
Posttranslational Modifications in Thyroid Cancer: Implications for Pathogenesis, Diagnosis, Classification, and Treatment

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Ubiquitination
2.1. Ubiquitination and Proteasomal Degradation of the Tumor Repressor PCBP1 Are Increased in Thyroid Cancer
2.2. The Activity of the Ubiquitin Ligase Smurf1 and Ubiquitin-Dependent Degradation of the Tumor Suppressor Kisspeptin-1 Are Increased in Thyroid Cancer
2.3. Ubiquitin-Dependent Degradation of VEGFR2 Decreases Angiogenesis and Aggressiveness of Poorly Differentiated PTC
2.4. Ubiquitin Staining May Help Differentiate PTC from NIFTP
3. Sumoylation
3.1. Decreased Nuclear Levels of Sumoylated PDGF-C in Thyroid Cancer
3.2. Sumoylation Inhibits the Tumor Suppressor CCDC6 in Thyroid Cancer Cells by Reducing Its Interaction with the Transcription Factor CREB-1
3.3. SUMO Inhibitors May Offer a Novel Approach to the Treatment of Anaplastic Thyroid Cancer
4. Acetylation
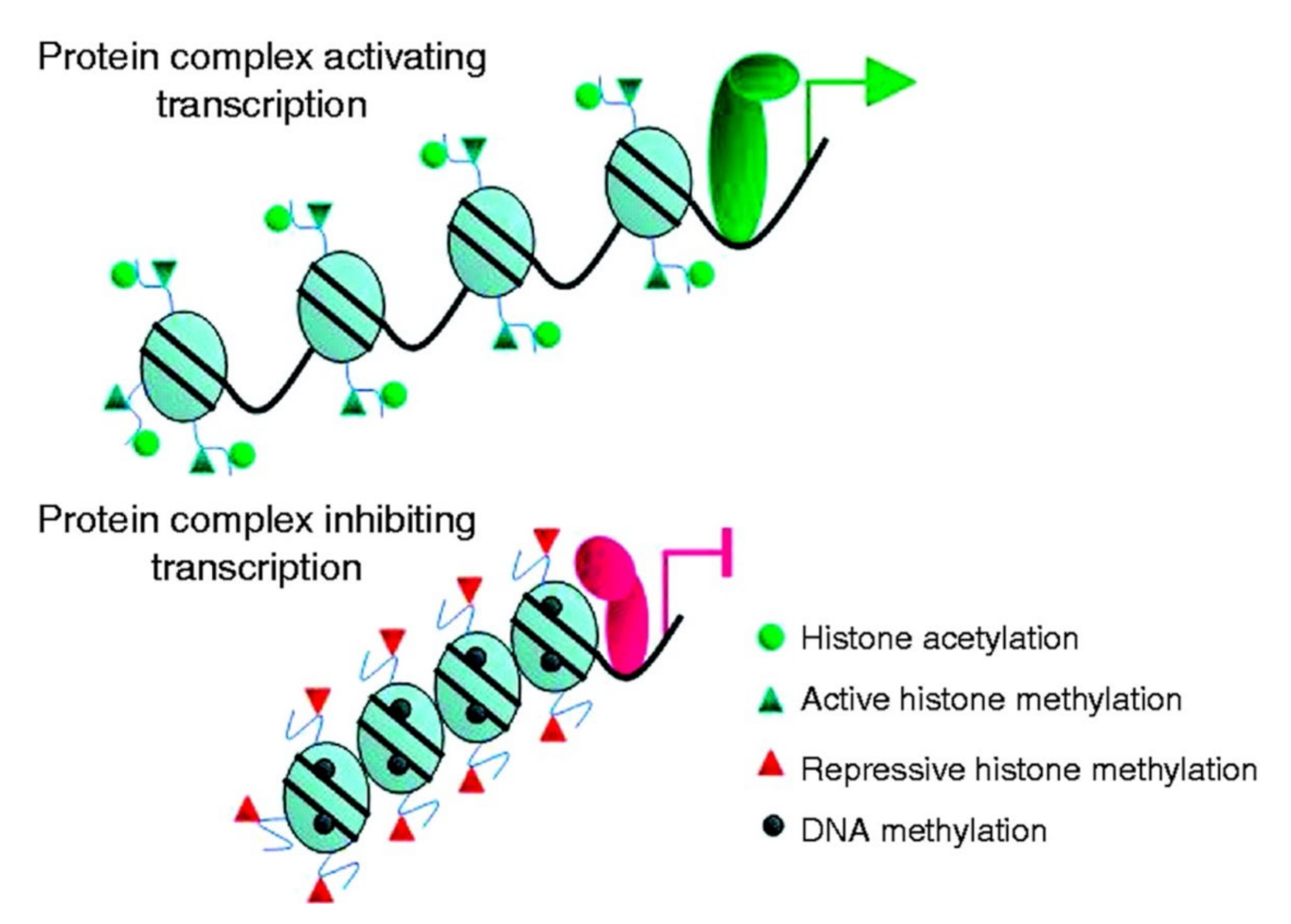
4.1. Histone Acetylation Is Differentially Regulated in Different Types of Thyroid Tumor
4.2. Treatment of Thyroid Cancer with HDAC and HAT Inhibitors
5. Phosphorylation
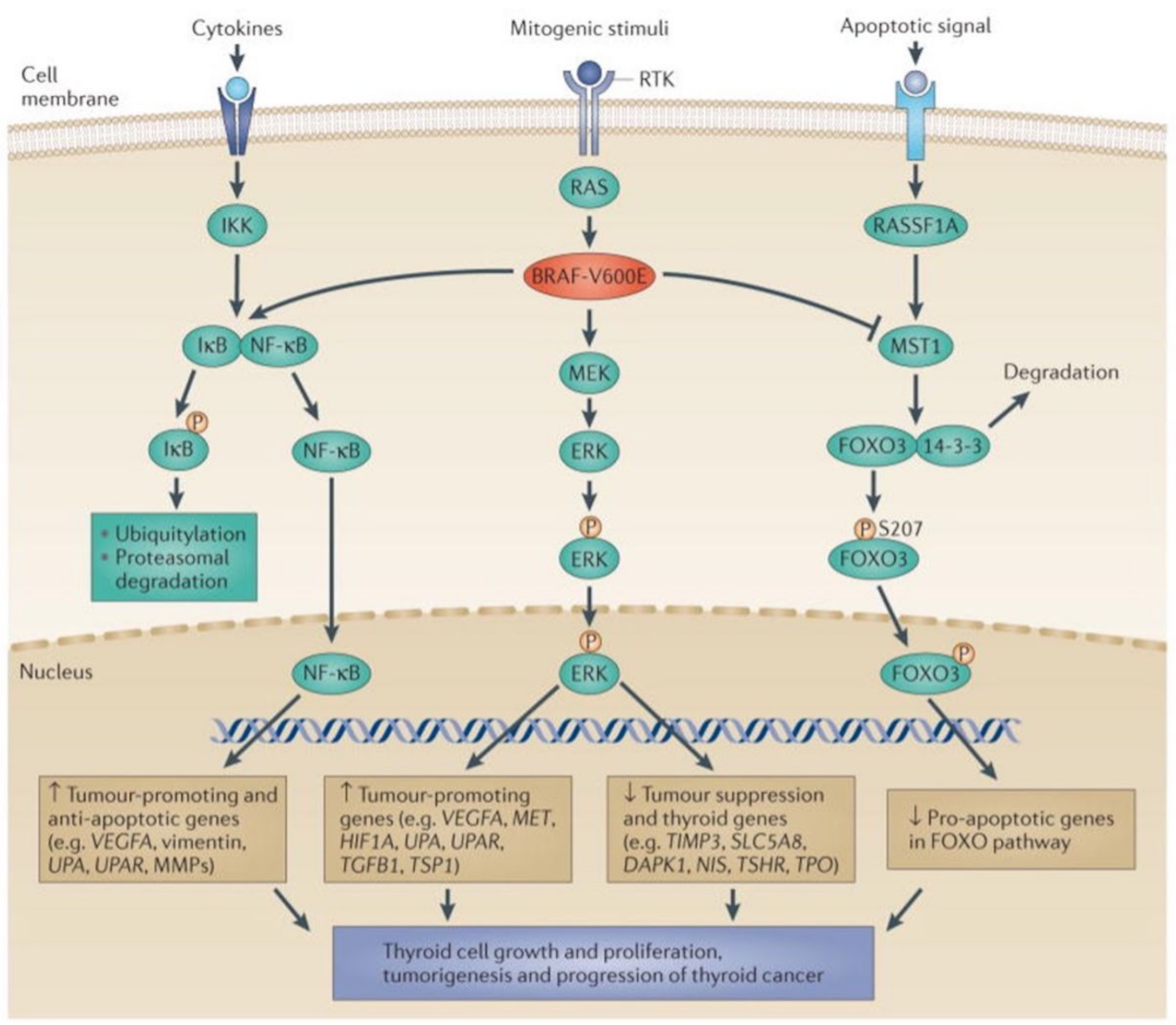
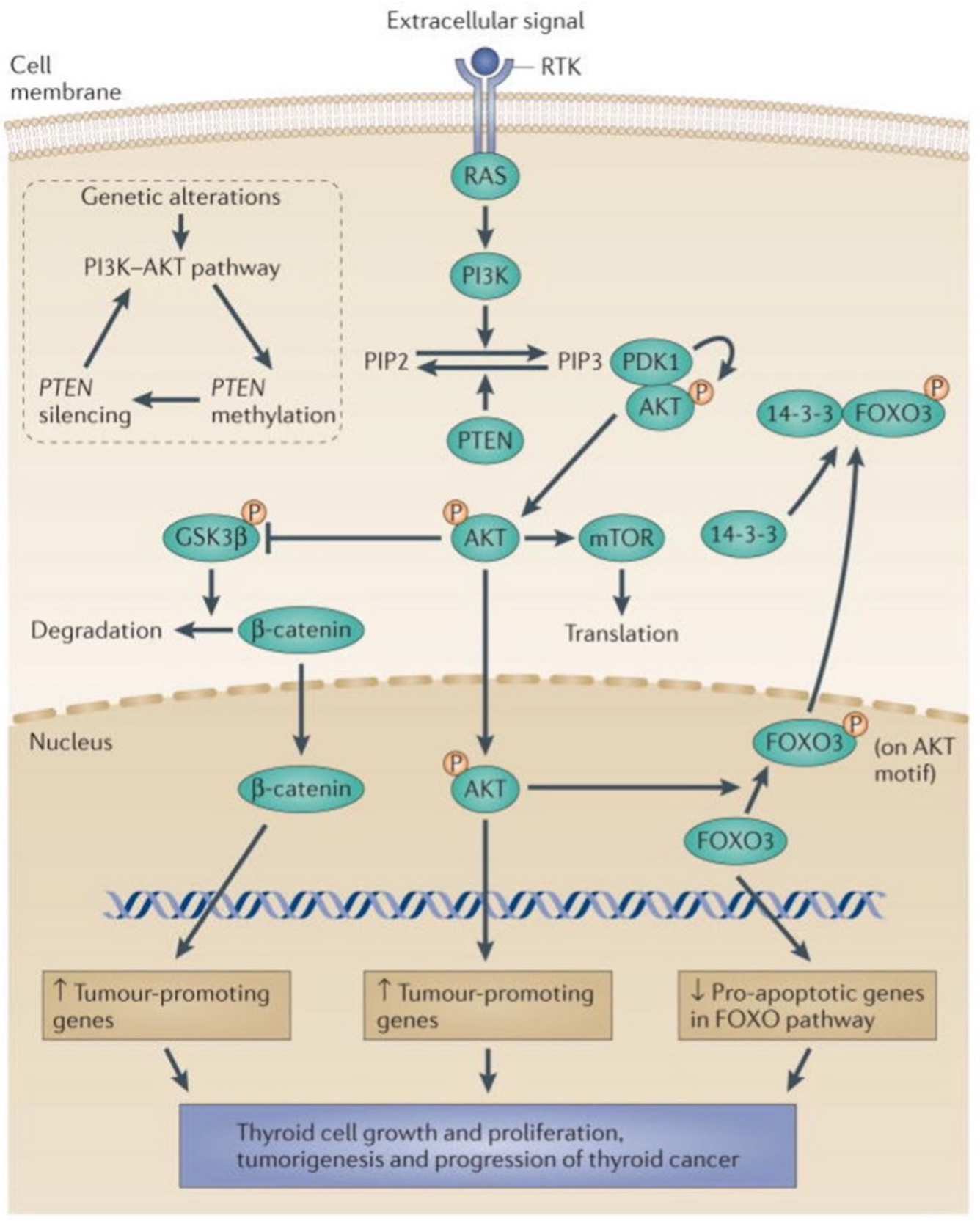
5.1. The MAPK and PI3K-Akt Signaling Pathways in Thyroid Cancer
5.2. Phosphorylation and Activation of Akt by Downregulated Expression of the Transcription Factor ZNF677 Is Involved in Thyroid Tumorigenesis
5.3. Glycosylation and Phosphorylation of AkT Promote Thyroid Malignancy
5.4. Deacetylation and Phosphorylation of Akt Promotes Thyroid Tumorigenesis, Illustrating the Role of Interactions between Different Posttranslational Modifications
5.5. Phosphorylation of the Tumor Suppressor RB (Retinoblastoma Protein) Is Involved in Anaplastic Thyroid Cancer
6. Methylation
Epigenetics and Thyroid Cancer
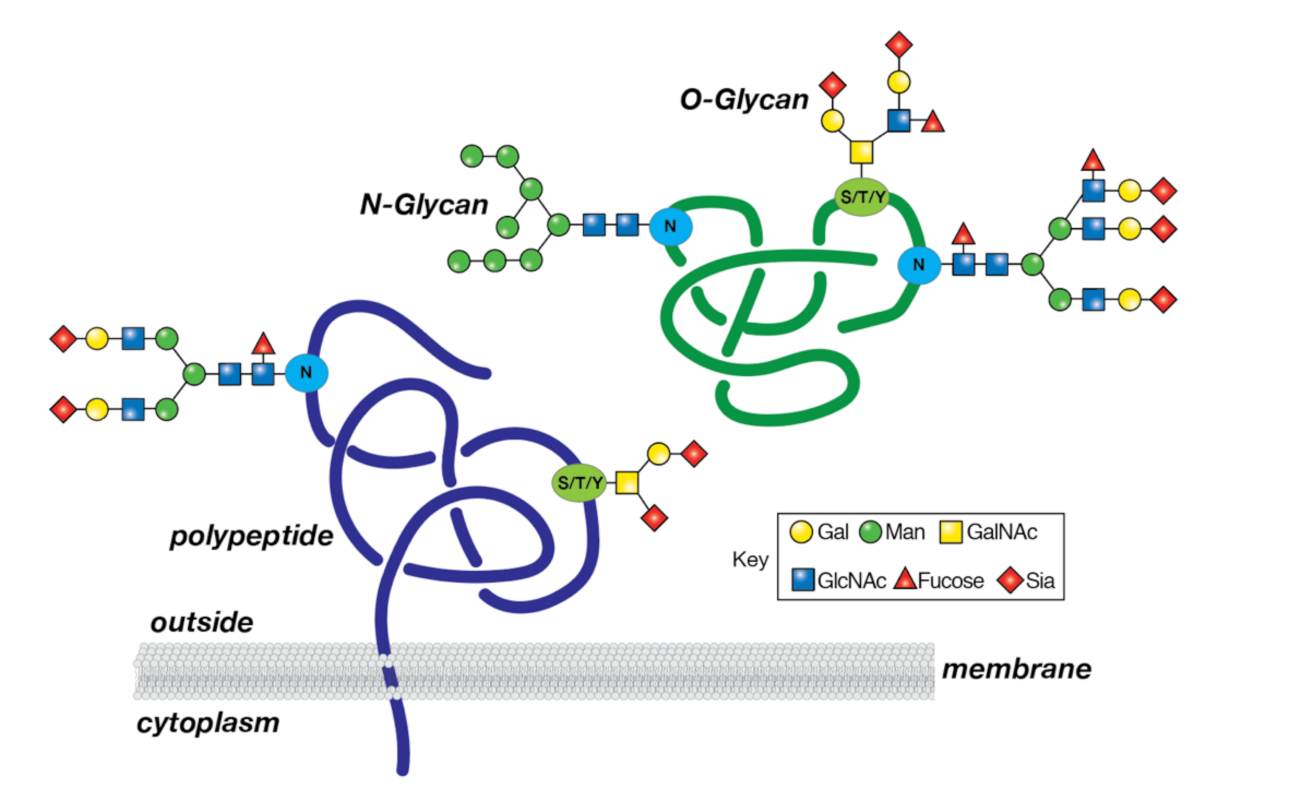
7. Glycosylation
7.1. Immunohistochemical Analysis Demonstrates Different Patterns of Sialylation and Fucosylation in Different Types of Thyroid Cancer
7.2. Fucosylation of Epidermal Growth Factor Receptor (EGFR) by Fucosyltransferase 7 Is Involved in Cellular Proliferation and Migration in FTC
7.3. β1,6 GlcNAc Side Chain Branching of Matriptase N-Glycans by GNT-V Is Implicated in Early Malignant Transformation of PTC
7.4. Thyroglobulin as a Biomarker
8. Succinylation
9. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Novelli, F.; Cappello, P.; Principe, M.; Bulfamante, S. Alpha-Enolase i ENO1 i a potential target in novel immunotherapies. Front. Biosci. 2017, 22, 944–959. [Google Scholar] [CrossRef]
