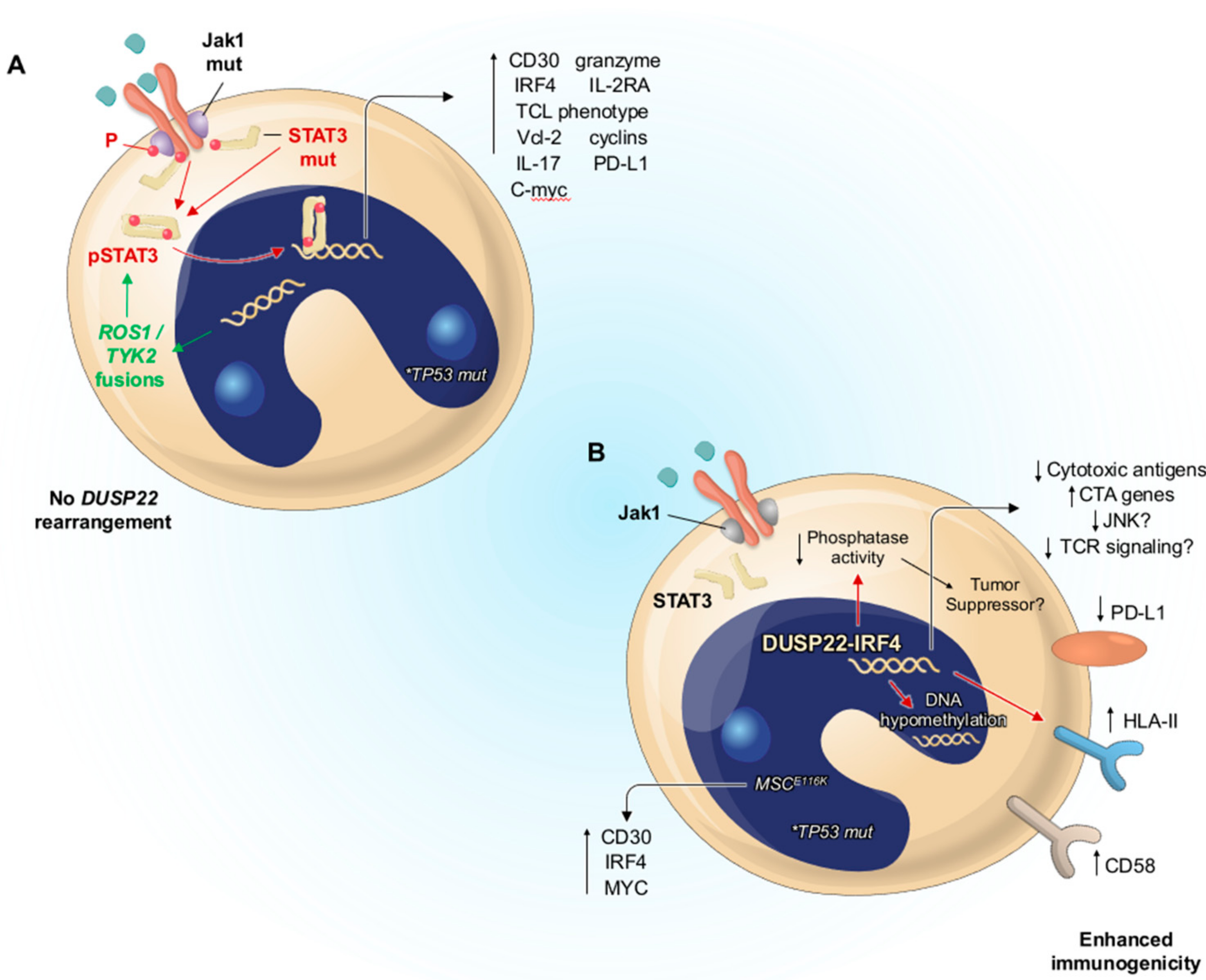
3.5. Molecular Pathogenesis
3.6. Management
4. Primary Cutaneous ALCL
4.1. Definition
4.2. History
4.3. Risk Factors
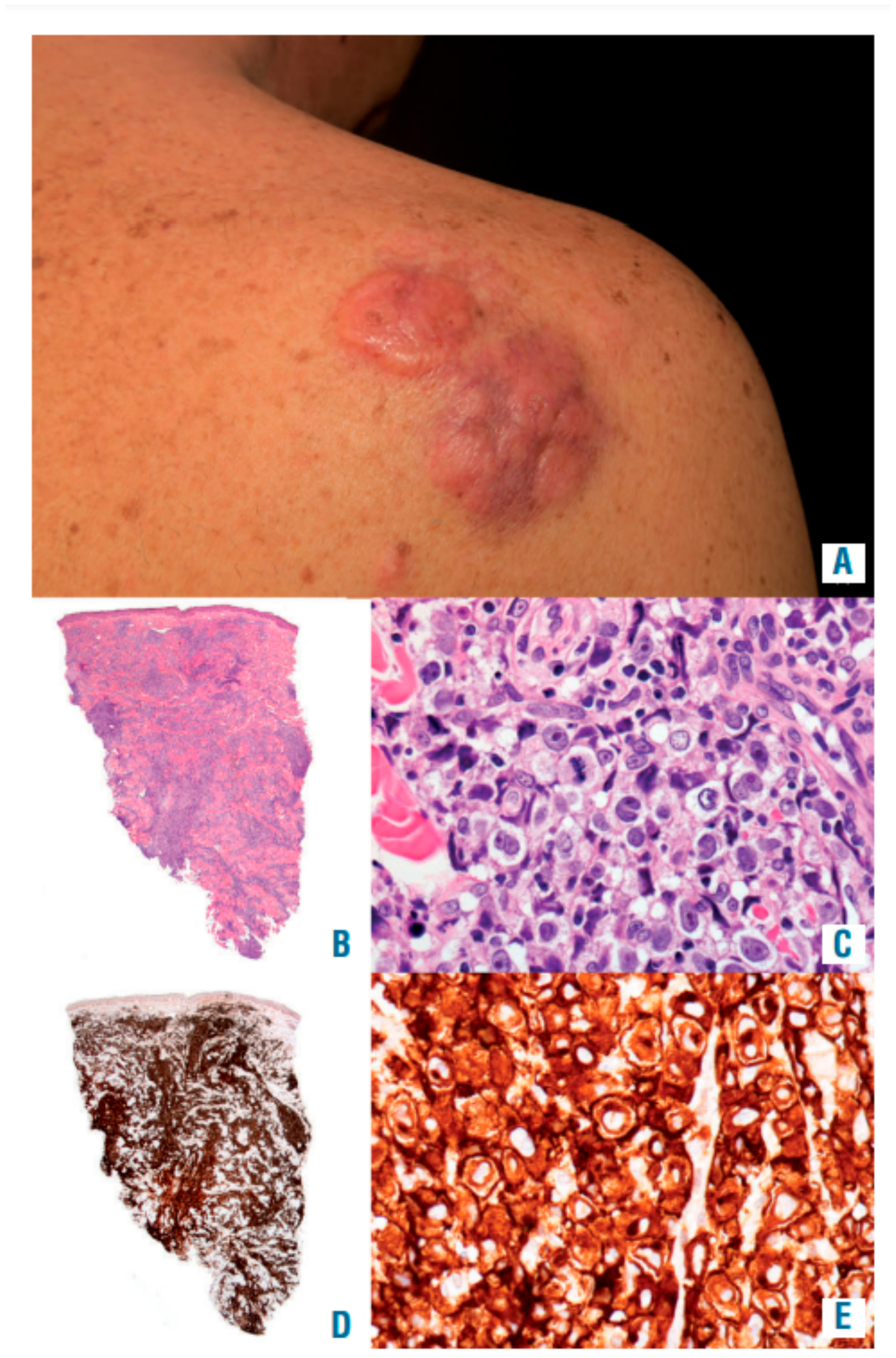
4.4. Clinical Features
4.5. Molecular Pathogenesis
4.6. Management
5. Breast-Implant-Associated ALCL
5.1. Definition
5.2. History
5.3. Risk Factors
5.4. Clinical Features
5.5. Molecular Pathogenesis
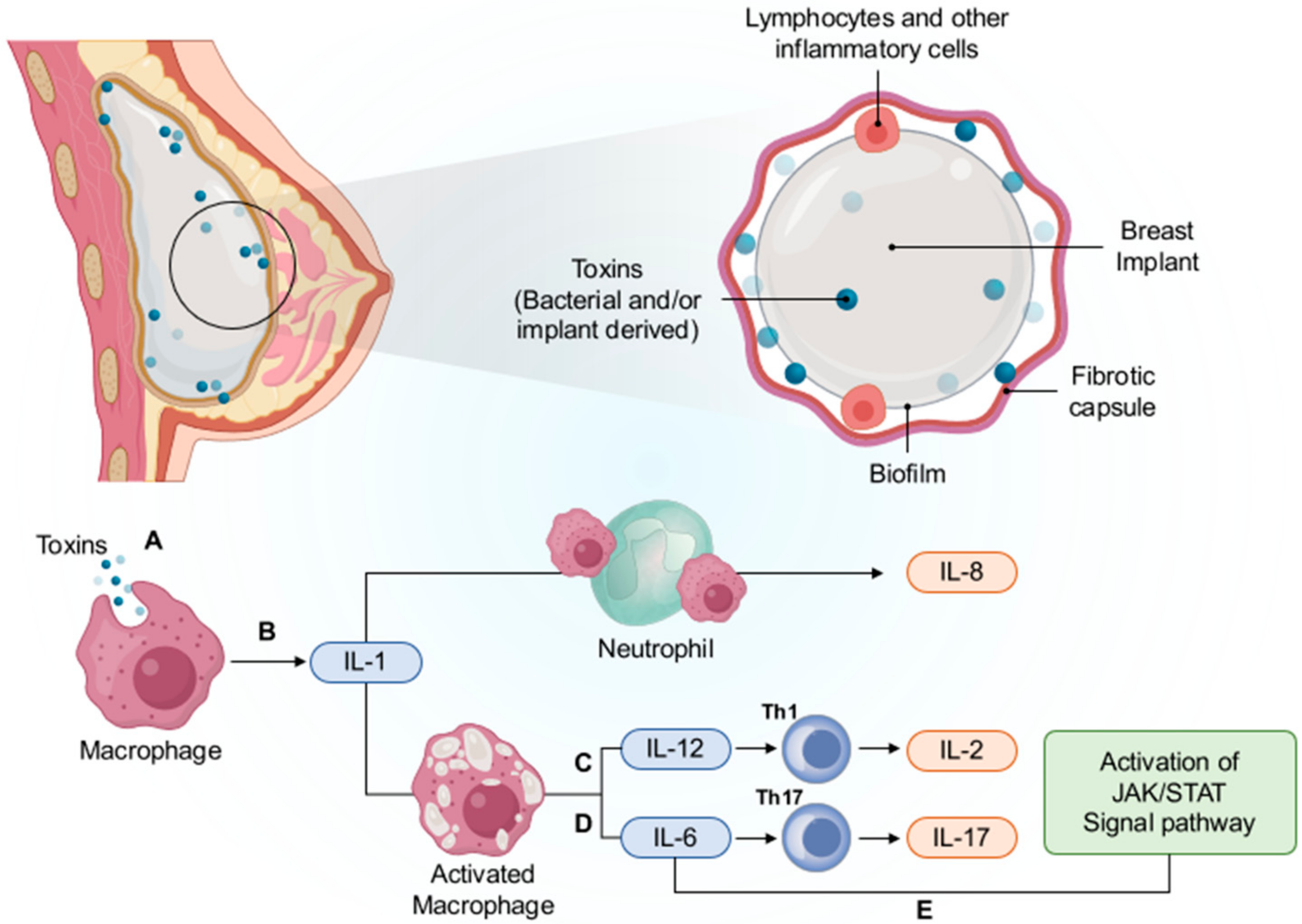
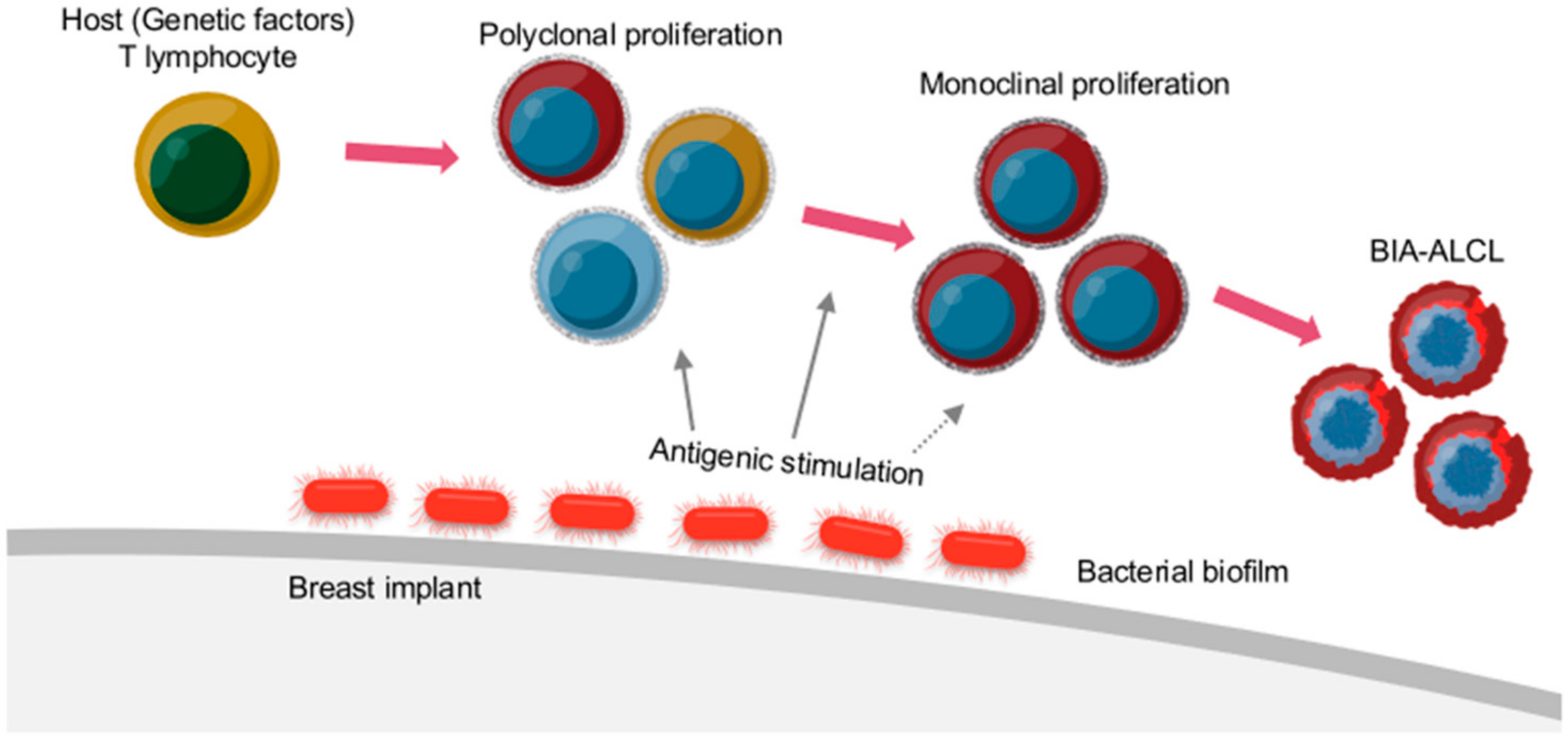
5.5.1. Chronic Inflammation and Immune Response
5.5.2. Subclinical Bacterial Infection
5.5.3. Genetic Alterations
5.6. Management
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Arber, D.A.; Hasserjian, R.P.; Le Beau, M.M.; et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; International Agency for Research on Cancer (IARC): Lyon, France, 2017; Volume 2, ISBN 9789283244943. [Google Scholar]
- Laurent, C.; Baron, M.; Amara, N.; Haioun, C.; Dandoit, M.; Maynadié, M.; Parrens, M.; Vergier, B.; Copie-Bergman, C.; Fabiani, B.; et al. Impact of Expert Pathologic Review of Lymphoma Diagnosis: Study of Patients from the French Lymphopath Network. J. Clin. Oncol. 2017, 35, 2008–2017. [Google Scholar] [PubMed]
- Armitage, J.; Vose, J.; Weisenburger, D. International peripheral T-cell and naturalkiller/T-cell lymphoma study: Pathology findings and clinical outcomes. J. Clin. Oncol. 2008, 26, 4124–4130. [Google Scholar]
- Philippe, E.; Creech, K.T.; Cook, N.; Segura, J. Recurrent Primary Cutaneous Anaplastic Large Cell Lymphoma with Systemic Involvement: A Case Report and Literature Review. Cureus 2021, 13, e14284. [Google Scholar] [CrossRef]
- Morton, L.M.; Wang, S.S.; Devesa, S.S.; Hartge, P.; Weisenburger, D.D.; Linet, M.S. Lymphomaincidence patterns by WHO subtype in the United States, 1992–2001. Blood 2006, 107, 265–276. [Google Scholar] [PubMed]
- Adams, S.V.; Newcomb, P.A.; Shustov, A.R. Racial patterns of peripheral T-cell lymphomaincidence and survival in the United States. J. Clin. Oncol. 2016, 34, 963–971. [Google Scholar]
- Stein, H.; Mason, D.Y.; Gerdes, J.; O’Connor, N.; Wainscoat, J.; Pallesen, G.; Gatter, K.; Falini, B.; Delsol, G.; Lemke, H.; et al. The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplasticlymphoid tissue: Evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activatedlymphoid cells. Blood 1985, 66, 848–858. [Google Scholar]
- Stansfeld, A.G.; Diebold, J.; Kapanci, Y.; Kelényi, G.; Lennert, K.; Mioduszewska, O.; Noel, H.; Rilke, F.; Sundstrom, C.; Van Unnik, J.; et al. Updated Kiel Classification for Lymphomas. Lancet 1988, 1, 292–293. [Google Scholar] [CrossRef]
- Harris, N.L.; Jaffe, E.S.; Stein, H.; Banks, P.M.; Chan, J.K.; Cleary, M.L.; Delsol, G.; De Wolf-Peeters, C.; Falini, B.; Gatter, K.C.; et al. A revised European-American classification of lymphoid neoplasms: A proposalfrom the International Lymphoma Study Group. Blood 1994, 84, 1361–1392. [Google Scholar]
- Jaffe, E.S.; Harris, N.L.; Stein, H.; Vardiman, J.W. (Eds.) World Health Organization Classification of Tumours, Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues; IARC Press: Lyon, France, 2001. [Google Scholar]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef]
- Ducray, S.P.; Natarajan, K.; Garland, G.D.; Turner, S.D.; Egger, G. The Transcriptional Roles of ALK Fusion Proteins in Tu-morigenesis. Cancers 2019, 11, 1074. [Google Scholar]
- Martorana, F.; Motta, G.; Pavone, G.; Motta, L.; Stella, S.; Vitale, S.R.; Manzella, L.; Vigneri, P. AKT Inhibitors: New Weapons in the Fight Against Breast Cancer? Front. Pharmacol. 2021, 12, 662232. [Google Scholar] [CrossRef]
- Catalina, A.; Andrew, L.F. How I Diagnose Anaplastic Large Cell Lymphoma. AJCP 2021, 155, 479–497. [Google Scholar]
- Mereu, E.; Pellegrino, E.; Scarfò, I.; Inghirami, G.; Piva, R. The heterogeneous landscape of ALK negative ALCL. Oncotarget 2017, 8, 18525–18536. [Google Scholar] [CrossRef] [PubMed]
- Schwab, U.; Stein, H.; Gerdes, J.; Lemke, H.; Kirchner, H.; Schaadt, M.; Diehl, V. Production of a monoclonal antibody specific for Hodgkin and Sternberg–Reed cells of Hodgkin’s disease and a subset of normal lymphoid cells. Nature 1982, 299, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Lennert, K.; Feller, A. Histopathology of Non-Hodgkin Lymphoma; Springer: New York, NY, USA, 1992. [Google Scholar]
- Delsol, G.; Falini, B.; Müller-Hermelink, H.K.; Campo, E.; Jaffe, E.S.; Gascoyne, R.D.; Jaffe, E.S.; Stein, H.; Campo, E. Anaplastic Large Cell Lymphoma, ALK-Positive. In World Health Organization (WHO) Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Vardiman, J.W., Eds.; IARC Press: Lyon, France, 2008; pp. 312–316. [Google Scholar]
- Gaulard, P.; Jaffe, E.; Krenacs, L.; Macon, W.R. Hepatosplenic T-Cell Lymphoma. In WHO Classification of Tumours of Hematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC: Lyon, France, 2008; pp. 292–293. [Google Scholar]
- Arber, D.A.; Chang, K.L.; Weiss, L.M. Peripheral T-cell lymphoma with Toutonlike tumor giant cells associated with HIV infection: Report of two cases. Am. J. Surg. Pathol. 1999, 23, 519–522. [Google Scholar]
- Burke, A.P.; Andriko, J.-A.; Virmani, R. Anaplastic Large Cell Lymphoma (CD 30+), T-Phenotype, in the Heart of an HIV-Positive Man. Cardiovasc. Pathol. 2000, 9, 49–52. [Google Scholar] [CrossRef]
- Burkhardt, B.; Zimmermann, M.; Oschlies, I.; Niggli, F.; Mann, G.; Parwaresch, R.; Riehm, H.; Schrappe, M.; Reiter, A.; The BFM Group. The impact of age and gender on biology, clinical features and treatment outcome of non-Hodgkin lymphoma in childhood and adolescence. Br. J. Haematol. 2005, 131, 39–49. [Google Scholar]
- Falini, B.; Pileri, S.; Zinzani, P.L.; Carbone, A.; Zagonel, V.; Wolf-Peeters, C.; Verhoef, G.; Menestrina, F.; Todeschini, G.; Paulli, M.; et al. ALK+ lymphoma: Clinico-pathological findings and outcome. Blood 1999, 93, 2697–2706. [Google Scholar]
- Stein, H.; Foss, H.D.; Durkop, H.; Marafioti, T.; Delsol, G.; Pulford, K.; Pileri, S.; Falini, B. CD30(+) anaplastic large cell lym-phoma: A review of its histopathologic, genetic, and clinical features. Blood 2000, 96, 3681–3695. [Google Scholar]
