
Targeting circDGKD Intercepts TKI’s Effects on Up-Regulation of Estrogen Receptor β and Vasculogenic Mimicry in Renal Cell Carcinoma

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
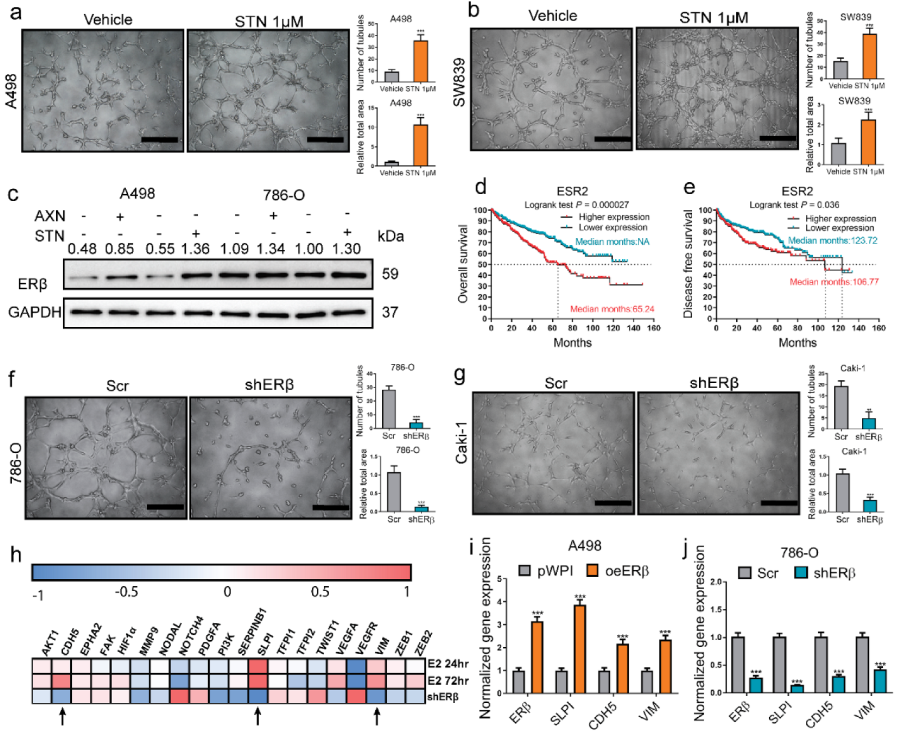
2.1. VM Formation in RCC Induced by Sunitinib Treatment via ERβ Up-Regulation Could Be Blocked by Silencing VE-Cadherin
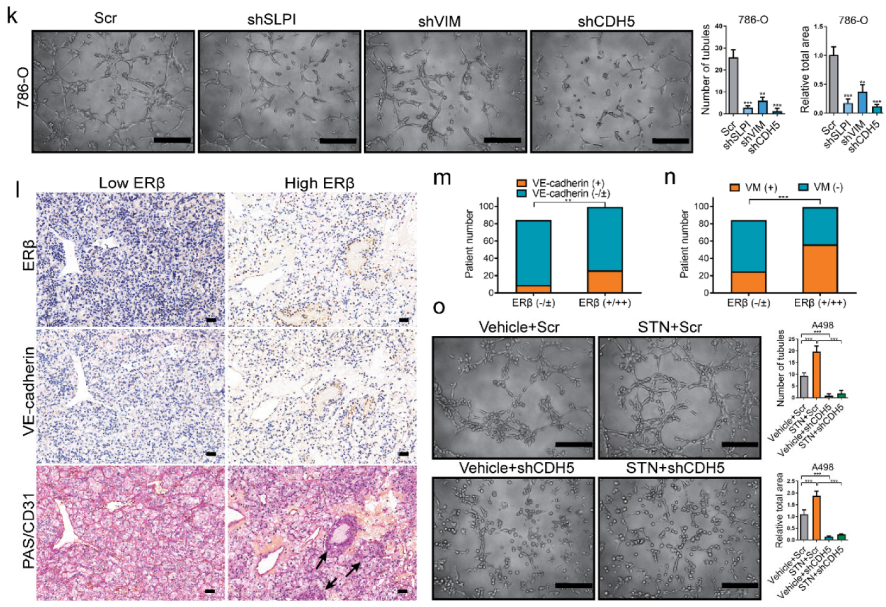
2.2. The miRNA-125-5p Is Involved in Mediating ERβ’s Modulation of VE-Cadherin
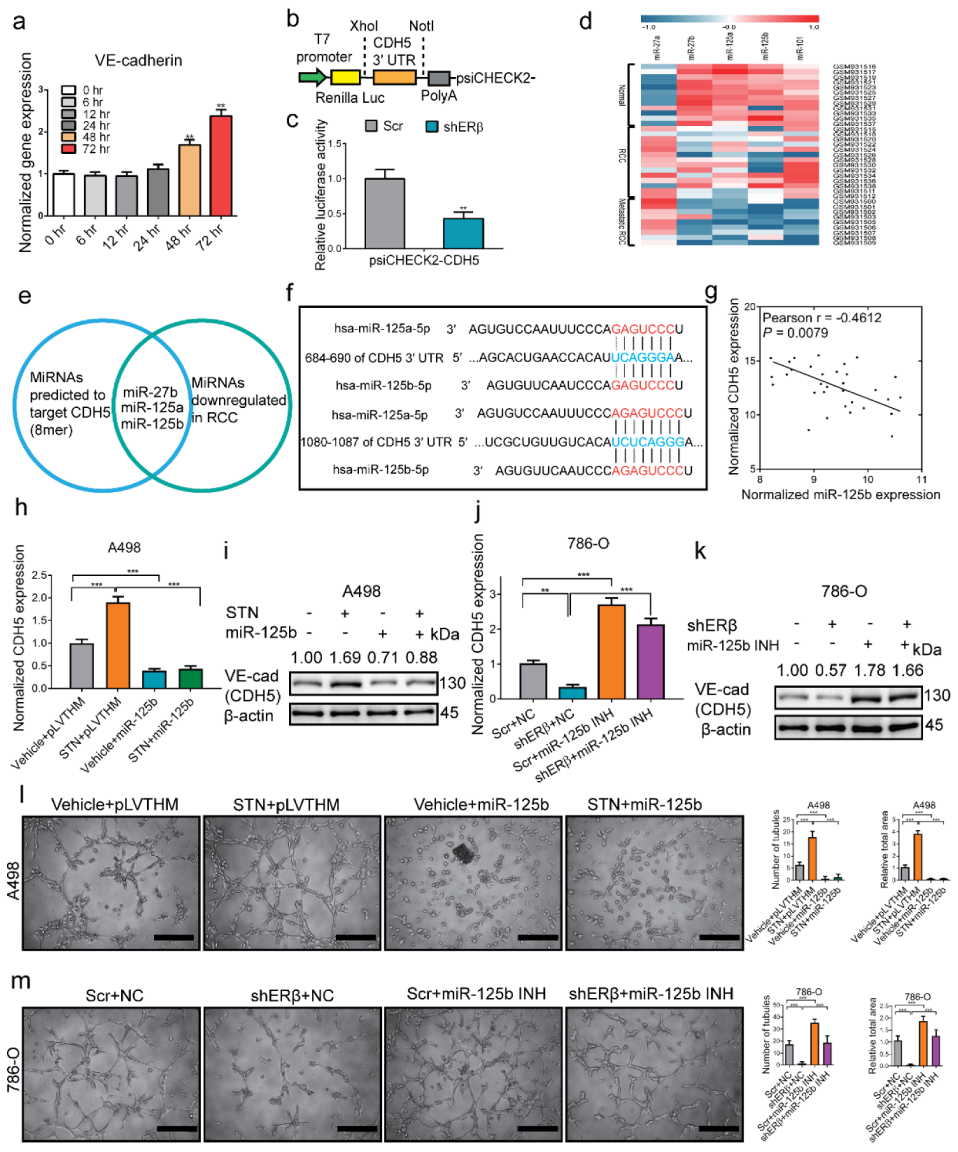
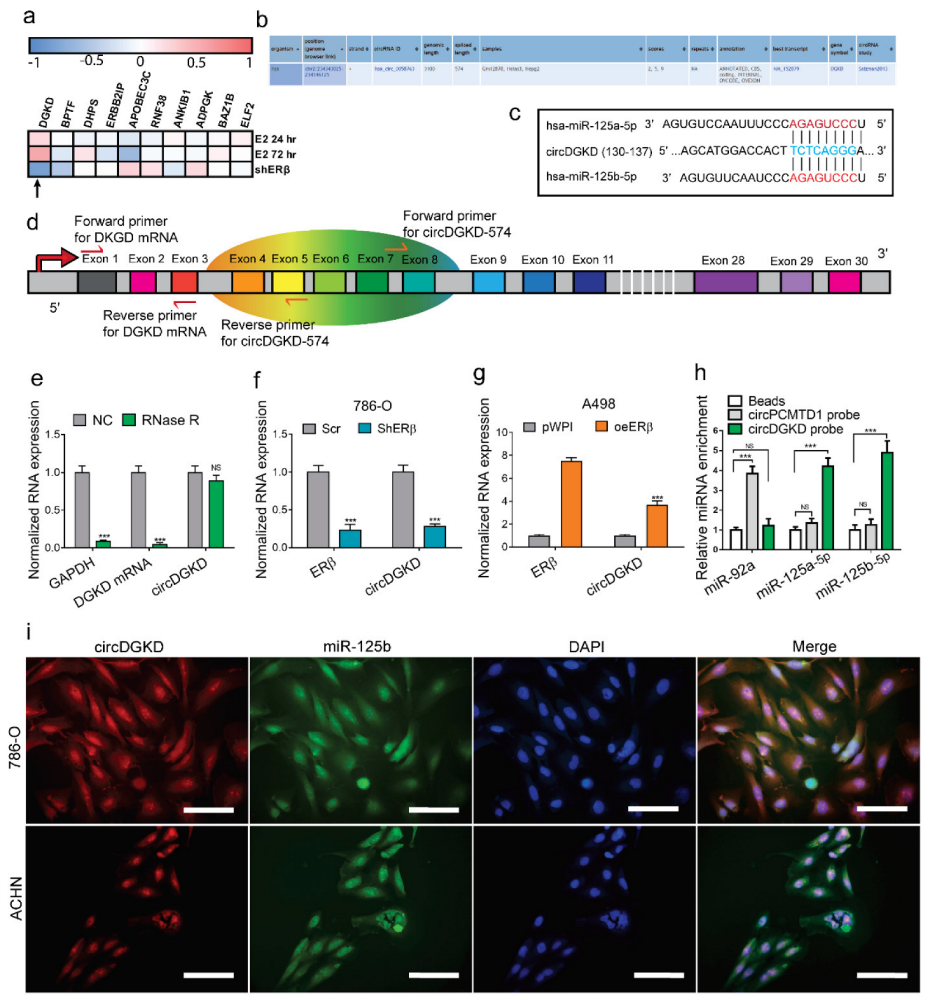
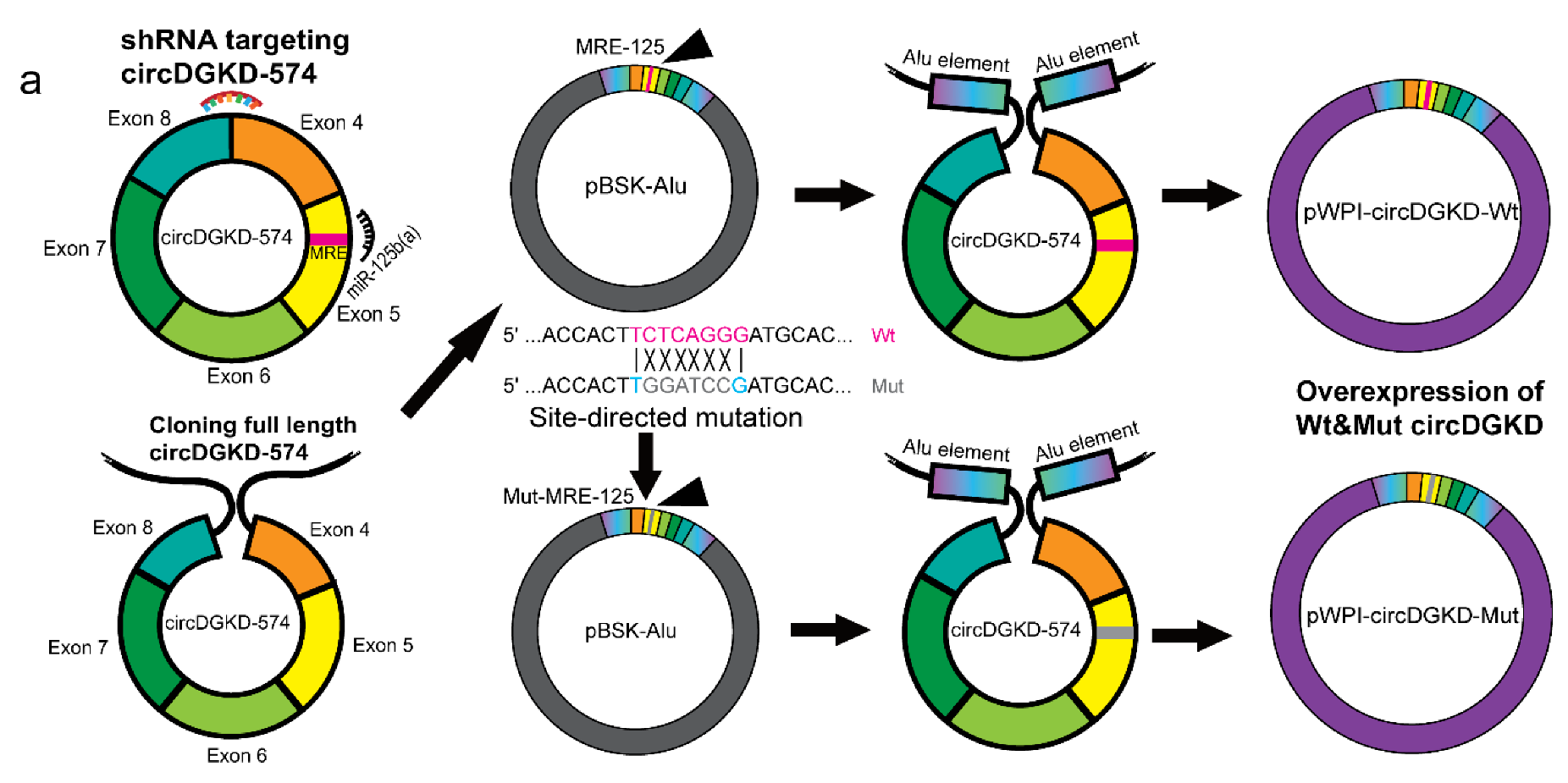
2.3. ERβ Increases circDGKD Which Sponges miRNA-125-5p in RCC
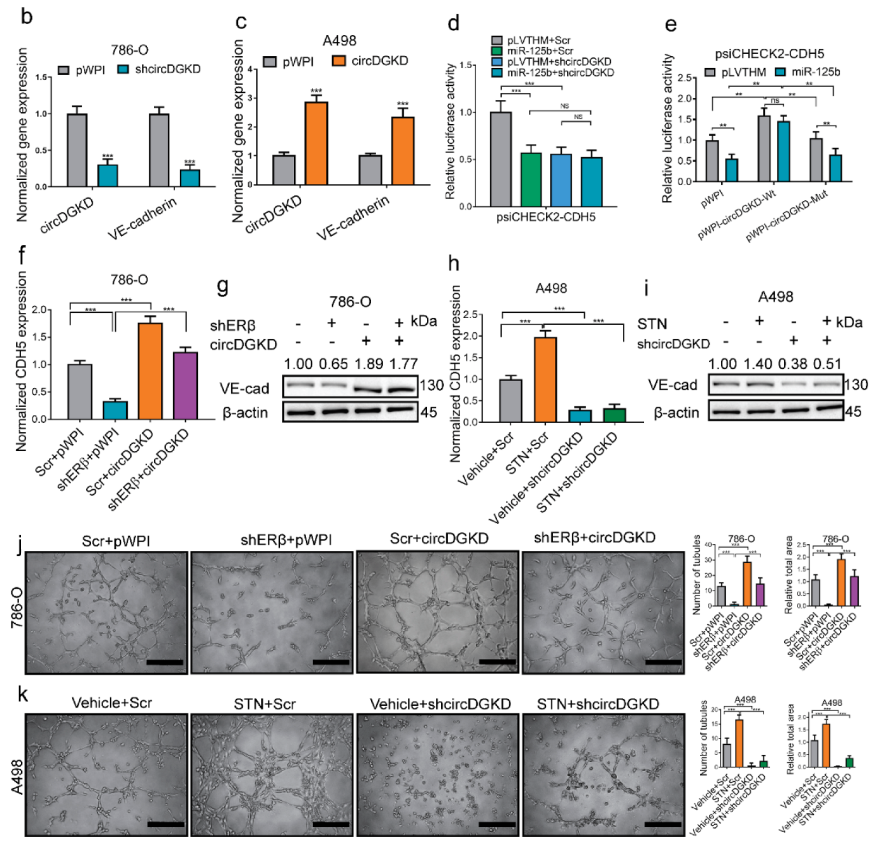
2.4. Knockdown of circDGKD Reverses ERβ’s Modulation of VE-Cadherin and VM Formation
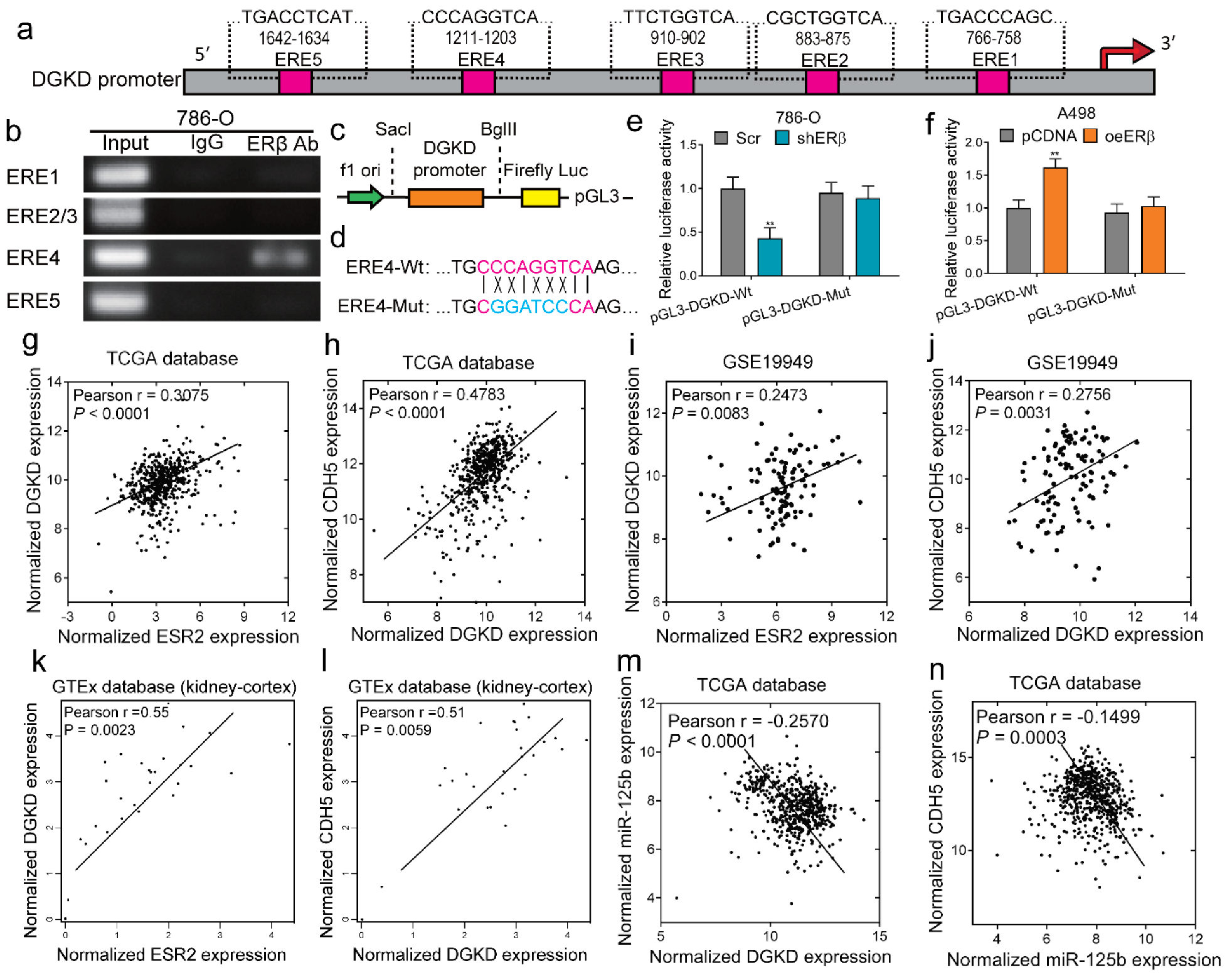
2.5. ERβ Can Bind to the Promoter of DGKD and Transcriptionally Regulates circDGKD Expression
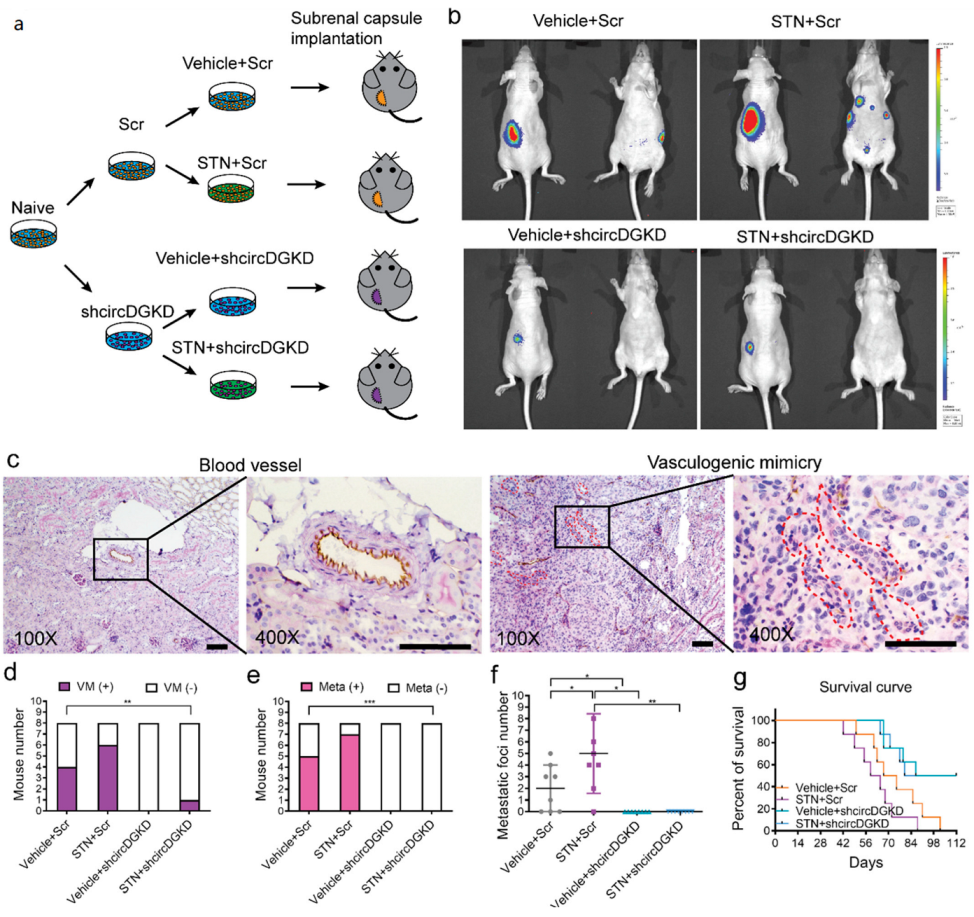
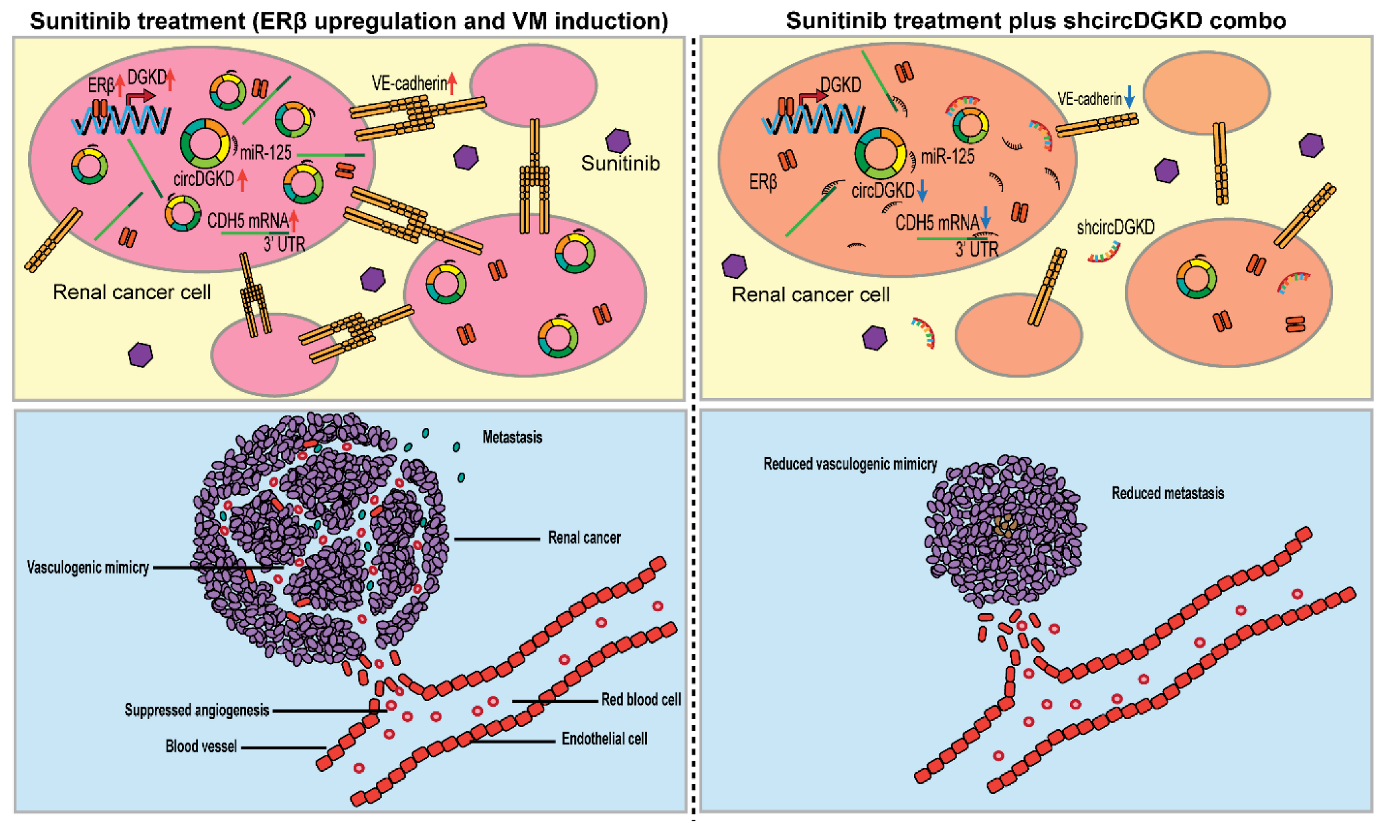
2.6. Orthotopic Mouse Model Studies Confirmed That shcircDGKD Intercepts Sunitinib-Pretreatment-Induced RCC VM Formation, Reduces Metastases, and Improves Survival
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Tube Formation Assay
4.3. RNA Extraction, Reverse Transcription, and Quantitative Real-Time PCR Analysis
4.4. Immunohistochemistry (IHC) and Florescence In Situ Hybridization (FISH)
4.5. Western Blot Analysis
4.6. Luciferase Reporter Gene Assay
4.7. Chromatin Immunoprecipitation Assay (ChIP)
4.8. RNA Construction and Transduction
4.9. Orthotopic Subrenal Capsule Implantation