- Yan, F.; Qian, M.; He, Q.; Zhu, H.; Yang, B. The posttranslational modifications of Hippo-YAP pathway in cancer. Biochim. Et. Biophys. Acta (BBA)-Gen. Subj. 2020, 1864, 129397. [Google Scholar] [CrossRef]
- Saji, M.; Ringel, M.D. The PI3K-Akt-mTOR pathway in initiation and progression of thyroid tumors. Mol. Cell Endocrinol. 2010, 321, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Celano, M.; Mio, C.; Sponziello, M.; Verrienti, A.; Bulotta, S.; Durante, C.; Damante, G.; Russo, D. Targeting post-translational histone modifications for the treatment of non-medullary thyroid cancer. Mol. Cell. Endocrinol. 2018, 469, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Tian, Z.; Qu, Y.; Yang, Q.; Guan, H.; Shi, B.; Ji, M.; Hou, P. SIRT7 promotes thyroid tumorigenesis through phosphor-ylation and activation of Akt and p70S6K1 via DBC1/SIRT1 axis. Oncogene 2019, 38, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Amit, M.; Na’ara, S.; Francis, D.; Matanis, W.; Zolotov, S.; Eisenhaber, B.; Eisenhaber, F.; Weiler Sagie, M.; Malkin, L.; Billan, S.; et al. Post-translational regulation of radioactive iodine therapy response in papillary thyroid carcinoma. J. Mol. Endocrinol. 2014, 52, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Hałasa, M.; Wawruszak, A.; Przybyszewska, A.; Jaruga, A.; Guz, M.; Kałafut, J.; Stepulak, A.; Cybulski, M. H3K18Ac as a Marker of Cancer Progression and Potential Target of Anti-Cancer Therapy. Cells 2019, 8, 485. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, E.; Ito, Y.; Miyoshi, Y. Involvement of Aberrant Glycosylation in Thyroid Cancer. J. Oncol. 2010, 2010, 816595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciechanover, A.; Elias, S.; Heller, H.; Ferber, S.; Hershko, A. Characterization of the heat-stable polypeptide of the ATP-dependent proteolytic system from reticulocytes. J. Biol. Chem. 1980, 255, 7525–7528. [Google Scholar] [CrossRef]
- Cappadocia, L.; Lima, C.D. Ubiquitin-like Protein Conjugation: Structures, Chemistry, and Mechanism. Chem. Rev. 2017, 118, 889–918. [Google Scholar] [CrossRef]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef] [PubMed]
- Pfoh, R.; Lacdao, I.K.; Saridakis, V. Deubiquitinases and the new therapeutic opportunities offered to cancer. Endocr.-Relat. Cancer 2015, 22, T35–T54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, J.W.; Ordureau, A.; Heo, J.-M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef]
- Waxman, S.; Germain, D. Targeting the Ubiquitin-Proteasome Pathway in Cancer Therapy. Anti-Cancer Agents Med. Chem. 2007, 7, 359–365. [Google Scholar] [CrossRef]