- Filippa, D.A.; Ladanyi, M.; Wollner, N.; Straus, D.J.; O’Brien, J.P.; Portlock, C.; Gangi, M.; Sun, M. CD30 (Ki-1)-positive ma-lignant lymphomas: Clinical, immunophenotypic, histologic, and genetic characteristics and differences with Hodgkin’s dis-ease. Blood 1996, 87, 2905–2917. [Google Scholar]
- Kadin, M.E.; Carpenter, C. Systemic and primary cutaneous anaplastic large cell lymphomas. Semin. Hematol. 2003, 40, 244–256. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a Kinase Gene, ALK, to a Nucleolar Protein Gene, NPM, in Non-Hodgkin’s Lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Lamant, L.; Gascoyne, R.D.; Duplantier, M.M.; Armstrong, F.; Raghab, A.; Chhanabhai, M.; Rajcan-Separovic, E.; Raghab, J.; Delsol, G.; Espinos, E. Non-muscle myosin heavy chain (MYH9): A new partner fused to ALK in anaplastic large cell lym-phoma. Genes Chromosom. Cancer 2003, 37, 427–432. [Google Scholar] [PubMed]
- Touriol, C.; Greenland, C.; Lamant, L.; Pulford, K.; Bernard, F.; Rousset, T.; Mason, D.Y.; Delsol, G. Further demonstration of the diversity of chromosomal changes involving 2p23 in ALK-positive lymphoma: 2 cases expressing ALK kinase fused to CLTCL (clathrin chain polypeptide-like). Blood 2000, 95, 3204–3207. [Google Scholar]
- Tort, F.; Pinyol, M.; Pulford, K.; Roncador, G.; Hernandez, L.; Nayach, I.; Kluin-Nelemans, H.C.; Kluin, P.; Touriol, C.; Delsol, G.; et al. Molecular characterization of a new ALK translocation involving moesin (MSN-ALK) in anaplastic large cell lym-phoma. Lab. Investig. 2001, 81, 419–426. [Google Scholar]
- Falini, B.; Pulford, K.; Pucciarini, A.; Carbone, A.; De Wolf-Peeters, C.; Cordell, J.; Fizzotti, M.; Santucci, A.; Pelicci, P.G.; Pileri, S.; et al. Lymphomas expressing ALK fusion protein(s) other than NPM-ALK. Blood 1999, 94, 3509–3515. [Google Scholar]
- Cools, J.; Wlodarska, I.; Somers, R.; Mentens, N.; Pedeutour, F.; Maes, B.; De Wolf-Peeters, C.; Pauwels, P.; Hagemeijer, A.; Marynen, P. Identification of novel fusion partners of ALK, the anaplastic lymphoma kinase, in anaplastic large-cell lymphoma and inflammatory myofibroblastic tumor. Genes Chromosom. Cancer 2002, 34, 354–362. [Google Scholar] [CrossRef]
- Feldman, A.L.; Vasmatzis, G.; Asmann, Y.W.; Davila, J.; Middha, S.; Eckloff, B.W.; Johnson, S.H.; Porcher, J.C.; Ansell, S.M.; Caride, A. Novel TRAF1-ALK fusion identified by deep RNA sequencing of anaplastic large cell lymphoma. Genes Chromosom. Cancer 2013, 52, 1097–1102. [Google Scholar]
- Hernández, L.; Pinyol, M.; Hernández, S.; Beà, S.; Pulford, K.; Rosenwald, A.; Lamant, L.; Falini, B.; Ott, G.; Mason, D.Y.; et al. TRK-Fused Gene (TFG) Is a New Partner of ALK in Anaplastic Large Cell Lymphoma Producing Two Structurally DifferentTFG-ALK Translocations. Blood 1999, 94, 3265–3268. [Google Scholar] [CrossRef]
- Lamant, L.; Dastugue, N.; Pulford, K.; Delsol, G.; Mariame, B. A new fusion gene TPM3-ALK in anaplastic large cell lymphoma created by a (1;2)(q25;p23) translocation. Blood 1999, 93, 3088–3095. [Google Scholar]
- Mason, D.Y.; Pulford, K.A.; Bischof, D.; Kuefer, M.U.; Butler, L.H.; Lamant, L.; Delsol, G.; Morris, S.W. Nucleolar localization of the nucleophosmin-anaplastic lymphoma kinase is not required for malignant transformation. Cancer Res. 1998, 58, 1057–1062. [Google Scholar] [PubMed]
- Rosenwald, A.; Ott, G.; Pulford, K.; Katzenberger, T.; Kühl, J.; Kalla, J.; Ott, M.M.; Mason, D.Y.; Müller-Hermelink, H.K. t(1;2)(q21;p23) and t(2;3)(p23;q21): Two Novel Variant Translocations of the t(2;5)(p23;q35) in Anaplastic Large Cell Lymphoma. Blood 1999, 94, 362–364. [Google Scholar] [CrossRef] [PubMed]
- Trinei, M.; Lanfrancone, L.; Campo, E.; Pulford, K.; Mason, D.Y.; Pelicci, P.G.; Falini, B. A new variant anaplastic lymphoma kinase (ALK)-fusion protein (ATIC-ALK) in a case of ALK-positive anaplastic large cell lymphoma. Cancer Res. 2000, 60, 793–798. [Google Scholar]
- Wlodarska, I.; De Wolf-Peeters, C.; Falini, B.; Verhoef, G.; Morris, S.W.; Hagemeijer, A.; Van den Berghe, H. The cryptic inv(2)(p23q35) defines a new molecular genetic subtype of ALK-positive anaplastic large-cell lymphoma. Blood 1998, 92, 2688–2695. [Google Scholar]
- Abate, F.; The European T-Cell Lymphoma Study Group; Todaro, M.; Van Der Krogt, J.-A.; Boi, M.; Landra, I.; Machiorlatti, R.; Tabbo, F.; Messana, K.; Abele, C.; et al. A novel patient-derived tumorgraft model with TRAF1-ALK anaplastic large-cell lymphoma translocation. Leukemia 2015, 29, 1390–1401. [Google Scholar] [CrossRef]
- Pulford, K.; Lamant, L.; Espinos, E.; Jiang, Q.; Xue, L.; Turturro, F.; Delsol, G.; Morris, S.W. The emerging normal and dis-ease-related roles of anaplastic lymphoma kinase. Cell. Mol. Life Sci. 2004, 61, 2939–2953. [Google Scholar] [PubMed]
- Pulford, K.; Morris, S.; Turturro, F. Anaplastic lymphoma kinase proteins in growth control and cancer. J. Cell. Physiol. 2004, 199, 330–358. [Google Scholar] [CrossRef]
- Duyster, J.; Bai, R.-Y.; Morris, S.W. Translocations involving anaplastic lymphoma kinase (ALK). Oncogene 2001, 20, 5623–5637. [Google Scholar] [CrossRef]
- Fujimoto, J.; Shiota, M.; Iwahara, T.; Seki, N.; Satoh, H.; Mori, S.; Yamamoto, T. Characterization of the transforming activity of p80, a hyperphosphorylated protein in a Ki-1 lymphoma cell line with chromosomal translocation t(2;5). Proc. Natl. Acad. Sci. USA 1996, 93, 4181–4186. [Google Scholar] [CrossRef]
- Bischof, D.; Pulford, K.; Mason, D.Y.; Morris, S.W. Role of the nucleophosmin (NPM) portion of the non-Hodgkin’s lympho-ma-associated NPM-anaplastic lymphoma kinase fusion protein in oncogenesis. Mol. Cell. Biol. 1997, 17, 2312–2325. [Google Scholar]
- Zamo, A.; Chiarle, R.; Piva, R.; Howes, J.; Fan, Y.; Chilosi, M.; Levy, D.E.; Inghirami, G. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene 2002, 21, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Anastasov, N.; Bonzheim, I.; Rudelius, M.; Klier, M.; Dau, T.; Angermeier, D.; Duyster, J.; Pittaluga, S.; Fend, F.; Raffeld, M.; et al. C/EBPβ expression in ALK-positive anaplastic large cell lymphomas is required for cell proliferation and is induced by the STAT3 signaling pathway. Haematologica 2010, 95, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Slupianek, A.; Nieborowska-Skorska, M.; Hoser, G.; Morrione, A.; Majewski, M.; Xue, L.; Morris, S.W.; Wasik, M.A.; Skorski, T. Role of phosphatidylinositol 3-kinase-Akt pathway in nucleophosmin/anaplastic lymphoma kinase-mediated lymphomagen-esis. Cancer Res. 2001, 61, 2194–2199. [Google Scholar] [PubMed]