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Chow, W.H.; Dong, L.M.; Devesa, S.S. Epidemiology and risk factors for kidney cancer. Nat. Rev. Urol. 2010, 7, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, A.; Solimando, A.G.; Kerscher, A.; Bittrich, M.; Kalogirou, C.; Kubler, H.; Rosenwald, A.; Bargou, R.; Kollmannsberger, P.; Schilling, B.; et al. Subgroup-independent mapping of renal cell carcinoma-machine learning reveals prognostic mitochondrial gene signature beyond histopathologic boundaries. Front. Oncol. 2021, 11, 621278. [Google Scholar] [CrossRef] [PubMed]
- Aziz, S.A.; Sznol, J.; Adeniran, A.; Colberg, J.W.; Camp, R.L.; Kluger, H.M. Vascularity of primary and metastatic renal cell carcinoma specimens. J. Transl. Med. 2013, 11, 15. [Google Scholar] [CrossRef] [Green Version]
- Méjean, A.; Ravaud, A.; Thezenas, S.; Colas, S.; Beauval, J.-B.; Bensalah, K.; Geoffrois, L.; Thiery-Vuillemin, A.; Cormier, L.; Lang, H. Sunitinib alone or after nephrectomy in metastatic renal-cell carcinoma. N. Engl. J. Med. 2018, 379, 417–427. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Cella, D.; Reeves, J.; Hawkins, R.; Guo, J.; Nathan, P.; Staehler, M.; de Souza, P.; Merchan, J.R.; et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N. Engl. J. Med. 2013, 369, 722–731. [Google Scholar] [CrossRef] [Green Version]
- Bedke, J.; Albiges, L.; Capitanio, U.; Giles, R.H.; Hora, M.; Lam, T.B.; Ljungberg, B.; Marconi, L.; Klatte, T.; Volpe, A.; et al. The 2021 updated European association of urology guidelines on renal cell carcinoma: Immune checkpoint inhibitor–based