- Datta, K.; Suman, S.; Kumar, S.; Fornacer, A.J. Colorectal carcinogenesis, radiation quality, and the ubiquitin-proteasome path-way. J. Cancer 2016, 7, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Dong, S.; Wang, H.; Li, L.; Ye, Q.; Li, Y.; Miao, J.; Jhiang, S.; Zhao, J.; Zhao, Y. Two distinct E3 ligases, SCF FBXL19 and HECW1, degrade thyroid transcription factor 1 in normal thyroid epithelial and follicular thyroid carcinoma cells, respectively. FASEB J. 2019, 33, 10538–10550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Vardy, L.A.; Tan, C.P.; Loo, J.M.; Guo, K.; Li, J.; Lim, S.G.; Zhou, J.; Chng, W.J.; Ng, S.B.; et al. PCBP1 Suppresses the Translation of Metastasis-Associated PRL-3 Phosphatase. Cancer Cell 2010, 18, 52–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.-J.; Jung, Y.-S.; Chen, X. Poly (C)-Binding Protein 1 Regulates p63 Expression through mRNA Stability. PLoS ONE 2013, 8, e71724. [Google Scholar] [CrossRef]
- Zhang, Z.-Z.; Shen, Z.-Y.; Shen, Y.-Y.; Zhao, E.-H.; Wang, M.; Wang, C.-J.; Cao, H.; Xu, J. HOTAIR Long Noncoding RNA Promotes Gastric Cancer Metastasis through Suppression of Poly r(C)-Binding Protein (PCBP) 1. Mol. Cancer Ther. 2015, 14, 1162–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.-P.; Zhang, W.-S.; Tan, J.; Zhao, M.-H.; Lian, L.-J.; Cai, J. Poly r(C) binding protein (PCBP) 1 expression is regulated at the post-translation level in thyroid carcinoma. Am. J. Transl. Res. 2017, 9, 708–714. [Google Scholar] [PubMed]
- Zhang, M.-P.; Zhang, W.-S.; Tan, J.; Zhao, M.-H.; Lian, L.-J.; Cai, J. Poly r(C) binding protein (PCBP) 1 expression is regulated by the E3 ligase UBE4A in thyroid carcinoma. Biosci. Rep. 2017, 37, BSR20170114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.; Su, H.; Song, X.; Cao, H.; Kong, L.; Cui, W. Smad Ubiquitination Regulatory Factor 1 (Smurf1) Promotes Thyroid Cancer Cell Proliferation and Migration via Ubiquitin-Dependent Degradation of Kisspeptin-1. Cell. Physiol. Biochem. 2018, 49, 2047–2059. [Google Scholar] [CrossRef] [PubMed]
- Ji, K.; Ye, L.; Mason, M.D.; Jiang, W.G. The Kiss-1/Kiss-1R complex as a negative regulator of cell motility and cancer metastasis (Review). Int. J. Mol. Med. 2013, 32, 747–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savvidis, C.; Papaoiconomou, E.; Petraki, C.; Msaouel, P.; Koutsilieris, M. The role of KISS1/KISS1R system in tumor growth and invasion of differentiated thyroid cancer. Anticancer Res. 2015, 35, 819–826. [Google Scholar]
- Gao, R.J.; Wang, N.P.; Hui, Y.E.; Gao, Q.J.; Zhou, Y.; Duan, H.S. Expression and clinical significance of tumor suppressor gene Kiss-1 in papillary thyroid carcinoma. Chin. J. Bases Clin. Gen. Surg. 2010, 8, 465–473. [Google Scholar]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef]
- Dvorak, H.F. Rous-Whipple Award Lecture. How Tumors Make Bad Blood Vessels and Stroma. Am. J. Pathol. 2003, 162, 1747–1757. [Google Scholar] [CrossRef] [Green Version]
