
E-Cadherin-Deficient Cells Are Sensitive to the Multikinase Inhibitor Dasatinib

 , , , and
, , , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Bioinformatics
2.1.1. Datasets and Processing
2.1.2. Differential Expression Analysis
2.1.3. Reactome Analysis
2.2. Cell Culture
2.3. Small Molecule Inhibitors and Chemicals
2.4. Nuclei Enumeration Assay
2.5. FACS
2.6. Organoid Culture
2.6.1. Mammary Organoid Generation
2.6.2. Gastric Organoid Generation
2.7. Drug Treatment of Organoids
2.8. Protein Extraction and Western Blotting
3. Results
3.1. Differential Gene Expression Analysis Results
3.1.1. AKT3 Expression Is Significantly Inversely Correlated with CDH1 Expression in Gastric Tumour Datasets
3.1.2. AKT3-Associated Genes Are Enriched for Extracellular Matrix Pathways
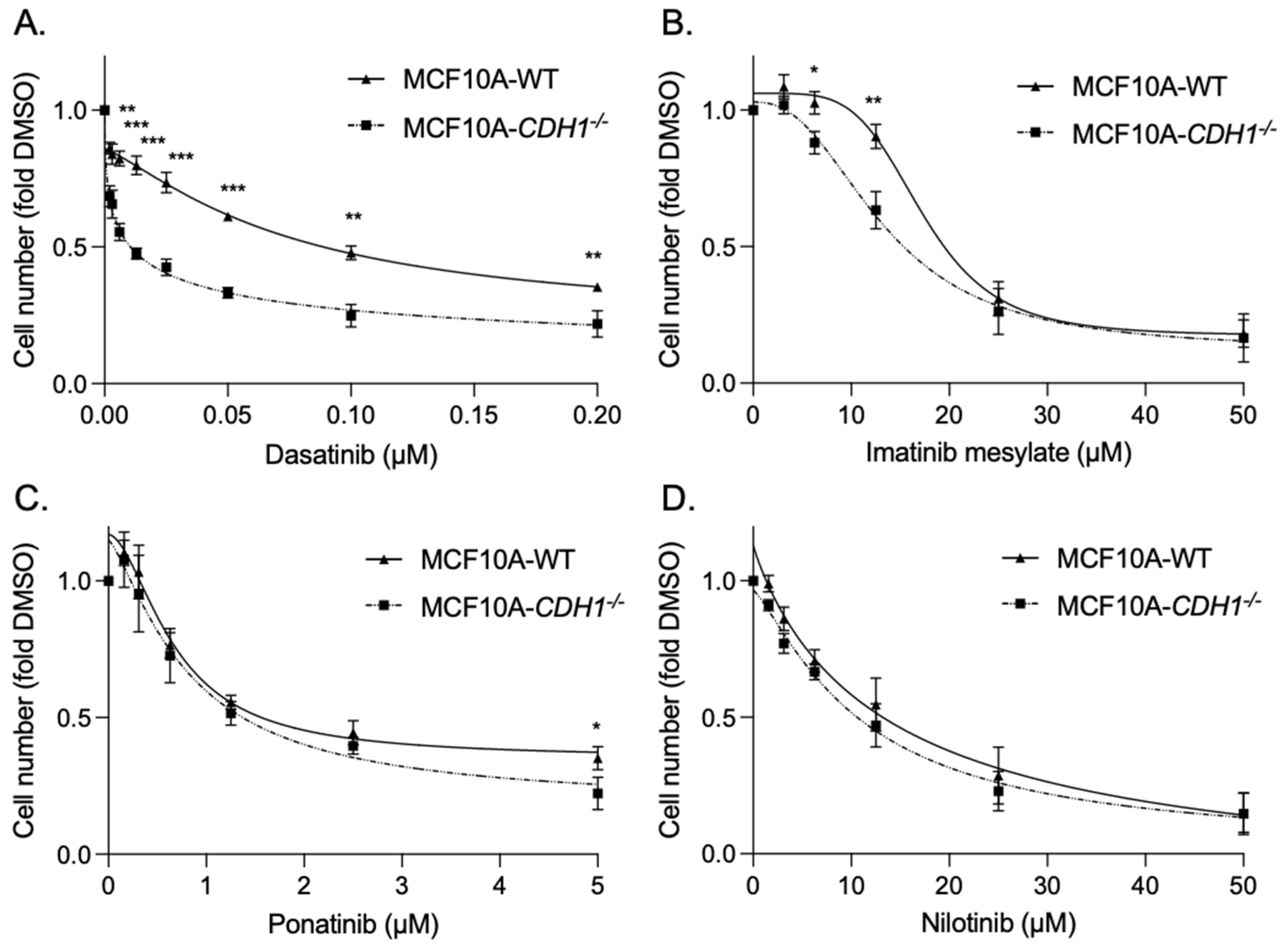
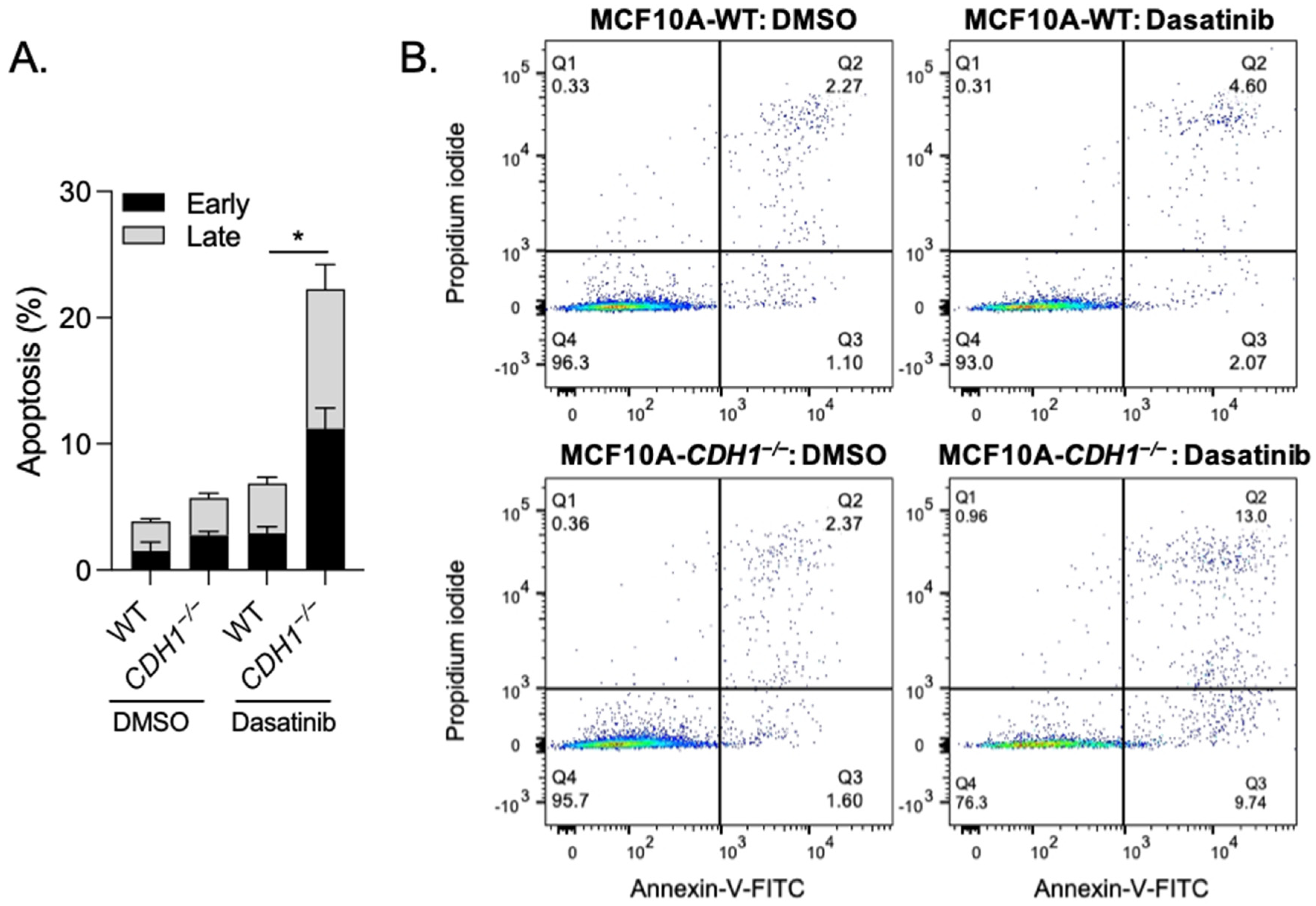
3.2. Breast Cells Lacking CDH1 Are More Sensitive to the Kinase Inhibitor Dasatinib Than Wild-Type Cells
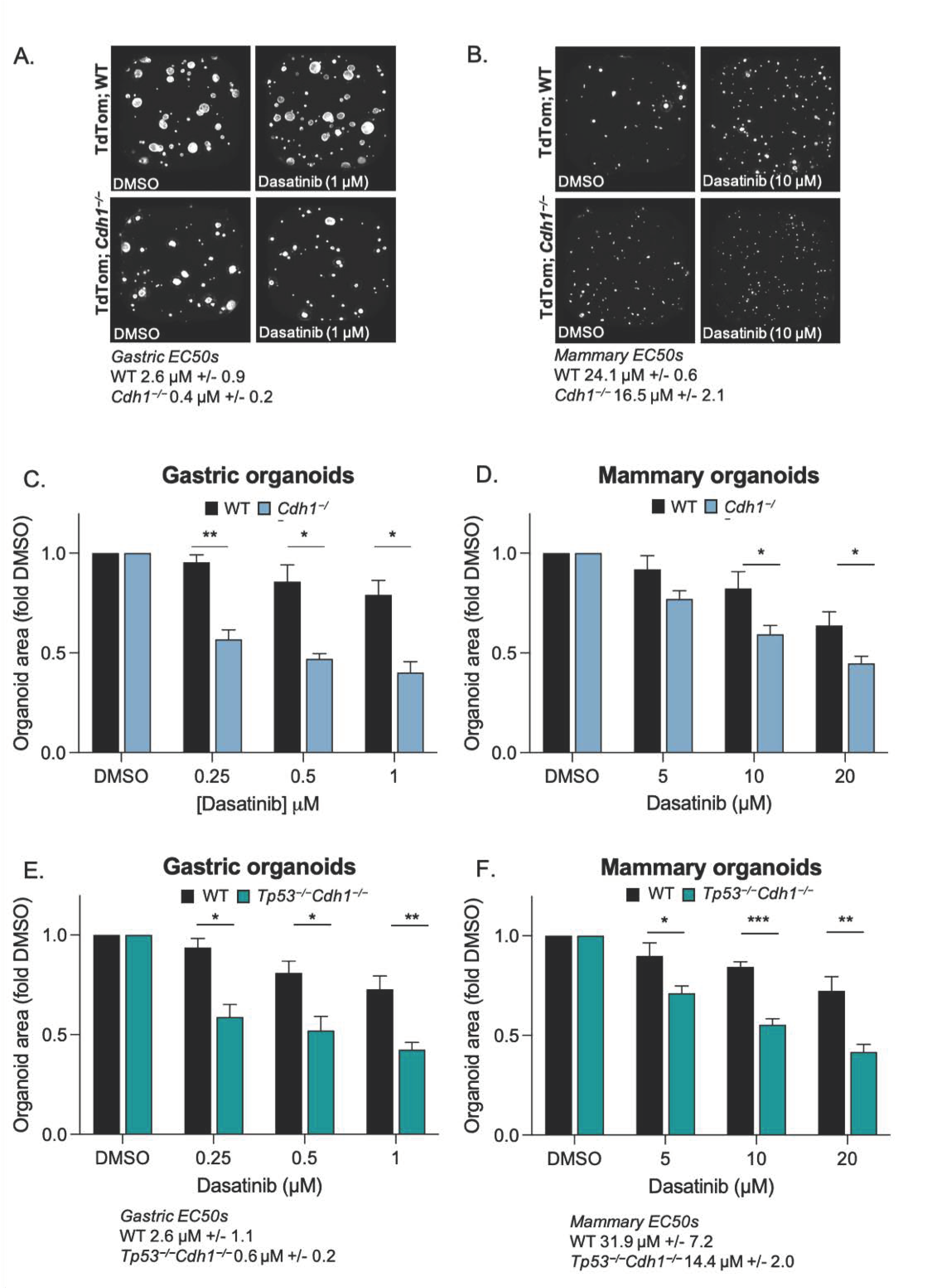
3.3. Conditional Knock-Out of Cdh1 Sensitises Gastric and Mammary Organoids to Dasatinib
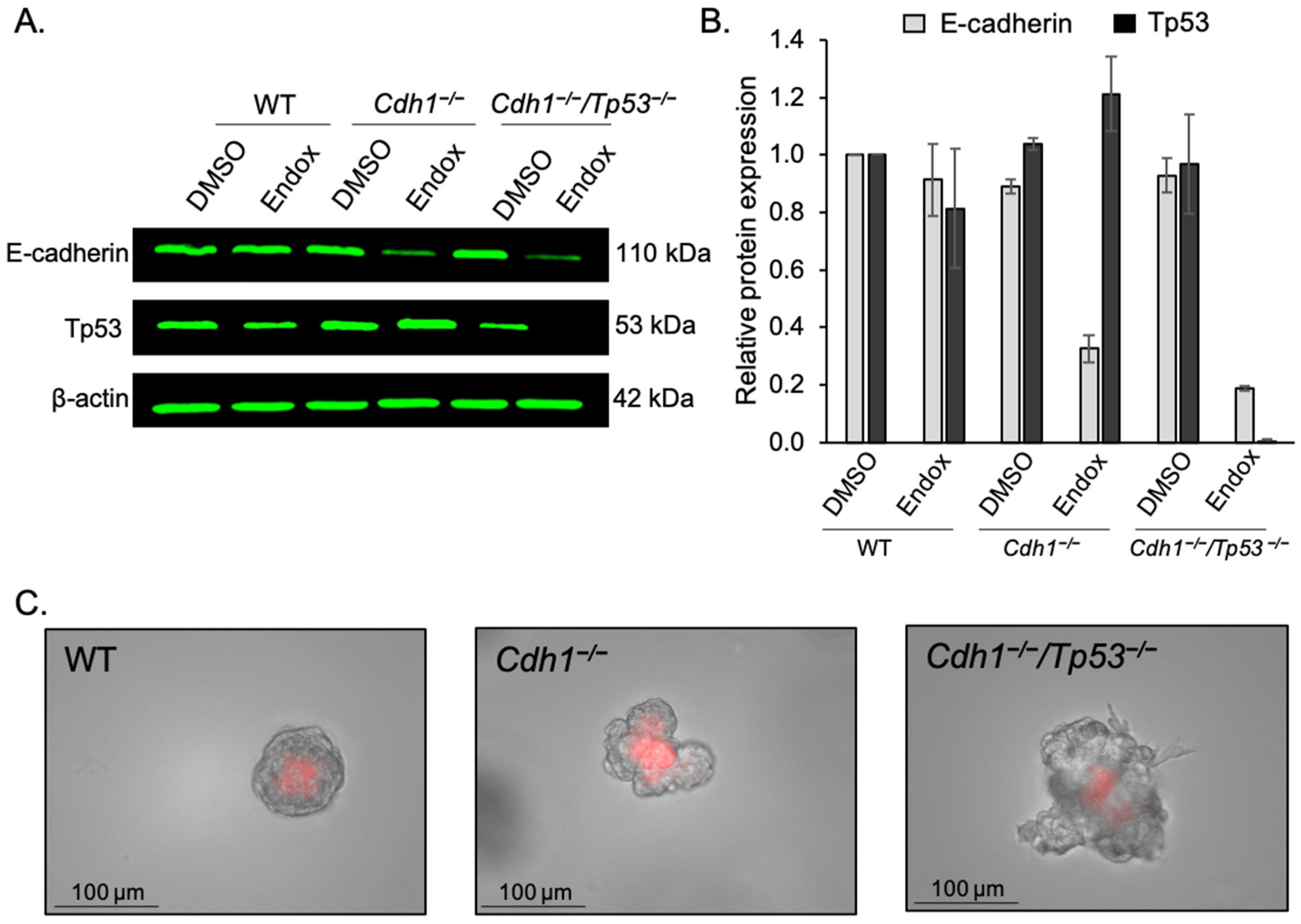
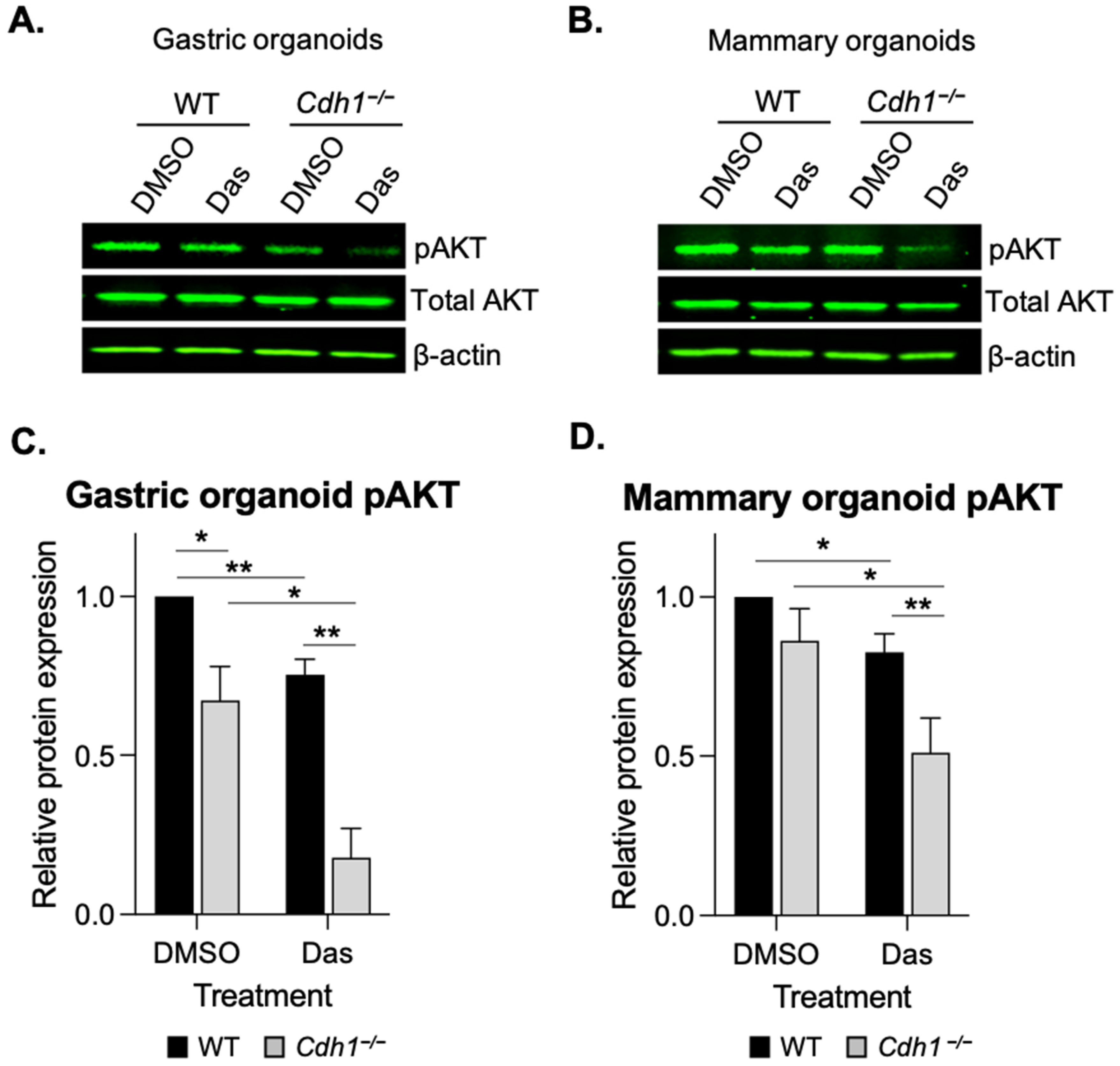
3.4. Dasatinib Inhibits AKT Phosphorylation in Cdh1-Deficient Organoids
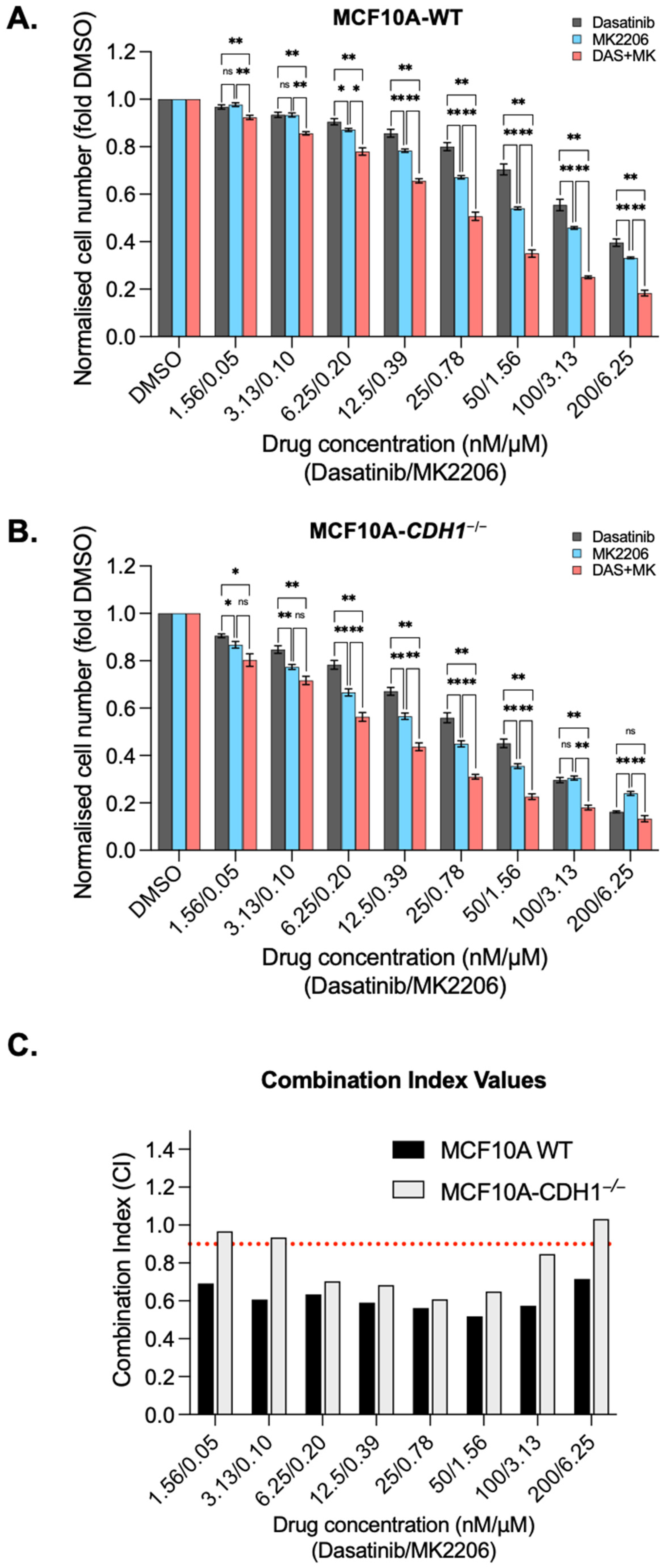
3.5. Dasatinib Is Synergistic with the Allosteric AKT Inhibitor MK2206 in MCF10A Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blair, V.R.; McLeod, M.; Carneiro, F.; Coit, D.G.; D’Addario, J.L.; van Dieren, J.M.; Harris, K.L.; Hoogerbrugge, N.; Oliveira, C.; van der Post, R.S.; et al. Hereditary Diffuse Gastric Cancer: Updated Clinical Practice Guidelines. Lancet Oncol. 2020, 21, e386–e397. [Google Scholar] [CrossRef]
- Guilford, P.; Hopkins, J.; Harraway, J.; McLeod, M.; McLeod, N.; Harawira, P.; Taite, H.; Scoular, R.; Miller, A.; Reeve, A.E. E-Cadherin Germline Mutations in Familial Gastric Cancer. Nature 1998, 392, 402–405. [Google Scholar] [CrossRef]
- Hansford, S.; Kaurah, P.; Li-Chang, H.; Woo, M.; Senz, J.; Pinheiro, H.; Schrader, K.A.; Schaeffer, D.F.; Shumansky, K.; Zogopoulos, G.; et al. Hereditary Diffuse Gastric Cancer Syndrome: CDH1 Mutations and Beyond. JAMA Oncol. 2015, 1, 23–32. [Google Scholar] [CrossRef]
- Xicola, R.M.; Li, S.; Rodriguez, N.; Reinecke, P.; Karam, R.; Speare, V.; Black, M.H.; LaDuca, H.; Llor, X. Clinical Features and Cancer Risk in Families with Pathogenic CDH1 Variants Irrespective of Clinical Criteria. J. Med. Genet. 2019, 56, 838–843. [Google Scholar] [CrossRef]
- Roberts, M.E.; Ranola, J.M.O.; Marshall, M.L.; Susswein, L.R.; Graceffo, S.; Bohnert, K.; Tsai, G.; Klein, R.T.; Hruska, K.S.; Shirts, B.H. Comparison of CDH1 Penetrance Estimates in Clinically Ascertained Families vs Families Ascertained for Multiple Gastric Cancers. JAMA Oncol. 2019, 5, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- Pharoah, P.D.P.; Guilford, P.; Caldas, C. Incidence of Gastric Cancer and Breast Cancer in CDH1 (E-Cadherin) Mutation Carriers from Hereditary Diffuse Gastric Cancer Families. Gastroenterology 2001, 121, 1348–1353. [Google Scholar] [CrossRef]
- Becker, K.F.; Atkinson, M.J.; Reich, U.; Becker, I.; Nekarda, H.; Siewert, J.R.; Höfler, H. E-Cadherin Gene Mutations Provide Clues to Diffuse Type Gastric Carcinomas. Cancer Res. 1994, 54, 3845–3852. [Google Scholar] [PubMed]