combination therapies for treatment-naive metastatic clear-cell renal cell carcinoma are standard of care. Eur. Urol. 2021, 80, 393–397. [Google Scholar] [CrossRef]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Dabrosin, C.; Yin, X.; Fuster, M.M.; Arreola, A.; Rathmell, W.K.; Generali, D.; Nagaraju, G.P.; El-Rayes, B.; Ribatti, D.; et al. Broad targeting of angiogenesis for cancer prevention and therapy. Semin. Cancer Biol. 2015, 35, S224–S243. [Google Scholar] [CrossRef]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef] [Green Version]
- Shirakawa, K.; Kobayashi, H.; Heike, Y.; Kawamoto, S.; Brechbiel, M.W.; Kasumi, F.; Iwanaga, T.; Konishi, F.; Terada, M.; Wakasugi, H. Hemodynamics in vasculogenic mimicry and angiogenesis of inflammatory breast cancer xenograft. Cancer Res. 2002, 62, 560–566. [Google Scholar] [PubMed]
- Meng, J.; Chen, S.; Lei, Y.-Y.; Han, J.-X.; Zhong, W.-L.; Wang, X.-R.; Liu, Y.-R.; Gao, W.-F.; Zhang, Q.; Tan, Q. Hsp90β promotes aggressive vasculogenic mimicry via epithelial–mesenchymal transition in hepatocellular carcinoma. Oncogene 2019, 38, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Baeten, C.I.; Hillen, F.; Pauwels, P.; de Bruine, A.P.; Baeten, C.G. Prognostic role of vasculogenic mimicry in colorectal cancer. Dis. Colon Rectum 2009, 52, 2028–2035. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Qie, S.; Zhang, S.; Sun, T.; Zhao, X.; Gao, S.; Ni, C.; Wang, X.; Liu, Y.; Zhang, L. Role and mechanism of vasculogenic mimicry in gastrointestinal stromal tumors. Hum. Pathol. 2008, 39, 444–451. [Google Scholar] [CrossRef]