- Bai, R.-Y.; Dieter, P.; Peschel, C.; Morris, S.W.; Duyster, J. Nucleophosmin-Anaplastic Lymphoma Kinase of Large-Cell Anaplastic Lymphoma Is a Constitutively Active Tyrosine Kinase That Utilizes Phospholipase C-γ To Mediate Its Mitogenicity. Mol. Cell. Biol. 1998, 18, 6951–6961. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.T.; Zhao, C.; Zhang, Q.; Wasik, M.A. Nucleophosmin-anaplastic lymphoma kinase: The ultimate oncogene and therapeutic target. Blood 2017, 129, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Inghirami, G.; Chiarle, R.; Simmons, W.J.; Piva, R.; Schlessinger, K.; Levy, D.E. New and old functions of STAT3: A pivotal target for individualized treatment of cancer. Cell Cycle 2005, 4, 1131–1133. [Google Scholar] [PubMed]
- Marzec, M.; Halasa, K.; Liu, X.; Wang, H.Y.; Cheng, M.; Baldwin, D.; Tobias, J.W.; Schuster, S.J.; Woetmann, A.; Zhang, Q.; et al. Malignant Transformation of CD4+T Lymphocytes Mediated by Oncogenic Kinase NPM/ALK Recapitulates IL-2–Induced Cell Signaling and Gene Expression Reprogramming. J. Immunol. 2013, 191, 6200–6207. [Google Scholar] [CrossRef]
- Chiarle, R.; Simmons, W.J.; Cai, H.; Dhall, G.; Zamò, A.; Raz, R.; Karras, J.G.; Levy, D.E.; Inghirami, G. Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat. Med. 2005, 11, 623–629. [Google Scholar] [CrossRef]
- Weilemann, A.; Grau, M.; Erdmann, T.; Merkel, O.; Sobhiafshar, U.; Anagnostopoulos, I.; Hummel, M.; Siegert, A.; Hayford, C.; Madle, H.; et al. Essential role of IRF4 and MYC signaling for survival of anaplastic large cell lymphoma. Blood 2015, 125, 124–132. [Google Scholar] [CrossRef]
- Bandini, C.; Pupuleku, A.; Spaccarotella, E.; Pellegrino, E.; Wang, R.; Vitale, N.; Duval, C.; Cantarella, D.; Rinaldi, A.; Provero, P.; et al. IRF4 Mediates the Oncogenic Effects of STAT3 in Anaplastic Large Cell Lymphomas. Cancers 2018, 10, 21. [Google Scholar] [CrossRef]
- Shaffer, A.L.; Emre, N.C.T.; Lamy, L.; Ngo, V.N.; Wright, G.; Xiao, W.; Powell, J.; Dave, S.; Yu, X.; Zhao, H.; et al. IRF4 addiction in multiple myeloma. Nature 2008, 454, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Chernyshova, E.V.; Abramov, D.S.; Konovalov, D.M.; Larin, S.S.; Myakova, N.V. Molecular biological characteristics of ALK-positive anaplastic large cell lymphoma. Oncohematology 2016, 11, 25–30. [Google Scholar]
- Lollies, A.; Hartmann, S.; Schneider, M.; Bracht, T.; Weiß, A.L.; Arnolds, J.; Klein-Hitpass, L.; Sitek, B.; Hansmann, M.-L.; Küppers, R.; et al. An oncogenic axis of STAT-mediated BATF3 upregulation causing MYC activity in classical Hodgkin lymphoma and anaplastic large cell lymphoma. Leukemia 2018, 32, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Wang, P.; Young, L.C.; Lai, R.; Li, L. Proteome-Wide Identification of Novel Binding Partners to the Oncogenic Fusion Gene Protein, NPM-ALK, using Tandem Affinity Purification and Mass Spectrometry. Am. J. Pathol. 2009, 174, 361–370. [Google Scholar] [CrossRef][Green Version]
- Marzec, M.; Liu, X.; Wong, W.; Yang, Y.; Pasha, T.L.; Kantekure, K.; Zhang, P.; Woetmann, A.; Cheng, M.; Odum, N.; et al. Oncogenic kinase NPM/ALK induces expression of HIF1α mRNA. Oncogene 2011, 30, 1372–1378. [Google Scholar] [CrossRef]
- Martinengo, C.; Poggio, T.; Menotti, M.; Scalzo, M.S.; Mastini, C.; Ambrogio, C.; Pellegrino, E.; Riera, L.; Piva, R.; Ribatti, D.; et al. ALK-Dependent Control of Hypoxia-Inducible Factors Mediates Tumor Growth and Metastasis. Cancer Res. 2014, 74, 6094–6106. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, J.D.; Wu, F.; Ye, X.; Sharon, D.; Hitt, M.; McMullen, T.P.; Hegazy, S.A.; Gélébart, P.; Yang, J.; et al. The expression and oncogenic effects of the embryonic stem cell marker SALL4 in ALK-positive anaplastic large cell lymphoma. Cell. Signal. 2012, 24, 1955–1963. [Google Scholar] [CrossRef]
- Gelebart, P.; Hegazy, S.A.; Wang, P.; Bone, K.M.; Anand, M.; Sharon, D.; Hitt, M.; Pearson, J.D.; Ingham, R.J.; Ma, Y.; et al. Aberrant expression and biological significance of Sox2, an embryonic stem cell transcriptional factor, in ALK-positive anaplastic large cell lymphoma. Blood Cancer J. 2012, 2, e82. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, P.; Wu, F.; Li, M.; Sharon, D.; Ingham, R.J.; Hitt, M.; McMullen, T.P.; Lai, R. Aberrant expression of the transcriptional factor Twist1 promotes invasiveness in ALK-positive anaplastic large cell lymphoma. Cell. Signal. 2012, 24, 852–858. [Google Scholar] [CrossRef]
- Crescenzo, R.; Abate, F.; Lasorsa, E.; Tabbo, F.; Gaudiano, M.; Chiesa, N.; Di Giacomo, F.; Spaccarotella, E.; Barbarossa, L.; Ercole, E.; et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lym-phoma. Cancer Cell 2015, 27, 516–532. [Google Scholar]
- Marzec, M.; Zhang, Q.; Goradia, A.; Raghunath, P.N.; Liu, X.; Paessler, M.; Wang, H.Y.; Wysocka, M.; Cheng, M.; Ruggeri, B.A.; et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc. Natl. Acad. Sci. USA 2008, 105, 20852–20857. [Google Scholar] [CrossRef] [PubMed]
- Kasprzycka, M.; Zhang, Q.; Witkiewicz, A.K.; Marzec, M.; Potoczek, M.; Liu, X.; Wang, H.Y.; Milone, M.C.; Basu, S.; Mauger, J.; et al. γc-Signaling Cytokines Induce a Regulatory T Cell Phenotype in Malignant CD4+ T Lymphocytes. J. Immunol. 2008, 181, 2506–2512. [Google Scholar] [CrossRef] [PubMed]
- Desjobert, C.; Renalier, M.-H.; Bergalet, J.; Dejean, E.; Joseph, N.; Kruczynski, A.; Soulier, J.; Espinos, E.; Meggetto, F.; Cavaillé, J.; et al. MiR-29a down-regulation in ALK-positive anaplastic large cell lymphomas contributes to apoptosis blockade through MCL-1 overexpression. Blood 2011, 117, 6627–6637. [Google Scholar] [CrossRef] [PubMed]
- Piva, R.; Pellegrino, E.; Mattioli, M.; Agnelli, L.; Lombardi, L.; Boccalatte, F.; Costa, G.; Ruggeri, B.A.; Cheng, M.; Chiarle, R.; et al. Functional validation of the anaplastic lymphoma kinase signature identifies CEBPB and Bcl2A1 as critical target genes. J. Clin. Investig. 2006, 116, 3171–3182. [Google Scholar] [CrossRef] [PubMed]
- Bonzheim, I.; Irmler, M.; Klier-Richter, M.; Steinhilber, J.; Anastasov, N.; Schäfer, S.; Adam, P.; Beckers, J.; Raffeld, M.; Fend, F.; et al. Identification of C/EBPβ Target Genes in ALK+ Anaplastic Large Cell Lymphoma (ALCL) by Gene Expression Profiling and Chromatin Immunoprecipitation. PLoS ONE 2013, 8, e64544. [Google Scholar] [CrossRef]