- Vos, C.B.; Cleton-Jansen, A.M.; Berx, G.; de Leeuw, W.J.; ter Haar, N.T.; van Roy, F.; Cornelisse, C.J.; Peterse, J.L.; van de Vijver, M.J. E-Cadherin Inactivation in Lobular Carcinoma in Situ of the Breast: An Early Event in Tumorigenesis. Br. J. Cancer 1997, 76, 1131–1133. [Google Scholar] [CrossRef]
- Barroso-Sousa, R.; Metzger-Filho, O. Differences between Invasive Lobular and Invasive Ductal Carcinoma of the Breast: Results and Therapeutic Implications. Ther. Adv. Med. Oncol. 2016, 8, 261–266. [Google Scholar] [CrossRef]
- Choi, I.J.; Kim, C.G.; Lee, J.Y.; Kim, Y.-I.; Kook, M.-C.; Park, B.; Joo, J. Family History of Gastric Cancer and Helicobacter Pylori Treatment. N. Engl. J. Med. 2020, 382, 427–436. [Google Scholar] [CrossRef]
- King, T.A.; Pilewskie, M.; Muhsen, S.; Patil, S.; Mautner, S.K.; Park, A.; Oskar, S.; Guerini-Rocco, E.; Boafo, C.; Gooch, J.C.; et al. Lobular Carcinoma in Situ: A 29-Year Longitudinal Experience Evaluating Clinicopathologic Features and Breast Cancer Risk. J. Clin. Oncol. 2015, 33, 3945–3952. [Google Scholar] [CrossRef] [PubMed]
- van Nes, J.G.H.; Fontein, D.B.Y.; Hille, E.T.M.; Voskuil, D.W.; van Leeuwen, F.E.; de Haes, J.C.J.M.; Putter, H.; Seynaeve, C.; Nortier, J.W.R.; van de Velde, C.J.H. Quality of Life in Relation to Tamoxifen or Exemestane Treatment in Postmenopausal Breast Cancer Patients: A Tamoxifen Exemestane Adjuvant Multinational (TEAM) Trial Side Study. Breast Cancer Res. Treat. 2012, 134, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Wills, S.M.; Zekman, R.; Bestul, D.; Kuwajerwala, N.; Decker, D. Tamoxifen Malabsorption after Roux-En-Y Gastric Bypass Surgery: Case Series and Review of the Literature. Pharmacotherapy 2010, 30, 217. [Google Scholar] [CrossRef]
- He, W.; Eriksson, M.; Eliasson, E.; Grassmann, F.; Bäcklund, M.; Gabrielson, M.; Hammarström, M.; Margolin, S.; Thorén, L.; Wengström, Y.; et al. CYP2D6 Genotype Predicts Tamoxifen Discontinuation and Drug Response: A Secondary Analysis of the KARISMA Trial. Ann. Oncol. 2021, 32, 1286–1293. [Google Scholar] [CrossRef] [PubMed]
- Al-Batran, S.-E.; Homann, N.; Pauligk, C.; Goetze, T.O.; Meiler, J.; Kasper, S.; Kopp, H.-G.; Mayer, F.; Haag, G.M.; Luley, K.; et al. Perioperative Chemotherapy with Fluorouracil plus Leucovorin, Oxaliplatin, and Docetaxel versus Fluorouracil or Capecitabine plus Cisplatin and Epirubicin for Locally Advanced, Resectable Gastric or Gastro-Oesophageal Junction Adenocarcinoma (FLOT4): A Randomised, Phase 2/3 Trial. Lancet 2019, 393, 1948–1957. [Google Scholar] [CrossRef]
- Bittoni, A.; Scartozzi, M.; Giampieri, R.; Faloppi, L.; Bianconi, M.; Mandolesi, A.; Del Prete, M.; Pistelli, M.; Cecchini, L.; Bearzi, I.; et al. Clinical Evidence for Three Distinct Gastric Cancer Subtypes: Time for a New Approach. PLoS ONE 2013, 8, e78544. [Google Scholar] [CrossRef]
- Park, S.H.; Sohn, T.S.; Lee, J.; Lim, D.H.; Hong, M.E.; Kim, K.-M.; Sohn, I.; Jung, S.H.; Choi, M.G.; Lee, J.H.; et al. Phase III Trial to Compare Adjuvant Chemotherapy With Capecitabine and Cisplatin Versus Concurrent Chemoradiotherapy in Gastric Cancer: Final Report of the Adjuvant Chemoradiotherapy in Stomach Tumors Trial, Including Survival and Subset Analyses. J. Clin. Oncol. 2015, 33, 3130–3136. [Google Scholar] [CrossRef]