- Li, M.; Gu, Y.; Zhang, Z.; Zhang, S.; Zhang, D.; Saleem, A.F.; Zhao, X.; Sun, B. Vasculogenic mimicry: A new prognostic sign of gastric adenocarcinoma. Pathol. Oncol. Res. POR 2010, 16, 259–266. [Google Scholar] [CrossRef]
- Sood, A.K.; Fletcher, M.S.; Zahn, C.M.; Gruman, L.M.; Coffin, J.E.; Seftor, E.A.; Hendrix, M.J. The clinical significance of tumor cell-lined vasculature in ovarian carcinoma: Implications for anti-vasculogenic therapy. Cancer Biol. Ther. 2002, 1, 661–664. [Google Scholar] [CrossRef] [Green Version]
- Yue, W.Y.; Chen, Z.P. Does vasculogenic mimicry exist in astrocytoma? J. Histochem. Cytochem. Off. J. Histochem. Soc. 2005, 53, 997–1002. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Zhang, S.; Zhao, X.; Zhang, W.; Hao, X. Vasculogenic mimicry is associated with poor survival in patients with mesothelial sarcomas and alveolar rhabdomyosarcomas. Int. J. Oncol. 2004, 25, 1609–1614. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, D.; Yao, Z.; Lin, X.; Liu, J.; Gu, Q.; Dong, X.; Liu, F.; Wang, Y.; Yao, N. Anti-angiogenic treatment promotes triple-negative breast cancer invasion via vasculogenic mimicry. Cancer Biol. Ther. 2017, 18, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Sun, B.; Zhao, X.; Ma, Y.; Ji, R.; Gu, Q.; Dong, X.; Li, J.; Liu, F.; Jia, X. Twist1 expression induced by sunitinib accelerates tumor cell vasculogenic mimicry by increasing the population of CD133+ cells in triple-negative breast cancer. Mol. Cancer 2014, 13, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribatti, D.; Annese, T.; Ruggieri, S.; Tamma, R.; Crivellato, E. Limitations of anti-angiogenic treatment of tumors. Transl. Oncol. 