- Yu, B.-H.; Zhang, Y.; Xue, T.; Shui, R.-H.; Lu, H.-F.; Zhou, X.-Y.; Zhu, X.-Z.; Li, X.-Q. The clinicopathological relevance of uniform CD56 expression in anaplastic large cell lymphoma: A retrospective analysis of 18 cases. Diagn. Pathol. 2021, 16, 1–8. [Google Scholar] [CrossRef]
- Sibon, D.; Fournier, M.; Brière, J.; Lamant, L.; Haioun, C.; Coiffier, B.; Bologna, S.; Morel, P.; Gabarre, J.; Hermine, O.; et al. Long-Term Outcome of Adults with Systemic Anaplastic Large-Cell Lymphoma Treated Within the Groupe d’Étude des Lymphomes de l’Adulte Trials. J. Clin. Oncol. 2012, 30, 3939–3946. [Google Scholar] [CrossRef]
- Piccaluga, P.P.; Fuligni, F.; De Leo, A.; Bertuzzi, C.; Rossi, M.; Bacci, F.; Sabattini, E.; Agostinelli, C.; Gazzola, A.; Laginestra, M.A.; et al. Molecular Profiling Improves Classification and Prognostication of Nodal Peripheral T-Cell Lymphomas: Results of a Phase III Diagnostic Accuracy Study. J. Clin. Oncol. 2013, 31, 3019–3025. [Google Scholar] [CrossRef]
- Schmitz, N.; Trümper, L.; Ziepert, M.; Nickelsen, M.; Ho, A.D.; Metzner, B.; Peter, N.; Loeffler, M.; Rosenwald, A.; Pfreundschuh, M. Treatment and prognosis of mature T-cell and NK-cell lymphoma: An analysis of patients with T-cell lymphoma treated in studies of the German High-Grade Non-Hodgkin Lymphoma Study Group. Blood 2010, 116, 3418–3425. [Google Scholar] [CrossRef]
- Sibon, D.; Nguyen, D.-P.; Schmitz, N.; Suzuki, R.; Feldman, A.L.; Gressin, R.; Lamant, L.; Weisenburger, D.D.; Rosenwald, A.; Nakamura, S.; et al. ALK-positive anaplastic large-cell lymphoma in adults: An individual patient data pooled analysis of 263 patients. Haematologica 2019, 104, e562–e565. [Google Scholar] [CrossRef]
- Ellin, F.; Landstrom, J.; Jerkeman, M.; Relander, T. Real-world data on prognostic factors and treatment in peripheral T-cell lymphomas: A study from the swedishlymphoma registry. Blood 2014, 124, 1570–1577. [Google Scholar] [PubMed]
- Moskowitz, C.H.; Bertino, J.R.; Glassman, J.R.; Hedrick, E.E.; Hunte, S.; Coady-Lyons, N.; Agus, D.B.; Goy, A.; Jurcic, J.; Noy, A.; et al. Ifosfamide, Carboplatin, and Etoposide: A Highly Effective Cytoreduction and Peripheral-Blood Progenitor-Cell Mobilization Regimen for Transplant-Eligible Patients with Non-Hodgkin’s Lymphoma. J. Clin. Oncol. 1999, 17, 3776–3785. [Google Scholar] [CrossRef]
- Press, O.W.; Livingston, R.; Mortimer, J.; Collins, C.; Appelbaum, F. Treatment of relapsed non-Hodgkin’s lymphomas with dexamethasone, high-dose cytarabine, and cisplatin before marrow transplantation. J. Clin. Oncol. 1991, 9, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.-Y.; Tang, Y.; Zhu, Q.; Zhuang, Y.; Cheng, Y.-M.; Wang, L.; Zou, L.-F. Gemcitabine, oxaliplatin and dexamethasone as salvage treatment for elderly patients with refractory and relapsed peripheral T-cell lymphoma. Leuk. Lymphoma 2013, 54, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Gambacorti-Passerini, C.; Messa, C.; Pogliani, E.M. Crizotinib in Anaplastic Large-Cell Lymphoma. N. Engl. J. Med. 2011, 364, 775–776. [Google Scholar] [CrossRef] [PubMed]
- Mossé, Y.P.; Lim, M.; Voss, S.D.; Wilner, K.; Ruffner, K.; Laliberte, J.; Rolland, D.; Balis, F.; Maris, J.M.; Weigel, B.J.; et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: A Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 2013, 14, 472–480. [Google Scholar] [CrossRef]
- Mosse, Y.P.; Voss, S.D.; Lim, M.S.; Rolland, D.; Minard, C.G.; Fox, E.; Adamson, P.; Wilner, K.; Blaney, S.M.; Weigel, B.J. Targeting ALK with Crizotinib in Pediatric Ana-plastic Large Cell Lymphoma and Inflammatory Myofibroblastic Tumor: A Children’s Oncology Group Study. J. Clin. Oncol. 2017, 35, 3215–3221. [Google Scholar]
- Gambacorti-Passerini, C.; Mussolin, L.; Brugieres, L. Abrupt Relapse of ALK-Positive Lymphoma after Discontinuation of Crizotinib. N. Engl. J. Med. 2016, 374, 95–96. [Google Scholar] [CrossRef]
- Gambacorti-Passerini, C.; Farina, F.; Stasia, A.; Redaelli, S.; Ceccon, M.; Mologni, L.; Messa, C.; Guerra, L.; Giudici, G.; Sala, E.; et al. Crizotinib in advanced, chemo-resistant anaplastic lymphoma kinase-positive lymphoma patients. J. Natl. Cancer Inst. 2014, 106, djt378. [Google Scholar]
- Ceccon, M.; Mologni, L.; Bisson, W.; Scapozza, L.; Passerini, C.G. Crizotinib-Resistant NPM-ALK Mutants Confer Differential Sensitivity to Unrelated Alk Inhibitors. Mol. Cancer Res. 2013, 11, 122–132. [Google Scholar] [CrossRef]
- Emmanouilides, C.; Colovos, C.; Pinter-Brown, L.; Hernandez, L.; Schiller, G.; Territo, M.; Rosen, P. Pilot study of fixed-infusion rate gemcitabine with cisplatin and dexamethasone in patients with relapsed or refractory lymphoma. Clin. Lymphoma 2004, 5, 45–49. [Google Scholar] [PubMed]
- Dhawale, T.M.; Shustov, A.R. Autologous and allogeneic hematopoietic cell transplantation in peripheral T/NK-cell lym-phomas: A histology-specific review. Hematol. Oncol. Clin. N. Am. 2017, 31, 335–357. [Google Scholar]
- Fanin, R.; The EBMT Lymphoma Working Party; De Elvira, M.C.R.; Sperotto, A.; Baccarani, M.; Goldstone, A. Autologous stem cell transplantation for T and null cell CD30-positive anaplastic large cell lymphoma: Analysis of 64 adult and paediatric cases reported to the European Group for Blood and Marrow Transplantation (EBMT). Bone Marrow Transplant. 1999, 23, 437–442. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Smith, S.M.; Burns, L.J.; van Besien, K.; LeRademacher, J.; He, W.; Fenske, T.S.; Suzuki, R.; Hsu, J.W.; Schouten, H.C.; Hale, G.A.; et al. Hematopoietic cell transplantation for systemic mature T-cell non-hodgkin lym-phoma. J. Clin. Oncol. 2013, 31, 3100–3109. [Google Scholar] [PubMed]
- Le Gouill, S.; Milpied, N.; Buzyn, A.; De Latour, R.P.; Vernant, J.-P.; Mohty, M.; Moles, M.-P.; Bouabdallah, K.; Bulabois, C.-E.; Dupuis, J.; et al. Graft-versus-lymphoma effect for aggressive T-cell lymphomas in adults: A study by the societefrancaise de greffe de moelle et de therapiecellulaire. J. Clin. Oncol. 2008, 26, 2264–2271. [Google Scholar] [PubMed]