- Schulz, C.; Kullmann, F.; Kunzmann, V.; Fuchs, M.; Geissler, M.; Vehling-Kaiser, U.; Stauder, H.; Wein, A.; Al-Batran, S.-E.; Kubin, T.; et al. NeoFLOT: Multicenter Phase II Study of Perioperative Chemotherapy in Resectable Adenocarcinoma of the Gastroesophageal Junction or Gastric Adenocarcinoma-Very Good Response Predominantly in Patients with Intestinal Type Tumors. Int. J. Cancer 2015, 137, 678–685. [Google Scholar] [CrossRef]
- Dessauvagie, B.; Thomas, A.; Thomas, C.; Robinson, C.; Combrink, M.; Budhavaram, V.; Kunjuraman, B.; Meehan, K.; Sterrett, G.; Harvey, J. Invasive Lobular Carcinoma of the Breast: Assessment of Proliferative Activity Using Automated Ki-67 Immunostaining. Pathology 2019, 51, 681–687. [Google Scholar] [CrossRef]
- Wilson, N.; Ironside, A.; Diana, A.; Oikonomidou, O. Lobular Breast Cancer: A Review. Front. Oncol. 2021, 10, 591399. [Google Scholar] [CrossRef]
- Delpech, Y.; Coutant, C.; Hsu, L.; Barranger, E.; Iwamoto, T.; Barcenas, C.H.; Hortobagyi, G.N.; Rouzier, R.; Esteva, F.J.; Pusztai, L. Clinical Benefit from Neoadjuvant Chemotherapy in Oestrogen Receptor-Positive Invasive Ductal and Lobular Carcinomas. Br. J. Cancer 2013, 108, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Metzger Filho, O.; Giobbie-Hurder, A.; Mallon, E.; Gusterson, B.; Viale, G.; Winer, E.P.; Thürlimann, B.; Gelber, R.D.; Colleoni, M.; Ejlertsen, B.; et al. Relative Effectiveness of Letrozole Compared With Tamoxifen for Patients With Lobular Carcinoma in the BIG 1-98 Trial. J. Clin. Oncol. 2015, 33, 2772–2779. [Google Scholar] [CrossRef] [PubMed]
- Brew, T.; Bougen-Zhukov, N.; Mitchell, W.; Decourtye, L.; Schulpen, E.; Nouri, Y.; Godwin, T.; Guilford, P. Loss of E-Cadherin Leads to Druggable Vulnerabilities in Sphingolipid Metabolism and Vesicle Trafficking. Cancers 2022, 14, 102. [Google Scholar] [CrossRef]
- Decourtye-Espiard, L.; Bougen-Zhukov, N.; Godwin, T.; Brew, T.; Schulpen, E.; Black, M.A.; Guilford, P. E-Cadherin-Deficient Epithelial Cells Are Sensitive to HDAC Inhibitors. Cancers 2022, 14, 175. [Google Scholar] [CrossRef]
- Bougen-Zhukov, N.; Nouri, Y.; Godwin, T.; Taylor, M.; Hakkaart, C.; Single, A.; Brew, T.; Permina, E.; Chen, A.; Black, M.A.; et al. Allosteric AKT Inhibitors Target Synthetic Lethal Vulnerabilities in E-Cadherin-Deficient Cells. Cancers 2019, 11, 1359. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, K.; Thompson, D.A.; Barthel, A.; Sakaue, H.; Liu, W.; Weigel, R.J.; Roth, R.A. Up-Regulation of Akt3 in Estrogen Receptor-Deficient Breast Cancers and Androgen-Independent Prostate Cancer Lines. J. Biol. Chem. 1999, 274, 21528–21532. [Google Scholar] [CrossRef]
- Hinz, N.; Jücker, M. Distinct Functions of AKT Isoforms in Breast Cancer: A Comprehensive Review. Cell Commun. Signal. 2019, 17, 154. [Google Scholar] [CrossRef] [PubMed]
- Buikhuisen, J.Y.; Gomez Barila, P.M.; Torang, A.; Dekker, D.; de Jong, J.H.; Cameron, K.; Vitale, S.; Stassi, G.; van Hooff, S.R.; Castro, M.A.A.; et al. AKT3 Expression in Mesenchymal Colorectal Cancer Cells Drives Growth and Is Associated with Epithelial-Mesenchymal Transition. Cancers 2021, 13, 801. [Google Scholar] [CrossRef]