- Bunone, G.; Vigneri, P.; Mariani, L.; But, S.; Collini, P.; Pilotti, S.; Pierotti, M.A.; Bongarzone, I. Expression of angiogenesis stimula-tors and inhibitors in human thyroid tumors and correlation with clinical pathological features. Am. J. Pathol. 1999, 155, 1967–1976. [Google Scholar] [CrossRef] [Green Version]
- Vieira, J.; Santos, S.C.R.; Espadinha, C.; Correia, I.; Vag, T.; Casalou, C.; Cavaco, B.; Catarino, A.L.; Dias, S.; Leite, V. Expression of vascular endothelial growth factor (VEGF) and its receptors in thyroid carcinomas of follicular origin: A potential autocrine loop. Eur. J. Endocrinol. 2005, 153, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Antona, C.; Pallares, J.; Montero-Conde, C.; Inglada-Pérez, L.; Castelblanco, E.; Landa, I.; Leskelä, S.; Leandro-García, L.J.; López-Jiménez, E.; Letón, R.; et al. Overexpression and activation of EGFR and VEGFR2 in medullary thyroid carcinomas is related to metastasis. Endocr. Relat. Cancer 2010, 17, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Shaik, S.; Nucera, C.; Inuzuka, H.; Gao, D.; Garnaas, M.; Frechette, G.; Harris, L.; Wan, L.; Fukushima, H.; Husain, A.; et al. SCF(β-TRCP) suppresses angiogenesis and thyroid cancer cell migra-tion by promoting ubiquitination and destruction of VEGF receptor 2. J. Exp. Med. 2012, 209, 1289–1307. [Google Scholar] [CrossRef] [PubMed]
- Gupta-Abramson, V.; Troxel, A.; Nellore, A.; Puttaswamy, K.; Redlinger, M.; Ransone, K.; Mandel, S.J.; Flaherty, K.T.; Loevner, L.A.; O’Dwyer, P.J.; et al. Phase II Trial of Sorafenib in Advanced Thyroid Cancer. J. Clin. Oncol. 2008, 26, 4714–4719. [Google Scholar] [CrossRef] [Green Version]
- Cabanillas, M.E.; Waguespack, S.G.; Bronstein, Y.; Williams, M.D.; Feng, L.; Hernandez, M.; Lopez, A.; Sherman, S.I.; Busaidy, N.L. Treatment with Tyrosine Kinase Inhibitors for Patients with Differentiated Thyroid Cancer: The, M.D. Anderson Experience. J. Clin. Endocrinol. Metab. 2010, 95, 2588–2595. [Google Scholar] [CrossRef] [Green Version]
- Nikiforov, Y.E.; Seethala, R.R.; Tallini, G.; Baloch, Z.W.; Basolo, F.; Thompson, L.D.; Barletta, J.A.; Wenig, B.M.; Al Ghuzlan, A.; Kakudo, K.; et al. Nomenclature revision for encapsulated follicular variant of papillary thyroid carcinoma: A paradigm shift to reduce overtreatment of indolent tumors. JAMA Oncol. 2016, 2, 1023–1029. [Google Scholar] [CrossRef] [Green Version]
- Lindeman, B.M.; Nehs, M.A.; Angell, T.E.; Alexander, E.K.; Gawande, A.A.; Moore, F.D., Jr.; Doherty, G.M.; Cho, N.L. Effect of noninva-sive follicular thyroid neoplasm with papillary-like nuclear features (NIFTP) on malignancy rates in thyroid nodules: How to counsel patients on extent of surgery. Ann. Surg. Oncol. 2019, 26, 93–97. [Google Scholar] [CrossRef]