2019, 12, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Takeichi, M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science 1991, 251, 1451–1455. [Google Scholar] [CrossRef] [PubMed]
- Giannotta, M.; Trani, M.; Dejana, E. VE-cadherin and endothelial adherens junctions: Active guardians of vascular integrity. Dev. Cell 2013, 26, 441–454. [Google Scholar] [CrossRef] [Green Version]
- Hendrix, M.J.; Seftor, E.A.; Meltzer, P.S.; Gardner, L.M.; Hess, A.R.; Kirschmann, D.A.; Schatteman, G.C.; Seftor, R.E. Expression and functional significance of VE-cadherin in aggressive human melanoma cells: Role in vasculogenic mimicry. Proc. Natl. Acad. Sci. USA 2001, 98, 8018–8023. [Google Scholar] [CrossRef] [Green Version]
- Shirakawa, K.; Furuhata, S.; Watanabe, I.; Hayase, H.; Shimizu, A.; Ikarashi, Y.; Yoshida, T.; Terada, M.; Hashimoto, D.; Wakasugi, H. Induction of vasculogenesis in breast cancer models. Br. J. Cancer 2002, 87, 1454–1461. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.G.; Xue, X.Y.; Wang, L.; Zhang, X.; Yan, M.; Tu, Y.Y.; Lin, W.; Jiang, X.F.; Ren, H.G.; Zhang, W.; et al. CDH5 is specifically activated in glioblastoma stemlike cells and contributes to vasculogenic mimicry induced by hypoxia. Neuro-oncology 2013, 15, 865–879. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.Z.; Mei, J.; Qian, Z.K.; Cai, X.S.; Jiang, Y.; Huang, W.D. The role of VE-cadherin in osteosarcoma cells. Pathol. Oncol. Res. POR 2010, 16, 111–117. [Google Scholar] [CrossRef]