- Lv, J.; Zhang, S.; Liu, Y.; Li, C.; Guo, T.; Zhang, S.; Li, Z.; Jiao, Z.; Sun, H.; Zhang, Y.; et al. NR2F1-AS1/MiR-190a/PHLDB2 Induces the Epithelial–Mesenchymal Transformation Process in Gastric Cancer by Promoting Phosphorylation of AKT3. Front. Cell Dev. Biol. 2021, 9, 688949. [Google Scholar] [CrossRef]
- Chin, Y.R.; Yoshida, T.; Marusyk, A.; Beck, A.H.; Polyak, K.; Toker, A. Targeting Akt3 Signaling in Triple-Negative Breast Cancer. Cancer Res. 2014, 74, 964–973. [Google Scholar] [CrossRef]
- Shrivastava, A.; Radziejewski, C.; Campbell, E.; Kovac, L.; McGlynn, M.; Ryan, T.E.; Davis, S.; Goldfarb, M.P.; Glass, D.J.; Lemke, G.; et al. An Orphan Receptor Tyrosine Kinase Family Whose Members Serve as Nonintegrin Collagen Receptors. Mol. Cell 1997, 1, 25–34. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive Molecular Characterization of Gastric Adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Lee, J.; Nebozhyn, M.; Kim, K.-M.; Ting, J.C.; Wong, S.S.; Liu, J.; Yue, Y.G.; Wang, J.; Yu, K.; et al. Molecular Analysis of Gastric Cancer Identifies Subtypes Associated with Distinct Clinical Outcomes. Nat. Med. 2015, 21, 449–456. [Google Scholar] [CrossRef]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision Weights Unlock Linear Model Analysis Tools for RNA-Seq Read Counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A Comparison of Normalization Methods for High Density Oligonucleotide Array Data Based on Variance and Bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene Ontology Analysis for RNA-Seq: Accounting for Selection Bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Single, A.; Beetham, H.; Telford, B.J.; Guilford, P.; Chen, A. A Comparison of Real-Time and Endpoint Cell Viability Assays for Improved Synthetic Lethal Drug Validation. J. Biomol. Screen. 2015, 20, 1286–1293. [Google Scholar] [CrossRef] [PubMed]
- Kühn, R.; Torres, R.M. Cre/LoxP Recombination System and Gene Targeting. Methods Mol. Biol. 2002, 180, 175–204. [Google Scholar] [CrossRef]
- Ewald, A.J. Isolation of Mouse Mammary Organoids for Long-Term Time-Lapse Imaging. Cold Spring Harb. Protoc. 2013, 2013, 130–133. [Google Scholar] [CrossRef]
- Luxenburger, A.; Bougen-Zhukov, N.; Fraser, M.G.; Beetham, H.; Harris, L.D.; Schmidt, D.; Cameron, S.A.; Guilford, P.J.; Evans, G.B. Discovery of AL-GDa62 as a Potential Synthetic Lethal Lead for the Treatment of Gastric Cancer. J. Med. Chem. 2021, 64, 18114–18142. [Google Scholar] [CrossRef]
- Leitinger, B. Discoidin Domain Receptor Functions in Physiological and Pathological Conditions. Int. Rev. Cell Mol. Biol. 2014, 310, 39–87. [Google Scholar] [CrossRef] [PubMed]
- Kurashige, J.; Hasegawa, T.; Niida, A.; Sugimachi, K.; Deng, N.; Mima, K.; Uchi, R.; Sawada, G.; Takahashi, Y.; Eguchi, H.; et al. Integrated Molecular Profiling of Human Gastric Cancer Identifies DDR2 as a Potential Regulator of Peritoneal Dissemination. Sci. Rep. 2016, 6, 22371. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-G.; Xu, L.; Jia, R.-R.; Wu, Q.; Wang, T.; Wei, J.; Ma, J.-L.; Shi, M.; Li, Z.-S. DDR2 Induces Gastric Cancer Cell Activities via Activating MTORC2 Signaling and Is Associated with Clinicopathological Characteristics of Gastric Cancer. Dig. Dis. Sci. 2016, 61, 2272–2283. [Google Scholar] [CrossRef]