- Hassler, M.R.; Pulverer, W.; Lakshminarasimhan, R.; Redl, E.; Hacker, J.; Garland, G.D.; Merkel, O.; Schiefer, A.-I.; Simonitsch-Klupp, I.; Kenner, L.; et al. Insights into the Pathogenesis of Anaplastic Large-Cell Lymphoma through Genome-wide DNA Methylation Profiling. Cell Rep. 2016, 17, 596–608. [Google Scholar] [CrossRef]
- Mason, D.Y.; Harris, N.L.; Delsol, G.; Stein, H.; Campo, E.; Kinney, M.C.; Jaffe, E.S.; Falini, B. Anaplastic Large Cell Lymphoma, ALK-Negative. In WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Vardiman, J.W., Eds.; IARC: Lyon, France, 2008; pp. 317–319. [Google Scholar]
- Feldman, A.L.; Harris, N.L.; Stein, H.; Campo, E.; Kinney, M.C.; Jaffe, E.S.; Falini, B.; Inghirami, G.G.; Pileri, S.A. Anaplastic Large Cell Lymphoma, ALK-Negative. In WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues, 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC: Lyon, France, 2017; pp. 418–421. [Google Scholar]
- Ferreri, A.J.; Govi, S.; Pileri, S.A.; Savage, K.J. Anaplastic large cell lymphoma, ALK-negative. Crit. Rev. Oncol. Hematol. 2013, 85, 206–215. [Google Scholar] [CrossRef]
- Kim, Y.C.; Yang, W.I.; Lee, M.-G.; Kim, S.N.; Cho, K.H.; Lee, S.J.; Lee, M.W.; Koh, J.K. Epstein-Barr virus in CD30+anaplastic large cell lymphoma involving the skin and lymphomatoid papulosis in South Korea. Int. J. Dermatol. 2006, 45, 1312–1316. [Google Scholar] [CrossRef]
- Ma, L.; Katz, Y.; Sharan, K.P.; Schwarting, R.; Kim, A.S. Epstein-Barr virus positive anaplastic large cell lymphoma: Myth or reality? Int. J. Clin. Exp. Pathol. 2010, 4, 100–110. [Google Scholar]
- Kinney, M.C.; Higgins, R.A.; Medina, E.A. Anaplastic large cell lymphoma: Twenty-five years of discovery. Arch. Pathol. Lab. Med. 2011, 135, 19–43. [Google Scholar]
- Tsuyama, N.; Sakamoto, K.; Sakata, S.; Dobashi, A.; Takeuchi, K. Anaplastic large cell lymphoma: Pathology, genetics, and clinical aspects. J. Clin. Exp. Hematop. 2017, 57, 120–142. [Google Scholar] [CrossRef] [PubMed]
- Savage, K.J.; Harris, N.L.; Vose, J.M.; Ullrich, F.; Jaffe, E.S.; Connors, J.M.; Rimsza, L.; Pileri, S.A.; Chhanabhai, M.; Gascoyne, R.D.; et al. ALK-anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: Report from the International Peripheral T-Cell Lymphoma Project. Blood 2008, 111, 5496–5504. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.C.-L.; Chan, J.K.-C.; Yan, K.W.; Kwong, Y.L. Anaplastic Large Cell Lymphoma Presenting as a Pleural Effusion and Mimicking Primary Effusion Lymphoma. A report of 2 cases. Acta Cytol. 2003, 47, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Grandhi, A.; Boros, A.L.; Berardo, N.; Reich, R.F.; Freedman, P.D. Two cases of CD30+, anaplastic lymphoma kinase (ALK)-negative anaplastic large cell lymphoma with oral manifestations. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2013, 115, e41–e47. [Google Scholar] [CrossRef]
- Kodama, K.; Hokama, M.; Kawaguchi, K.; Tanaka, Y.; Hongo, K. Primary ALK-1-negative anaplastic large cell lymphoma of the brain: Case report and review of the literature. Neuropathology 2009, 29, 166–171. [Google Scholar] [CrossRef]
- Lagmay, J.; Termuhlen, A.; Fung, B.; Ranalli, M. Primary testicular presentation of ALK-1-negative anaplastic large cell lym-phoma in a pediatric patient. J. Pediatr. Hematol. Oncol. 2009, 31, 330–332. [Google Scholar]
- Lannon, M.; Lu, J.-Q.; Chum, M.; Wang, B.H. ALK-negative CNS anaplastic large cell lymphoma: Case report and review of literature. Br. J. Neurosurg. 2020, 30, 1–6. [Google Scholar] [CrossRef]
- Lobo, J.; Henrique, R.; Monteiro, P.; Lobo, C. ALK-negative anaplastic large cell lymphoma with urinary bladder involvement diagnosed in urine cytology: A case report and literature review. Diagn. Cytopathol. 2017, 45, 354–358. [Google Scholar] [CrossRef]
- Saikia, U.N.; Sharma, N.; Duseja, A.; Bhalla, A.; Joshi, K. Anaplastic large cell lymphoma presenting as acute liver failure: A report of two cases with review of literature. Ann. Hepatol. 2010, 9, 457–461. [Google Scholar] [CrossRef]
- Sanka, R.K.; Eagle, R.C., Jr.; Wojno, T.H.; Neufeld, K.R.; Grossniklaus, H.E. Spectrum of CD30+ Lymphoid Proliferations in the Eyelid: Lymphomatoid Papulosis, Cutaneous Anaplastic Large Cell Lymphoma, and Anaplastic Large Cell Lymphoma. Ophthalmology 2010, 117, 343–351. [Google Scholar] [CrossRef]
- Karki, N.R.; Badin, K.; Savage, N.; Bryan, L. Leukaemic relapse of anaplastic large cell lymphoma, ALK negative. BMJ Case Rep. 2021, 14, e239213. [Google Scholar] [CrossRef]
- Kasinathan, G. Leukemic phase of ALK-negative anaplastic large cell lymphoma in a patient who is on androgenic steroids: A case report. Ann. Med. Surg. 2020, 49, 1–4. [Google Scholar] [CrossRef]
- Lu, Y.; Zhao, X.; Wang, E.; Chen, W.; Huang, Q. ALK-negative anaplastic large cell lymphoma with extensive peripheral blood and bone marrow involvements manifested as “leukemic phase”. Leuk. Res. 2010, 34, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.S.; Liu, B.W.F.; Lam, F.S.C.; Wong, K.F. ALK-negative anaplastic large cell lymphoma in leukemic phase with near-pentaploidy. Leuk. Lymphoma 2010, 51, 1927–1930. [Google Scholar] [CrossRef] [PubMed]
- Hapgood, G.; Savage, K.J. The biology and management of systemic anaplastic large cell lymphoma. Blood 2015, 126, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Pina-Oviedo, S.; Ortiz-Hidalgo, C.; Carballo-Zarate, A.A.; Zarate-Osorno, A. ALK-Negative Anaplastic Large Cell Lymphoma: Current Concepts and Molecular Pathogenesis of a Heterogeneous Group of Large T-Cell Lymphomas. Cancers 2021, 13, 4667. [Google Scholar]
- Luchtel, R.A.; Dasari, S.; Oishi, N.; Pedersen, M.B.; Hu, G.; Rech, K.L.; Ketterling, R.P.; Sidhu, J.; Wang, X.; Katoh, R.; et al. Molecular profiling reveals immunogenic cues in anaplastic large cell lymphomas with DUSP22 rearrangements. Blood 2018, 132, 1386–1398. [Google Scholar] [CrossRef]
- Sciallis, A.P.; Law, M.E.; Inwards, D.J.; McClure, R.F.; Macon, W.R.; Kurtin, P.J.; Dogan, A.; Feldman, A.L. Mucosal CD30-positive T-cell lymphoproliferations of the head and neck show a clinicopathologic spectrum similar to cutaneous CD30-positive T-cell lymphoproliferative disorders. Mod. Pathol. 2012, 25, 983–992. [Google Scholar] [CrossRef]
- Feldman, A.L.; Law, M.; Remstein, E.D.; Macon, W.R.; Erickson, L.A.; Grogg, K.L.; Kurtin, P.J.; Dogan, A. Recurrent translocations involving the IRF4 oncogene locus in peripheral T-cell lymphomas. Leukemia 2008, 23, 574–580. [Google Scholar] [CrossRef]