- Higgins, S.; James, B.; Sacks, B.; Mowschenson, P.; Nishino, M.; Hasselgren, P.O. Can cytologic and sonographic features guide ex-tent of surgery and prevent “overtreatment” of thyroid nodules classified as suspicious for malignancy (Bethesda V)? J. Surg. Res. 2021, 268, 112–118. [Google Scholar] [CrossRef]
- Brandler, T.C.; Zhou, F.; Liu, C.Z.; Cho, M.; Lau, R.P.; Simsir, A.; Patel, K.N.; Sun, W. Can noninvasive follicular thyroid neoplasm with papillary-like nuclear features be distinguished from classic papillary thyroid carcinoma and follicular adenomas by fi-ne-needle aspiration? Cancer Cytopathol. 2017, 125, 378–388. [Google Scholar] [CrossRef]
- Strickland, K.C.; Eszlinger, M.; Paschke, R.; Angell, T.E.; Alexander, E.K.; Marqusee, E.; Nehs, M.A.; Jo, V.Y.; Lowe, A.; Vivero, M.; et al. Molecular Testing of Nodules with a Suspicious or Malignant Cytologic Diagnosis in the Setting of Non-Invasive Follicular Thyroid Neoplasm with Papil-lary-Like Nuclear Features (NIFTP). Endocr. Pathol. 2018, 29, 68–74. [Google Scholar] [CrossRef]
- Al-Brahim, N.; Asa, S.L. Papillary thyroid carcinoma: An overview. Arch. Pathol. Lab. Med. 2006, 130, 1057–1062. [Google Scholar] [CrossRef]
- Ip, Y.T.; Filho, M.A.D.; Chan, J.K.C. Nuclear inclusions and pseudoinclusions: Friends or foes of the surgical pathologist? Int. J. Surg. Pathol. 2010, 18, 465–481. [Google Scholar] [CrossRef]
- Seethala, R.R.; Baloch, Z.W.; Barletta, J.A.; Khanafshar, E.; Mete, O.; Sadow, P.M.; LiVolsi, V.A.; Nikiforov, Y.E.; Tallini, G.; Thompson, L. Noninvasive follicular thyroid neoplasm with papillary-like nuclear features: A review for pathologists. Mod. Pathol. 2018, 31, 39–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bizzarro, T.; Martini, M.; Capodimonti, S.; Straccia, P.; Lombardi, C.P.; Pontecorvi, A.; Larocca, L.M.; Rossi, E.D. Young investigator challenge: The morphologic analysis of noninvasive follicular thyroid neoplasm with papillary-like nuclear features on liq-uid-based cytology: Some insights into their identification. Cancer Cytopathol. 2016, 124, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Howitt, B.E.; Chang, S.; Eszlinger, M.; Paschke, R.; Drage, M.G.; Krane, J.F.; Barletta, J.A. Fine-needle aspiration diagnoses of noninva-sive follicular variant of papillary thyroid carcinoma. Am. J. Clin. Pathol. 2015, 144, 850–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cracolici, V.; Krausz, T.; Cipriani, N.A. Ubiquitin Immunostaining in Thyroid Neoplasms Marks True Intranuclear Cytoplasmic Pseudoinclusions and May Help Differentiate Papillary Carcinoma from NIFTP. Head Neck Pathol. 