- Chen, P.-F.; Wang, F.; Nie, J.-Y.; Feng, J.-R.; Liu, J.; Zhou, R.; Wang, H.-L.; Zhao, Q. Co-Expression Network Analysis Identified CDH11 in Association with Progression and Prognosis in Gastric Cancer. OncoTargets Ther. 2018, 11, 6425–6436. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zang, D.; Cheng, Y.; Li, Z.; Yang, B.; Guo, T.; Liu, Y.; Qu, X.; Che, X. Identification of Key Gene and Pathways for the Prediction of Peritoneal Metastasis of Gastric Cancer by Co-Expression Analysis. J. Cancer 2020, 11, 3041–3051. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Ding, Y.; Wu, N.; Jiang, J.; Huang, Y.; Zhang, F.; Wang, H.; Zhou, Q.; Yang, Y.; Zhuo, W.; et al. FSTL1 Promotes Growth and Metastasis in Gastric Cancer by Activating AKT Related Pathway and Predicts Poor Survival. Am. J. Cancer Res. 2021, 11, 712–728. [Google Scholar] [PubMed]
- Chen, A.; Beetham, H.; Black, M.A.; Priya, R.; Telford, B.J.; Guest, J.; Wiggins, G.A.R.; Godwin, T.D.; Yap, A.S.; Guilford, P.J. E-Cadherin Loss Alters Cytoskeletal Organization and Adhesion in Non-Malignant Breast Cells but Is Insufficient to Induce an Epithelial-Mesenchymal Transition. BMC Cancer 2014, 14, 552. [Google Scholar] [CrossRef]
- Rix, U.; Hantschel, O.; Dürnberger, G.; Remsing Rix, L.L.; Planyavsky, M.; Fernbach, N.V.; Kaupe, I.; Bennett, K.L.; Valent, P.; Colinge, J.; et al. Chemical Proteomic Profiles of the BCR-ABL Inhibitors Imatinib, Nilotinib, and Dasatinib Reveal Novel Kinase and Nonkinase Targets. Blood 2007, 110, 4055–4063. [Google Scholar] [CrossRef]
- Day, E.; Waters, B.; Spiegel, K.; Alnadaf, T.; Manley, P.W.; Buchdunger, E.; Walker, C.; Jarai, G. Inhibition of Collagen-Induced Discoidin Domain Receptor 1 and 2 Activation by Imatinib, Nilotinib and Dasatinib. Eur. J. Pharmacol. 2008, 599, 44–53. [Google Scholar] [CrossRef]
- Canning, P.; Tan, L.; Chu, K.; Lee, S.W.; Gray, N.S.; Bullock, A.N. Structural Mechanisms Determining Inhibition of the Collagen Receptor DDR1 by Selective and Multi-Targeted Type II Kinase Inhibitors. J. Mol. Biol. 2014, 426, 2457–2470. [Google Scholar] [CrossRef] [PubMed]
- Grither, W.R.; Longmore, G.D. Inhibition of Tumor–Microenvironment Interaction and Tumor Invasion by Small-Molecule Allosteric Inhibitor of DDR2 Extracellular Domain. Proc. Natl. Acad. Sci. USA 2018, 115, E7786–E7794. [Google Scholar] [CrossRef] [PubMed]
- Godwin, T.D.; Kelly, S.T.; Brew, T.P.; Bougen-Zhukov, N.M.; Single, A.B.; Chen, A.; Stylianou, C.E.; Harris, L.D.; Currie, S.K.; Telford, B.J.; et al. E-Cadherin-Deficient Cells Have Synthetic Lethal Vulnerabilities in Plasma Membrane Organisation, Dynamics and Function. Gastric Cancer 2018, 22, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Kataoka, S.; Nakayama, M.; Ali, M.A.E.; Oshima, H.; Yamamoto, D.; Park, J.-W.; Takegami, Y.; An, T.; Jenkins, N.A.; et al. CRISPR-Cas9–Mediated Gene Knockout in Intestinal Tumor Organoids Provides Functional Validation for Colorectal Cancer Driver Genes. Proc. Natl. Acad. Sci. USA 2019, 116, 15635–15644. [Google Scholar] [CrossRef]
- Ranzani, G.N.; Luinetti, O.; Padovan, L.S.; Calistri, D.; Renault, B.; Burrel, M.; Amadori, D.; Fiocca, R.; Solcia, E. P53 Gene Mutations and Protein Nuclear Accumulation Are Early Events in Intestinal Type Gastric Cancer but Late Events in Diffuse Type. Cancer Epidemiol. Biomark. Prev. 1995, 4, 223–231. [Google Scholar]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef]