- Feldman, A.; Dogan, A.; Smith, D.I.; Law, M.E.; Ansell, S.M.; Johnson, S.H.; Porcher, J.C.; Özsan, N.; Wieben, E.D.; Eckloff, B.W.; et al. Discovery of recurrent t(6;7)(p25.3;q32.3) translocations in ALK-negative anaplastic large cell lymphomas by massively parallel genomic sequencing. Blood 2011, 117, 915–919. [Google Scholar] [CrossRef]
- Wada, D.A.; Law, M.E.; Hsi, E.D.; DiCaudo, D.J.; Ma, L.; Lim, M.; De Souza, A.; Comfere, N.I.; Weenig, R.H.; Macon, W.R.; et al. Specificity of IRF4 translocations for primary cutaneous anaplastic large cell lymphoma: A multicenter study of 204 skin biopsies. Mod. Pathol. 2010, 24, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Karai, L.J.; Kadin, M.E.; Hsi, E.D.; Sluzevich, J.C.; Ketterling, R.P.; Knudson, R.A.; Feldman, A.L. Chromosomal rearrangements of 6p25.3 define a new subtype of lymphomatoid pap-ulosis. Am. J. Surg. Pathol. 2013, 37, 1173–1181. [Google Scholar] [PubMed]
- Pham-Ledard, A.; Prochazkova-Carlotti, M.; Laharanne, E.; Vergier, B.; Jouary, T.; Beylot-Barry, M.; Merlio, J.-P. IRF4 gene rearrangements define a subgroup of CD30-positive cutaneous T-cell lymphoma: A study of 54 cases. J. Investig. Dermatol. 2010, 130, 816–825. [Google Scholar] [PubMed]
- Kiran, T.; Demirkesen, C.; Eker, C.; Kumusoglu, H.; Tuzuner, N. The significance of MUM1/IRF4 protein expression and IRF4 translocation of CD30(+) cutaneous T-cell lymphoproliferative disorders: A study of 53 cases. Leuk. Res. 2013, 37, 396–400. [Google Scholar]
- Kluk, J.; Child, F.; Robson, A. Lymphomatoid papulosis with 6p25.3 rearrangement: A further case of the newly described variant. Br. J. Dermatol. 2014, 171, 1590–1592. [Google Scholar] [CrossRef]
- Chen, A.J.; Zhou, G.; Juan, T.; Colicos, S.M.; Cannon, J.P.; Cabriera-Hansen, M.; Meyer, C.F.; Jurecic, R.; Copeland, N.G.; Gilbert, D.J.; et al. The Dual Specificity JKAP Specifically Activates the c-Jun N-terminal Kinase Pathway. J. Biol. Chem. 2002, 277, 36592–36601. [Google Scholar] [CrossRef]
- Li, J.-P.; Yang, C.-Y.; Chuang, H.-C.; Lan, J.-L.; Chen, D.-Y.; Chen, Y.-M.; Wang, X.; Chen, A.J.; Belmont, J.W.; Tan, T.-H. The phosphatase JKAP/DUSP22 inhibits T-cell receptor signalling and autoimmunity by inactivating Lck. Nat. Commun. 2014, 5, 3618. [Google Scholar] [CrossRef]
- Sekine, Y.; Tsuji, S.; Ikeda, O.; Sato, N.; Aoki, N.; Aoyama, K.; Sugiyama, K.; Matsuda, T. Regulation of STAT3-mediated signaling by LMW-DSP2. Oncogene 2006, 25, 5801–5806. [Google Scholar] [CrossRef]
- Kyriakou, C.; Canals, C.; Goldstone, A.; Caballero, D.; Metzner, B.; Kobbe, G.; Kolb, H.-J.; Kienast, J.; Reimer, P.; Finke, J.; et al. High-dose therapy and autologous stem-cell transplantation in angioim-munoblastic lymphoma: Complete remission at transplantation is the major determinant of outcome-lymphoma working party of the european group for blood and marrow transplantation. J. Clin. Oncol. 2008, 26, 218–224. [Google Scholar]
- Chen, J.; Zhang, Y.; Petrus, M.N.; Xiao, W.; Nicolae, A.; Raffeld, M.; Pittaluga, S.; Bamford, R.N.; Nakagawa, M.; Ouyang, S.T.; et al. Cytokine receptor signaling is required for the survival of ALK- anaplastic large cell lymphoma, even in the presence of JAK1/STAT3 mutations. Proc. Natl. Acad. Sci. USA 2017, 114, 3975–3980. [Google Scholar]
- Martin, J.M.; Wu, H.; Barta, S.K. CD30+ T-cell lymphoproliferative disorders. Chin. Clin. Oncol. 2019, 8, 4. [Google Scholar] [CrossRef]
- Willemze, R.; Paulli, M.; Kadin, M.E. Primary Cutaneous CD30-Positive T-Cell Lymphoproliferative Disorders. In WHO Classi-fication of Tumors of Hematopoietic and Lymphoid Tissues, 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC: Lyon, France, 2017; pp. 392–396. [Google Scholar]
- Bekkenk, M.W.; Geelen, F.A.; van Voorst Vader, P.C.; Heule, F.; Geerts, M.L.; van Vloten, W.A.; Meijer, C.J.L.M.; Willemze, R. Primary and secondary cutaneous CD30(+) lymphoproliferative disorders: A report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood 2000, 95, 3653–3661. [Google Scholar] [PubMed]
- Willemze, R.; Cerroni, L.; Kempf, W.; Berti, E.; Facchetti, F.; Swerdlow, S.H.; Jaffe, E.S. The 2018 update of the WHO-EORTC classification for primary cutaneous lym-phomas. Blood 2019, 133, 1703–1714. [Google Scholar] [PubMed]
- Liu, H.L.; Hoppe, R.T.; Kohler, S.; Harvell, J.D.; Reddy, S.; Kim, Y.H. CD30+ cutaneous lymphoproliferative disorders: The Stanford experience in lympho-matoid papulosis and primary cutaneous anaplastic large cell lymphoma. J. Am. Acad. Dermatol. 2003, 49, 1049–1058. [Google Scholar]
- Berti, E.; Gianotti, R.; Alessi, E. Primary Anaplastic Large Cell Lymphoma of the Skin. Dermatologica 1989, 178, 225–227. [Google Scholar] [CrossRef]
- Hruska, C.J.; Bertoli, R.J.; Young, Y.D.; Burkhart, P.H.; Googe, P.B. Primary cutaneous anaplastic large cell lymphoma in a patient receiving adalimumab. JAAD Case Rep. 2015, 1, 56–59. [Google Scholar] [CrossRef]
- Papathemeli, D.; Gräfe, R.; Hildebrandt, U.; Zettl, U.K.; Ulrich, J. Development of a primary cutaneous CD30(+) anaplastic large-cell T-cell lymphoma during treatment of multiple sclerosis with fingolimod. Mult. Scler. J. 2016, 22, 1888–1890. [Google Scholar] [CrossRef]
- De Bruin, P.; Beljaards, R.; Van Heerde, P.; Van Der Valk, P.; Noorduyn, L.; Van Krieken, J.; Kluin-Nelemans, J.C.; Willemze, R.; Meijer, C. Differences in clinical behaviour and immunophenotype between primary cutaneous and primary nodal anaplastic large cell lymphoma of T-cell or null cell phenotype. Histopathology 1993, 23, 127–135. [Google Scholar] [CrossRef]
- Seo, A.N.; Lee, S.J.; Choi, Y.H.; Chung, H.Y.; Huh, J.; Yoon, G.S. Congenital primary cutaneous anaplastic large-cell lymphoma: A case report. Am. J. Dermatopathol. 2015, 37, 398–400. [Google Scholar]
- Kempf, W.; Kazakov, D.; Paredes, B.E.; Laeng, H.R.; Palmedo, G.; Kutzner, H. Primary Cutaneous Anaplastic Large Cell Lymphoma with Angioinvasive Features and Cytotoxic Phenotype: A Rare Lymphoma Variant within the Spectrum of CD30+ Lymphoproliferative Disorders. Dermatology 2013, 227, 346–352. [Google Scholar] [CrossRef]