- Ding, J.; Yeh, C.R.; Sun, Y.; Lin, C.; Chou, J.; Ou, Z.; Chang, C.; Qi, J.; Yeh, S. Estrogen receptor beta promotes renal cell carcinoma progression via regulating LncRNA HOTAIR-miR-138/200c/204/217 associated CeRNA network. Oncogene 2018, 37, 5037–5053. [Google Scholar] [CrossRef]
- He, M.; Yang, H.; Shi, H.; Hu, Y.; Chang, C.; Liu, S.; Yeh, S. Sunitinib increases the cancer stem cells and vasculogenic mimicry formation via modulating the lncRNA-ECVSR/ERβ/Hif2-α signaling. Cancer Lett. 2022, 524, 15–28. [Google Scholar] [CrossRef]
- Pu, M.; Chen, J.; Tao, Z.; Miao, L.; Qi, X.; Wang, Y.; Ren, J. Regulatory network of miRNA on its target: Coordination between transcriptional and post-transcriptional regulation of gene expression. Cell. Mol. Life Sci. 2019, 76, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Klinge, C.M. miRNAs and estrogen action. Trends Endocrinol. Metab. TEM 2012, 23, 223–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piwecka, M.; Glažar, P.; Hernandez-Miranda, L.R.; Memczak, S.; Wolf, S.A.; Rybak-Wolf, A.; Filipchyk, A.; Klironomos, F.; Jara, C.A.C.; Fenske, P. Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science 2017, 357, eaam8526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontemaggi, G.; Turco, C.; Esposito, G.; Di Agostino, S. New molecular mechanisms and clinical impact of circRNAs in human cancer. Cancers 2021, 13, 3154. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhu, Q. Mechanisms regulating abnormal circular RNA biogenesis in cancer. Cancers 2021, 13, 4185. [Google Scholar] [CrossRef]