- McCart Reed, A.E.; Foong, S.; Kutasovic, J.R.; Nones, K.; Waddell, N.; Lakhani, S.R.; Simpson, P.T. The Genomic Landscape of Lobular Breast Cancer. Cancers 2021, 13, 1950. [Google Scholar] [CrossRef]
- Chou, T.-C. Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Valiathan, R.R.; Marco, M.; Leitinger, B.; Kleer, C.G.; Fridman, R. Discoidin domain receptor tyrosine kinases: New players in cancer progression. Cancer Metastasis Rev. 2012, 31, 295–321. [Google Scholar] [CrossRef]
- Liang, Y.; Xia, W.; Zhang, T.; Chen, B.; Wang, H.; Song, X.; Zhang, Z.; Xu, L.; Dong, G.; Jiang, F. Upregulated Collagen COL10A1 Remodels the Extracellular Matrix and Promotes Malignant Progression in Lung Adenocarcinoma. Front. Oncol. 2020, 10, 573534. [Google Scholar] [CrossRef]
- Chen, L.; Kong, X.; Fang, Y.; Paunikar, S.; Wang, X.; Brown, J.A.L.; Bourke, E.; Li, X.; Wang, J. Recent Advances in the Role of Discoidin Domain Receptor Tyrosine Kinase 1 and Discoidin Domain Receptor Tyrosine Kinase 2 in Breast and Ovarian Cancer. Front. Cell Dev. Biol. 2021, 9, 47314. [Google Scholar] [CrossRef] [PubMed]
- Alves, F.; Vogel, W.; Mossie, K.; Millauer, B.; Höfler, H.; Ullrich, A. Distinct Structural Characteristics of Discoidin I Subfamily Receptor Tyrosine Kinases and Complementary Expression in Human Cancer. Oncogene 1995, 10, 609–618. [Google Scholar] [PubMed]
- Taube, J.H.; Herschkowitz, J.I.; Komurov, K.; Zhou, A.Y.; Gupta, S.; Yang, J.; Hartwell, K.; Onder, T.T.; Gupta, P.B.; Evans, K.W.; et al. Core Epithelial-to-Mesenchymal Transition Interactome Gene-Expression Signature Is Associated with Claudin-Low and Metaplastic Breast Cancer Subtypes. Proc. Natl. Acad. Sci. USA 2010, 107, 15449–15454. [Google Scholar] [CrossRef]
- Zhang, K.; Corsa, C.A.; Ponik, S.M.; Prior, J.L.; Piwnica-Worms, D.; Eliceiri, K.W.; Keely, P.J.; Longmore, G.D. The Collagen Receptor Discoidin Domain Receptor 2 Stabilizes Snail1 Protein to Facilitate Breast Cancer Metastasis. Nat. Cell Biol. 2013, 15, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The Transcription Factor Snail Controls Epithelial-Mesenchymal Transitions by Repressing E-Cadherin Expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Denny, W.A.; Flanagan, J.U. Inhibitors of Discoidin Domain Receptor (DDR) Kinases for Cancer and Inflammation. Biomolecules 2021, 11, 1671. [Google Scholar] [CrossRef]
- Telford, B.J.; Chen, A.; Beetham, H.; Frick, J.; Brew, T.P.; Gould, C.M.; Single, A.; Godwin, T.; Simpson, K.J.; Guilford, P. Synthetic Lethal Screens Identify Vulnerabilities in GPCR Signaling and Cytoskeletal Organization in E-Cadherin-Deficient Cells. Mol. Cancer Ther. 2015, 14, 1213–1223. [Google Scholar] [CrossRef]
- Ikeda, K.; Wang, L.-H.; Torres, R.; Zhao, H.; Olaso, E.; Eng, F.J.; Labrador, P.; Klein, R.; Lovett, D.; Yancopoulos, G.D.; et al. Discoidin Domain Receptor 2 Interacts with Src and Shc Following Its Activation by Type I Collagen. J. Biol. Chem. 2002, 277, 19206–19212. [Google Scholar] [CrossRef]
- Yang, K.; Kim, J.H.; Kim, H.J.; Park, I.-S.; Kim, I.Y.; Yang, B.-S. Tyrosine 740 Phosphorylation of Discoidin Domain Receptor 2 by Src Stimulates Intramolecular Autophosphorylation and Shc Signaling Complex Formation. J. Biol. Chem. 2005, 280, 39058–39066. [Google Scholar] [CrossRef]