- Booken, N.; Goerdt, S.; Klemke, C.-D. Clinical spectrum of primary cutaneous CD30-positive anaplastic large cell lymphoma: An analysis of the Mannheim Cutaneous Lymphoma Registry. JDDG J. Dtsch. Dermatol. Ges. 2012, 10, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Kempf, W.; Pfaltz, K.; Vermeer, M.H.; Cozzio, A.; Ortiz-Romero, P.L.; Bagot, M.; Olsen, E.; Kim, Y.H.; Dummer, R.; Pimpinelli, N.; et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of pri-mary cutaneous CD30-positive lymphoproliferative disorders: Lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood 2011, 118, 4024–4035. [Google Scholar] [PubMed]
- Brown, R.A.; Fernandez-Pol, S.; Kim, J. Primary cutaneous anaplastic large cell lymphoma. J. Cutan. Pathol. 2017, 44, 570–577. [Google Scholar] [PubMed]
- Melchers, R.C.; Willemze, R.; Daniëls, L.A.; Neelis, K.J.; Bekkenk, M.W.; de Haas, E.R.; Horvath, B.; van Rossum, M.M.; Sanders, C.J.; Velstra, B.; et al. Recommendations for the Optimal Radiation Dose in Patients with Primary Cutaneous Anaplastic Large Cell Lymphoma: A Report of the Dutch Cutaneous Lymphoma Group. Int. J. Radiat. Oncol. 2017, 99, 1279–1285. [Google Scholar] [CrossRef]
- Artemi, P.; Wong, D.A.; Mann, S.; Regan, W. CD30 (Ki-1)-positive primary cutaneous T-cell lymphoma: Report of spontaneous resolution. Australas. J. Dermatol. 1997, 38, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.; Tan, B.B. Spontaneous regression of a childhood tumour with anaplastic histological features. Clin. Exp. Dermatol. 2012, 38, 318–320. [Google Scholar] [CrossRef]
- Prieto-Torres, L.; Rodriguez-Pinilla, S.M.; Onaindia, A.; Ara, M.; Requena, L.; A Piris, M. CD30-positive primary cutaneous lymphoproliferative disorders: Molecular alterations and targeted therapies. Haematologica 2019, 104, 226–235. [Google Scholar] [CrossRef]
- Sugaya, M.; Fujita, H.; Izutsu, K.; Oshima, K.; Takazawa, Y.; Ohmatsu, H.; Yoshimi, A.; Takahashi, T.; Kanda, Y.; Kurokawa, M.; et al. Primary cutaneous anaplastic large cell lymphoma with leg involvement: A case report and review of 11 cases. J. Dermatol. 2011, 38, 1009–1012. [Google Scholar] [CrossRef]
- Woo, D.K.; Jones, C.R.; Vanoli-Storz, M.N.; Kohler, S.; Reddy, S.; Advani, R.; Hoppe, R.T.; Kim, Y.H. Prognostic factors in primary cutaneous anaplastic large cell lymphoma: Characterization of clinical subset with worse outcome. Arch. Dermatol. 2009, 145, 667–674. [Google Scholar]
- Lee, W.J.; Moon, I.J.; Lee, S.H.; Won, C.H.; Chang, S.E.; Choi, J.H.; Moon, K.C.; Park, C.-S.; Huh, J.; Lee, M.W. Cutaneous anaplastic large-cell lymphoma (ALCL): A comparative clinical feature and survival outcome analysis of 52 cases according to primary tumor site. J. Am. Acad. Dermatol. 2016, 74, 1135–1143. [Google Scholar] [CrossRef]
- MacGrogan, G.; Vergier, B.; Dubus, P.; Beylot-Barry, M.; Belleannee, G.; Delaunay, M.M.; Eghbali, H.; Beylot, C.; Rivel, J.; Trojani, M.; et al. CD30-Positive Cutaneous Large Cell Lymphomas:A Comparative Study of Clinicopathologic and Molecular Features of 16 Cases. Am. J. Clin. Pathol. 1996, 105, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Orchard, G.; Lillington, D.M.; Russell-Jones, R.; Young, B.D.; Whittaker, S. Genetic alterations in primary cutaneous CD30+ anaplastic large cell lymphoma. Genes Chromosom. Cancer 2003, 37, 176–185. [Google Scholar] [CrossRef]
- Szuhai, K.; Van Doorn, R.; Tensen, C.P.; Kester, V. Array-CGH Analysis of Cutaneous Anaplastic Large Cell Lymphoma. Methods Pharmacol. Toxicol. 2013, 973, 197–212. [Google Scholar] [CrossRef]
- Csikes, C.R.; Knudson, R.A.; Greipp, P.T.; Feldman, A.L.; Kadin, M. Primary cutaneous CD30-positive T-cell lymphoprolifer-ative disorders with biallelic rearrangements of DUSP22. J. Investig. Dermatol. 2013, 133, 1680–1682. [Google Scholar]
- Ellis, D.; Chan, J. Extranodal lymphoproliferative disorders with emphasis on skin. Phathol. Int. 2004, 54, S318–S341. [Google Scholar]
- Oka, T.; Sugaya, M.; Cury-Martins, J.; Vasconcelos-Berg, R.; Suga, H.; Miyagaki, T.; Scheinberg, P.; Fujita, H.; Izutsu, K.; Sato, S.; et al. Hematopoietic stem cell transplantation for cutaneous T-cell lymphoma: Summary of 11 cases from two facilities in Japan and Brazil. J. Dermatol. 2015, 43, 638–642. [Google Scholar] [CrossRef]
- Duvic, M.; Reddy, S.A.; Pinter-Brown, L.; Korman, N.J.; Zic, J.; Kennedy, D.A.; Lorenz, J.; Sievers, E.; Kim, Y.H. A Phase II Study of SGN-30 in Cutaneous Anaplastic Large Cell Lymphoma and Related Lymphoproliferative Disorders. Clin. Cancer Res. 2009, 15, 6217–6224. [Google Scholar] [CrossRef]
- Kaffenberger, B.H.; Winardi, F.K.; Frederickson, J.; Porcu, P.; Wong, H.K. Periocular cutaneous anaplastic large cell lymphoma clearance with brentuximab vedotin. J. Clin. Aesthet. Dermatol. 2013, 6, 29–31. [Google Scholar]
- Desai, A.; Telang, G.H.; Olszewski, A.J. Remission of primary cutaneous anaplastic large cell lymphoma after a brief course of brentuximab vedotin. Ann. Hematol. 2012, 92, 567–568. [Google Scholar] [CrossRef]
- Milan, E.; Miceli, P.; Sernicola, A.; Marino, D.; Alaibac, M. Complete remission of primary coetaneous anaplastic large cell lymphoma after a short course of brentuximab vedotin. Mol. Clin. Oncol. 2021, 14, 121. [Google Scholar]
- Duvic, M.; Tetzlaff, M.T.; Gangar, P.; Clos, A.L.; Sui, D.; Talpur, R. Results of a Phase II Trial of Brentuximab Vedotin for CD30+ Cutaneous T-Cell Lymphoma and Lymphomatoid Papulosis. J. Clin. Oncol. 2015, 33, 3759–3765. [Google Scholar] [CrossRef] [PubMed]
- Keech, J.A., Jr.; Creech, B.J. Anaplastic T-Cell Lymphoma in Proximity to a Saline-Filled Breast Implant. Plast. Reconstr. Surg. 1997, 100, 554–555. [Google Scholar] [CrossRef] [PubMed]
- Miranda, R.N.; Feldman, A.L.; Soares, F.A. Breast implant-associated anaplastic large cell lymphoma. In Breast Tumors, 5th ed.; WHO Classification of Tumors Editorial Board, Ed.; IARC: Lyon, France, 2019; pp. 245–248. [Google Scholar]
- Doren, E.L.; Miranda, R.N.; Selber, J.C.; Garvey, P.B.; Liu, J.; Medeiros, L.J.; Butler, C.E.; Clemens, M.W. U.S. Epidemiology of Breast Implant–Associated Anaplastic Large Cell Lymphoma. Plast. Reconstr. Surg. 2017, 139, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- United States Food and Drug Administration. The FDA Requests Allergan Voluntarily Recall Natrelle BIOCELL Textured Breast Implants and Tissue Expanders from Themarket to Protect Patients: FDA Safety Communication; FDA: Silver Spring, MD, USA.