2018, 12, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Inabnet, W.B.; Palazzo, F.; Sosa, J.A.; Kriger, J.; Aspinall, S.; Barczynski, M.; Doherty, G.; Iacobone, M.; Nordenstrom, E.; Scott-Coombes, D.; et al. Correlating the Bethesda System for Reporting Thyroid Cytopathology with Histology and Extent of Surgery: A Review of 21,746 Patients from Four Endocrine Surgery Registries Across Two Continents. World J. Surg. 2019, 44, 426–435. [Google Scholar] [CrossRef]
- Yang, Y.; He, Y.; Wang, X.; Liang, Z.; He, G.; Zhang, P.; Zhu, H.; Xu, N.; Liang, S. Protein SUMOylation modification and its associa-tions with disease. Open Biol. 2017, 7, 170167. [Google Scholar] [CrossRef]
- Han, Z.J.; Feng, Y.H.; Gu, B.H.; Li, Y.M.; Chen, H. The post-translational modification, SUMOylation, and cancer (Review). Int. J. Oncol. 2018, 52, 1081–1094. [Google Scholar] [CrossRef] [Green Version]
- Reigstad, L.J.; Martinez, A.; Varhaug, J.E.; Lillehaug, J.R. Nuclear localisation of endogenous SUMO-1-modified PDGF-C in human thyroid tissue and cell lines. Exp. Cell Res. 2006, 312, 782–795. [Google Scholar] [CrossRef]
- De Andrade, J.P.; Lorenzen, A.W.; Wu, V.T.; Bogachek, M.V.; Park, J.M.; Gu, V.W.; Sevenich, C.M.; Cassady, V.C.; Beck, A.C.; Kulak, M.V.; et al. Targeting the SUMO pathway as a novel treatment for anaplastic thyroid cancer. Oncotarget 2017, 8, 114801–114815. [Google Scholar] [CrossRef] [Green Version]
- Luise, C.; Merolla, F.; Leone, V.; Paladino, S.; Sarnataro, D.; Fusco, A.; Celetti, A. Identification of Sumoylation Sites in CCDC6, the First Identified RET Partner Gene in Papillary Thyroid Carcinoma, Uncovers a Mode of Regulating CCDC6 Function on CREB1 Transcriptional Activity. PLoS ONE 2012, 7, e49298. [Google Scholar] [CrossRef] [Green Version]
- Tuccilli, C.; Baldini, E.; Sorrenti, S.; Di Gioia, C.; Bosco, D.; Ascoli, V.; Mian, C.; Barollo, S.; Rendina, R.; Coccaro, C.; et al. Papillary thyroid cancer is characterized by altered expression of gens involed in the sumoylation process. J. Biol. Regul. Homeost. Agents 2015, 29, 655–662. [Google Scholar] [PubMed]
- Li, X.; Pontén, A.; Aase, K.; Karlsson, L.; Abramsson, A.; Uutela, M.; Bäckström, G.; Hellström, M.; Boström, H.; Li, H.; et al. PDGF-C is a new protease-activated ligand for the PDGF al-pha-receptor. Nat. Cell Biol. 2000, 2, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Eriksson, U. Novel PDGF family members: PDGF-C and PDGF-D. Cytokine Growth Factor Rev. 2003, 14, 91–98. [Google Scholar] [CrossRef]