- Ahmed, S.B.M.; Prigent, S.A. Insights into the Shc Family of Adaptor Proteins. J. Mol. Signal. 2017, 12, 2. [Google Scholar] [CrossRef]
- Jiang, T.; Qiu, Y. Interaction between Src and a C-Terminal Proline-Rich Motif of Akt Is Required for Akt Activation. J. Biol. Chem. 2003, 278, 15789–15793. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Kim, O.; Yang, J.; Sato, K.; Eisenmann, K.M.; McCarthy, J.; Chen, H.; Qiu, Y. Regulation of Akt/PKB Activation by Tyrosine Phosphorylation. J. Biol. Chem. 2001, 276, 31858–31862. [Google Scholar] [CrossRef]
- Terai, H.; Tan, L.; Beauchamp, E.M.; Hatcher, J.M.; Liu, Q.; Meyerson, M.; Gray, N.S.; Hammerman, P.S. Characterization of DDR2 Inhibitors for the Treatment of DDR2 Mutated Nonsmall Cell Lung Cancer. ACS Chem. Biol. 2015, 10, 2687–2696. [Google Scholar] [CrossRef] [PubMed]
- Conchon, M.; Freitas, C.M.B.D.M.; Rego, M.A.D.C.; Braga Junior, J.W.R. Dasatinib—Clinical Trials and Management of Adverse Events in Imatinib Resistant/Intolerant Chronic Myeloid Leukemia. Rev. Bras. Hematol. Hemoter. 2011, 33, 131–139. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ackerman, A.B. The Breast Is Not an Organ. Am. J. Dermatopathol. 2008, 30, 304. [Google Scholar] [CrossRef] [PubMed]
- Suami, H.; Pan, W.-R.; Mann, G.B.; Taylor, G.I. The Lymphatic Anatomy of the Breast and Its Implications for Sentinel Lymph Node Biopsy: A Human Cadaver Study. Ann. Surg. Oncol. 2008, 15, 863–871. [Google Scholar] [CrossRef]
- Lee, O.; Khan, S.A. Novel Routes for Administering Chemoprevention: Local Transdermal Therapy to the Breasts. Semin. Oncol. 2016, 43, 107–115. [Google Scholar] [CrossRef]
- Bathara, M.; Date, T.; Chaudhari, D.; Ghadi, R.; Kuche, K.; Jain, S. Exploring the Promising Potential of High Permeation Vesicle-Mediated Localized Transdermal Delivery of Docetaxel in Breast Cancer To Overcome the Limitations of Systemic Chemotherapy. Mol. Pharm. 2020, 17, 2473–2486. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, Y.; Zeng, X.; Pei, W.; Fan, R.; Wang, Y.; Wang, X.; Li, J. A Combined Self-Assembled Drug Delivery for Effective Anti-Breast Cancer Therapy. Int. J. Nanomed. 2021, 16, 2373–2388. [Google Scholar] [CrossRef]
- Lee, O.; Ivancic, D.; Allu, S.; Shidfar, A.; Kenney, K.; Helenowski, I.; Sullivan, M.E.; Muzzio, M.; Scholtens, D.; Chatterton, R.T.; et al. Local Transdermal Therapy to the Breast for Breast Cancer Prevention and DCIS Therapy: Preclinical and Clinical Evaluation. Cancer Chemother. Pharmacol. 2015, 76, 1235–1246. [Google Scholar] [CrossRef]
- Lee, O.; Pilewskie, M.; Karlan, S.; Tull, M.B.; Benante, K.; Xu, Y.; Blanco, L.; Helenowski, I.; Kocherginsky, M.; Yadav, S.; et al. Local Transdermal Delivery of Telapristone Acetate Through Breast Skin, Compared With Oral Treatment: A Randomized Double-Blind, Placebo-Controlled Phase II Trial. Clin. Pharmacol. Ther. 2021, 109, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Neupane, R.; Boddu, S.H.S.; Abou-Dahech, M.S.; Bachu, R.D.; Terrero, D.; Babu, R.J.; Tiwari, A.K. Transdermal Delivery of Chemotherapeutics: Strategies, Requirements, and Opportunities. Pharmaceutics 2021, 13, 960. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | TCGA FC | GEO FC | TCGA Adj p-Value | GEO Adj p-Value |