- Yang, J.P.; Liao, Y.D.; Mai, D.M.; Xie, P.; Qiang, Y.Y.; Zheng, L.S.; Wang, M.Y.; Mei, Y.; Meng, D.F.; Xu, L.; et al. Tumor vasculogenic mimicry predicts poor prognosis in cancer patients: A meta-analysis. Angiogenesis 2016, 19, 191–200. [Google Scholar] [CrossRef]
- Shirakawa, K.; Kobayashi, H.; Sobajima, J.; Hashimoto, D.; Shimizu, A.; Wakasugi, H. Inflammatory breast cancer: Vasculogenic mimicry and its hemodynamics of an inflammatory breast cancer xenograft model. Breast Cancer Res. BCR 2003, 5, 136–139. [Google Scholar] [CrossRef] [Green Version]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Qu, S.; Yang, X.; Li, X.; Wang, J.; Gao, Y.; Shang, R.; Sun, W.; Dou, K.; Li, H. Circular RNA: A new star of noncoding RNAs. Cancer Lett. 2015, 365, 141–148. [Google Scholar] [CrossRef]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Argentiero, A.; Solimando, A.G.; Krebs, M.; Leone, P.; Susca, N.; Brunetti, O.; Racanelli, V.; Vacca, A.; Silvestris, N. Anti-angiogenesis and immunotherapy: Novel paradigms to envision tailored approaches in renal cell-carcinoma. J. Clin. Med. 2020, 9, 1594. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulieres, D.; Melichar, B.; et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Jridi, I.; Catacchio, I.; Majdoub, H.; Shahbazzadeh, D.; El Ayeb, M.; Frassanito, M.A.; Solimando, A.G.; Ribatti, D.; Vacca, A.; Borchani, L. The small subunit of Hemilipin 2, a new heterodimeric phospholipase A2 from Hemiscorpius lepturus scorpion venom, mediates the antiangiogenic effect of the whole protein. Toxicon 2017, 126, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Rao, S.S.; Wang, Z.X.; Cao, J.; Tan, Y.J.; Luo, J.; Li, H.M.; Zhang, W.S.; Chen, C.Y.; Xie, H. Exosomes from human umbilical cord blood accelerate cutaneous wound healing through miR-21-3p-mediated promotion of angiogenesis and fibroblast function. Theranostics 2018, 8, 169–184. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, J.; Cui, X.-G.; Chen, H.-J.; Sun, Y.; Yu, W.-W.; Luo, J.; Xiao, G.-Q.; Chang, C.; Qi, J.; Yeh, S. Targeting circDGKD Intercepts TKI’s Effects on Up-Regulation of Estrogen Receptor β and Vasculogenic Mimicry in Renal Cell Carcinoma. Cancers 2022, 14, 1639. https://doi.org/10.3390/cancers14071639
Ding J, Cui X-G, Chen H-J, Sun Y, Yu W-W, Luo J, Xiao G-Q, Chang C, Qi J, Yeh S. Targeting circDGKD Intercepts TKI’s Effects on Up-Regulation of Estrogen Receptor β and Vasculogenic Mimicry in Renal Cell Carcinoma. Cancers. 2022; 14(7):1639. https://doi.org/10.3390/cancers14071639
Chicago/Turabian StyleDing, Jie, Xin-Gang Cui, Hao-Jie Chen, Yin Sun, Wei-Wei Yu, Jie Luo, Guang-Qian Xiao, Chawnshang Chang, Jun Qi, and Shuyuan Yeh. 2022. "Targeting circDGKD Intercepts TKI’s Effects on Up-Regulation of Estrogen Receptor β and Vasculogenic Mimicry in Renal Cell Carcinoma" Cancers 14, no. 7: 1639. https://doi.org/10.3390/cancers14071639
APA StyleDing, J., Cui, X.-G., Chen, H.-J., Sun, Y., Yu, W.-W., Luo, J., Xiao, G.-Q., Chang, C., Qi, J., & Yeh, S. (2022). Targeting circDGKD Intercepts TKI’s Effects on Up-Regulation of Estrogen Receptor β and Vasculogenic Mimicry in Renal Cell Carcinoma. Cancers, 14(7), 1639. https://doi.org/10.3390/cancers14071639