- Leone, V.; Mansueto, G.; Pierantoni, G.M.; Tornincasa, M.; Merolla, F.; Cerrato, A.; Santoro, M.; Grieco, M.; Scaloni, A.; Celetti, A.; et al. CCDC6 represses CREB1 activity by recruiting histone deacetylase 1 and protein phosphatase 1. Oncogene 2010, 29, 4341–4351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Are, C.; Shaha, A.R. Anaplastic Thyroid Carcinoma: Biology, Pathogenesis, Prognostic Factors, and Treatment Approaches. Ann. Surg. Oncol. 2006, 13, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.D.; Baker, T.A.; Gann, A.; Levine, M.; Losik, R. Molecular Biology of the Gene; Pearson/CSH Press: Boston, MA, USA, 2014. [Google Scholar]
- Verdone, L.; Agricola, E.; Caserta, M.; Di Mauro, E. Histone acetylation in gene regulation. Brief Funct. Genom. Proteomic 2006, 5, 209–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-J.; Seto, E. Lysine Acetylation: Codified Crosstalk with Other Posttranslational Modifications. Mol. Cell 2008, 31, 449–461. [Google Scholar] [CrossRef] [Green Version]
- De Ruijter, A.J.M.; Van Gennip, A.H.; Caron, H.N.; Kemp, S.; Van Kuilenburg, A.B.P. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Barnes, P.J.; Adcock, I.; Ito, K. Histone acetylation and deacetylation: Importance in inflammatory lung diseases. Eur. Respir. J. 2005, 25, 552–563. [Google Scholar] [CrossRef]
- Cao, D.J.; Wang, Z.V.; Battiprolu, P.K.; Jiang, N.; Morales, C.R.; Kong, Y.; Rothermel, B.A.; Gillette, T.G.; Hill, J.A. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc. Natl. Acad. Sci. USA 2011, 108, 4123–4128. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, H.; Lehmann, L.H.; Worst, B.C.; Stanmore, D.A.; Backs, J. Histone deacetylase signaling in cardioprotection. Cell Mol. Life Sci. 2014, 71, 1673–1690. [Google Scholar]
- Hasselgren, P.O. Ubiquitination, phosphorylation, and acetylation--triple threat in muscle wasting. J. Cell Physiol. 2007, 213, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Alamdari, N.; Aversa, Z.; Castillero, E.; Hasselgren, P.-O. Acetylation and deacetylation—novel factors in muscle wasting. Metabolism 2012, 62, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Hwang, Y.J.; Kim, K.Y.; Kowall, N.W.; Ryu, H. Epigenetic Mechanisms of Neurodegeneration in Huntington’s Disease. Neurotherapeutics 2013, 10, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, I.; Porƒôba, E.; Kamieniarz, K.; Schneider, R. Histone modifiers in cancer: Friends or foes? Genes Cancer 2011, 2, 631–647. [Google Scholar] [CrossRef] [Green Version]
- Puppin, C.; Passon, N.; Lavarone, E.; Di Loreto, C.; Frasca, F.; Vella, V.; Vigneri, R.; Damante, G. Levels of histone acetylation in thyroid tumors. Biochem. Biophys. Res. Commun. 2011, 411, 679–683. [Google Scholar] [CrossRef]
- Russo, D.; Durante, C.; Bulotta, S.; Puppin, C.; Puxeddu, E.; Filetti, S.; Damante, G. Targeting histone deacetylase in thyroid cancer. Expert Opin. Ther. Targets 2012, 17, 179–193. [Google Scholar] [CrossRef]