|---|---|---|---|---|
| DDR2 | 5.13 | 2.04 | 6.84 × 10−51 | 2.89 × 10−35 |
| CDH11 | 3.14 | 2.33 | 1.99 × 10−43 | 1.26 × 10−31 |
| AOC3 | 4.65 | 2.45 | 2.91 × 10−43 | 1.57 × 10−30 |
| MSRB3 | 4.12 | 2.06 | 9.74 × 10−43 | 6.82 × 10−30 |
| FSTL1 | 2.61 | 2.07 | 9.96 × 10−43 | 4.76 × 10−29 |
| DPYSL3 | 4.28 | 2.07 | 4.95 × 10−42 | 1.46 × 10−28 |
| COL8A1 | 5.33 | 2.02 | 8.41 × 10−42 | 9.60 × 10−28 |
| TIMP3 | 3.16 | 2.16 | 9.55 × 10−40 | 1.96 × 10−27 |
| CCDC80 | 5.29 | 2.28 | 9.89 × 10−39 | 5.48 × 10−27 |
| LTBP1 | 3.00 | 2.10 | 1.54 × 10−38 | 4.74 × 10−26 |
| PDLIM3 | 4.40 | 2.50 | 2.10 × 10−38 | 1.85 × 10−25 |
| C1S | 2.87 | 2.37 | 3.61 × 10−36 | 2.66 × 10−25 |
| GREM1 | 6.27 | 2.35 | 9.83 × 10−36 | 3.64 × 10−25 |
| GLT8D2 | 2.78 | 2.18 | 2.28 × 10−35 | 1.88 × 10−24 |
| GAS1 | 5.25 | 3.11 | 2.37 × 10−35 | 2.77 × 10−24 |
| TNS1 | 3.67 | 2.35 | 5.32 × 10−35 | 5.75 × 10−24 |
| KCNMA1 | 7.31 | 3.77 | 3.20 × 10−34 | 1.16 × 10−23 |
| MYLK | 4.59 | 2.61 | 3.98 × 10−34 | 1.31 × 10−23 |
| C1R | 2.76 | 2.14 | 6.15 × 10−34 | 2.33 × 10−23 |
| ADAMTS1 | 2.75 | 2.08 | 1.82 × 10−33 | 3.75 × 10−23 |
| Reactome Pathway | Adj p-Value | Differentially Expressed Genes in Pathway | No. of Differentially Expressed Genes in Pathway | Total No. of Genes in Pathway |
|---|---|---|---|---|
| Extracellular matrix organization | 1.40 × 10−6 | COL6A3, COL8A1, COL15A1, FBLN1, FN1, TNC, LTBP1, MMP2, DDR2, ASPN, SDC2, ADAMTS1 | 12 | 289 |
| Regulation of IGF transport and uptake by IGFBPs | 1.00 × 10−4 | FSTL1, FN1, TNC, IGFBP5, LTBP1, MMP2, SDC2 | 7 | 120 |
| Smooth Muscle Contraction | 1.00 × 10−4 | MYH11, MYLK, ACTA2, ACTG2, CALD1 | 5 | 38 |
| Post-translational protein phosphorylation | 0.00058 | FSTL1, FN1, TNC, IGFBP5, LTBP1, SDC2 | 6 | 103 |
| Muscle contraction | 0.0022 | MYH11, MYLK, PLN, ACTA2, ACTG2, VIM, CALD1 | 7 | 194 |
| Degradation of the extracellular matrix | 0.0045 | COL6A3, COL8A1, COL15A1, FN1, MMP2, ADAMTS1 | 6 | 133 |
| Collagen degradation | 0.025 | COL6A3, COL8A1, COL15A1, MMP2 | 4 | 59 |
| Non-integrin membrane-ECM interactions | 0.045 | FN1, TNC, DDR2, SDC2 | 4 | 59 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bougen-Zhukov, N.; Decourtye-Espiard, L.; Mitchell, W.; Redpath, K.; Perkinson, J.; Godwin, T.; Black, M.A.; Guilford, P. E-Cadherin-Deficient Cells Are Sensitive to the Multikinase Inhibitor Dasatinib. Cancers 2022, 14, 1609. https://doi.org/10.3390/cancers14071609
Bougen-Zhukov N, Decourtye-Espiard L, Mitchell W, Redpath K, Perkinson J, Godwin T, Black MA, Guilford P. E-Cadherin-Deficient Cells Are Sensitive to the Multikinase Inhibitor Dasatinib. Cancers. 2022; 14(7):1609. https://doi.org/10.3390/cancers14071609
Chicago/Turabian StyleBougen-Zhukov, Nicola, Lyvianne Decourtye-Espiard, Wilson Mitchell, Kieran Redpath, Jacqui Perkinson, Tanis Godwin, Michael A. Black, and Parry Guilford. 2022. "E-Cadherin-Deficient Cells Are Sensitive to the Multikinase Inhibitor Dasatinib" Cancers 14, no. 7: 1609. https://doi.org/10.3390/cancers14071609
APA StyleBougen-Zhukov, N., Decourtye-Espiard, L., Mitchell, W., Redpath, K., Perkinson, J., Godwin, T., Black, M. A., & Guilford, P. (2022). E-Cadherin-Deficient Cells Are Sensitive to the Multikinase Inhibitor Dasatinib. Cancers, 14(7), 1609. https://doi.org/10.3390/cancers14071609