- Quesada, A.E.; Medeiros, L.J.; Clemens, M.W.; Ferrufino-Schmidt, M.C.; Pina-Oviedo, S.; Miranda, R.N. Breast implant-associated anaplastic large cell lymphoma: A review. Mod. Pathol. 2019, 32, 166–188. [Google Scholar] [PubMed]
- Laurent, C.; Delas, A.; Gaulard, P.; Haioun, C.; Moreau, A.; Xerri, L.; Traverse-Glehen, A.; Rousset, T.; Quintin-Roue, I.; Pet-rella, T.; et al. Breast implant-associated anaplastic large cell lymphoma: Two distinct clinicopathological variants with different outcomes. Ann. Oncol. 2016, 27, 306–314. [Google Scholar]
- Ferrufino-Schmidt, M.C.; Medeiros, L.J.; Liu, H.; Clemens, M.W.; Hunt, K.K.; Laurent, C.; Lofts, J.; Amin, M.B.; Chai, S.M.; Morine, A.; et al. Clinicopathologic Features and Prognostic Impact of Lymph Node Involvement in Patients with Breast Implant-associated Anaplastic Large Cell Lymphoma. Am. J. Surg. Pathol. 2018, 42, 293–305. [Google Scholar] [CrossRef]
- Scapini, P.; Lapinet-Vera, J.A.; Gasperini, S.; Calzetti, F.; Bazzoni, F.; Cassatella, M.A. The neutrophil as a cellular source of chemokines. Immunol. Rev. 2000, 177, 195–203. [Google Scholar] [CrossRef]
- Kobayashi, S.D.; Voyich, J.M.; Whitney, A.R.; DeLeo, F.R. Spontaneous neutrophilapoptosis and regulation of cell survival by granulocyte macrophage-colonystimulating factor. J. Leukoc. Biol. 2005, 78, 1408–1418. [Google Scholar]
- Rondón-Lagos, M.; Rangel, N.; Camargo-Villalba, G.; Forero-Castro, M. Biological and genetic landscape of breast im-plant-associated anaplastic large cell lymphoma (BIA-ALCL). Eur. J. Surg. Oncol. 2021, 47, 942–951. [Google Scholar]
- Grulich, A.E.; Vajdic, C.; Cozen, W. Altered Immunity as a Risk Factor for Non-Hodgkin Lymphoma. Cancer Epidemiol. Biomark. Prev. 2007, 16, 405–408. [Google Scholar] [CrossRef]
- Igney, F.H.; Krammer, P.H. Immune escape of tumors: Apoptosis resistance andtumor counterattack. J. Leukoc. Biol. 2002, 71, 907–920. [Google Scholar] [PubMed]
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Pajkos, A.; Deva, A.K.; Vickery, K.; Cope, C.; Chang, L.; Cossart, Y.E. Detection of subclinical infection in significant breast implant capsules. Plast. Reconstr. Surg. 2003, 111, 1605–1611. [Google Scholar] [PubMed]
- Lechner, M.G.; Lade, S.; Liebertz, D.J.; Prince, H.M.; Brody, G.S.; Webster, H.R.; Epstein, A.L. Breast implant-associated, ALKnegative, T-cell, anaplastic, large-cell lymphoma: Establishment and characterization of a model cell line (TLBR-1) for this newlyemerging clinical entity. Cancer 2011, 117, 1478–1489. [Google Scholar]
- Los-de Vries, G.T.; de Boer, M.; van Dijk, E.; Stathi, P.; Hijmering, N.J.; Roemer, M.G.M.; Mendeville, M.; Miedema, D.M.; de Boer, J.P.; Rakhorst, H.A.; et al. Chromosome 20 loss is characteristic of breast implant-associated anaplastic large cell lym-phoma. Blood 2020, 136, 2927–2932. [Google Scholar]
- Blombery, P.; Thompson, E.; Ryland, G.L.; Joyce, R.; Byrne, D.J.; Khoo, C.; Lade, S.; Hertzberg, M.; Hapgood, G.; Marlton, P.; et al. Frequent activating STAT3 mutations and novel recurrent genomic abnormalities detected in breast implant-associated anaplasticlarge cell lymphoma. Oncotarget 2018, 9, 36126–36136. [Google Scholar]
- Oishi, N.; Brody, G.S.; Ketterling, R.P.; Viswanatha, D.S.; He, R.; Dasari, S.; Mai, M.; Benson, H.K.; Sattler, C.A.; Boddicker, R.L.; et al. Genetic subtyping of breast implant–associated anaplastic large cell lymphoma. Blood 2018, 132, 544–547. [Google Scholar] [CrossRef]
- Di Napoli, A.; Jain, P.; Duranti, E.; Margolskee, E.; Arancio, W.; Facchetti, F.; Alobeid, B.; di Pompeo, F.S.; Mansukhani, M.; Bhagat, G. Targeted next generation sequencing of breast implant-associated anaplastic large cell lymphoma reveals mutations inJAK/STAT signalling pathway genes, TP53 and DNMT3A. Br. J. Haematol. 2018, 180, 741–744. [Google Scholar]
- Oishi, N.; Miranda, R.N.; Feldman, A.L. Genetics of Breast Implant-Associated Anaplastic Large Cell Lymphoma (BIA-ALCL). Aesthetic Surg. J. 2019, 39, S14–S20. [Google Scholar]
- Quesada, A.E.; Zhang, Y.; Ptashkin, R.; Ho, C.; Horwitz, S.; Benayed, R.; Dogan, A.; Arcila, M.E. Next generation sequencing ofbreast implant-associated anaplastic large cell lymphomas reveals a novel STAT3-JAK2 fusion among other activating ge-neticalterations within the JAK-STAT pathway. Breast. J. 2021, 27, 314–321. [Google Scholar]
- Laurent, C.; Nicolae, A.; Laurent, C.; Le Bras, F.; Haioun, C.; Fataccioli, V.; Amara, N.; Adélaïde, J.; Guille, A.; Schiano, J.-M.; et al. Gene alterations in epigenetic modifiers and JAK-STAT signaling are frequent in breast implant-associated ALCL. Blood 2020, 135, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. The JAK-STAT Signaling Pathway: Input and Output Integration. J. Immunol. 2007, 178, 2623–2629. [Google Scholar] [CrossRef] [PubMed]
- Deva, A.K.; Turner, S.D.; Kadin, M.E.; Magnusson, M.R.; Prince, H.M.; Miranda, R.N.; Inghirami, G.G.; Adams, W.P., Jr. Eti-ology of Breast Implant-Associated Anaplastic Large Cell Lymphoma (BIA-ALCL): Current Directions in Research. Cancers 2020, 12, 3861. [Google Scholar]
- Clemens, M.W.; Medeiros, L.J.; Butler, C.E.; Hunt, K.K.; Fanale, M.A.; Horwitz, S.; Weisenburger, D.D.; Liu, J.; Morgan, E.A.; Kanagal-Shamanna, R.; et al. Complete surgical excision isessential for the management of patients with breast implant-associated anaplasticlarge-cell lymphoma. J. Clin. Oncol. 2016, 34, 160–168. [Google Scholar]
- Estes, C.F.; Zhang, D.; Reyes, R.; Korentager, R.; McGinness, M.; Lominska, C. Locallyadvanced breast implant-associated anaplastic large-cell lymphoma: A case report ofsuccessful treatment with radiation and chemotherapy. Front. Oncol. 2015, 5, 26. [Google Scholar]
- Vaklavas, C.; Forero-Torres, A. Safety and efficacy of brentuximab vedotin in patients with Hodgkin lymphoma or systemic anaplastic large cell lymphoma. Ther. Adv. Hematol. 2012, 3, 209–225. [Google Scholar] [CrossRef]
- Zinzani, P.L.; Sasse, S.; Radford, J.; Gautam, A.; Bonthapally, V. Brentuximab vedotin inrelapsed/refractory Hodgkin lym-phoma: An updated review of published data fromthe named patient program. Crit. Rev. Oncol. Hematol. 2016, 104, 65–70. [Google Scholar]
- Horwitz, S.; O’Connor, O.A.; Pro, B.; Illidge, T.; Fanale, F.; Advani, R.; Bartlett, N.L.; Christensen, J.H.; Morschhauser, F.; Domingo-Domenech, E.; et al. Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma(ECHELON-2): A global, double-blind, randomised, phase 3 trial. Lancet 2019, 393, 229–240. [Google Scholar]
- Barranco, P.; Phillips-Angles, E.; Dominguez-Ortega, J.; Quirce, S. Dupilumab in the management of moderate-to-severe asthma: The data so far. Ther. Clin. Risk Manag. 2017, 13, 1139–1149. [Google Scholar] [CrossRef]
- Xu, X.; Zheng, Y.; Zhang, X.; He, Y.; Li, C. Efficacy and safety of dupilumab for the treatment of moderate-to-severe atopic dermatitis in adults. Oncotarget 2017, 8, 108480–108491. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.-R.; Chien, P.-N.; Nam, S.-Y.; Heo, C.-Y. Anaplastic Large Cell Lymphoma: Molecular Pathogenesis and Treatment. Cancers 2022, 14, 1650. https://doi.org/10.3390/cancers14071650
Zhang X-R, Chien P-N, Nam S-Y, Heo C-Y. Anaplastic Large Cell Lymphoma: Molecular Pathogenesis and Treatment. Cancers. 2022; 14(7):1650. https://doi.org/10.3390/cancers14071650
Chicago/Turabian StyleZhang, Xin-Rui, Pham-Ngoc Chien, Sun-Young Nam, and Chan-Yeong Heo. 2022. "Anaplastic Large Cell Lymphoma: Molecular Pathogenesis and Treatment" Cancers 14, no. 7: 1650. https://doi.org/10.3390/cancers14071650
APA StyleZhang, X.-R., Chien, P.-N., Nam, S.-Y., & Heo, C.-Y. (2022). Anaplastic Large Cell Lymphoma: Molecular Pathogenesis and Treatment. Cancers, 14(7), 1650. https://doi.org/10.3390/cancers14071650