- Lin, C.L.; Tsai, M.L.; Lin, C.Y.; Hsu, K.W.; Hsieh, W.S.; Chi, W.M.; Huang, L.C.; Lee, C.H. HDAC1 and HDAC2 Double Knockout Trig-gers Cell Apoptosis in Advanced Thyroid Cancer. Int. J. Mol. Sci. 2019, 20, 454. [Google Scholar] [CrossRef] [Green Version]
- Barneda-Zahonero, B.; Parra, M. Histone deacetylases and cancer. Mol. Oncol. 2012, 6, 579–589. [Google Scholar] [CrossRef] [Green Version]
- Manzo, F.; Tambaro, F.P.; Mai, A.; Altucci, L. Histone acetyltransferase inhibitors and preclinical studies. Expert Opin. Ther. Patents 2009, 19, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. The regulation of protein function by multisite phosphorylation–a 25 year update. Trends Biochem. Sci. 2000, 25, 596–601. [Google Scholar] [CrossRef]
- Singh, V.; Ram, M.; Kumar, R.; Prasad, R.; Roy, B.K.; Singh, K.K. Phosphorylation: Implications in Cancer. J. Protein Chem. 2017, 36, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hunter, T. Protein kinases and phosphatases: The Yin and Yang of protein phosphorylation and signaling. Cell 1995, 80, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Wu, C.; Hart, G.W. Analytical and biochemical perspectives of protein O-GlcNAcylation. Chem. Rev. 2021, 121, 1513–1581. [Google Scholar] [CrossRef] [PubMed]
- Xing, M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat. Cancer 2013, 13, 184–199. [Google Scholar] [CrossRef]
- Xing, M. Genetic Alterations in the Phosphatidylinositol-3 Kinase/Akt Pathway in Thyroid Cancer. Thyroid 2010, 20, 697–706. [Google Scholar] [CrossRef]
- Cabanillas, M.E.; Ryder, M.; Jimenez, C. Targeted Therapy for Advanced Thyroid Cancer: Kinase Inhibitors and Beyond. Endocr. Rev. 2019, 40, 1573–1604. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yang, Q.; Guan, H.; Shi, B.; Ji, M.; Hou, P. ZNF677 Suppresses Akt Phosphorylation and Tumorigenesis in Thyroid Cancer. Cancer Res. 2018, 78, 5216–5228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jen, J.; Wang, Y.C. Zinc finger proteins in cancer progression. J. Biomed. Sci. 2016, 23, 53–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, K.; Mahajan, N.P. PI3K-independent AKT activation in cancers: A treasure trove for novel therapeutics. J. Cell. Physiol. 2012, 227, 3178–3184. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Wang, C.; Ma, T.; You, S. O-GlcNAcylation enhances the invasion of thyroid anaplastic cancer cells partially by PI3K/Akt1 pathway. Onco. Targets Ther. 2015, 8, 3305–3313. [Google Scholar] [PubMed] [Green Version]
- Krześlak, A.; Jóźwiak, P.; Lipińska, A. Down-regulation of β-N-acetyl-D-glucosaminidase increases akt1 activity in thyroid an-aplastic cancer cells. Oncol. Rep. 2011, 26, 743–749. [Google Scholar]
- Cheng, Y.U.; Li, H.; Li, J.; Li, J.; Gao, Y.; Liu, B. O-GlcNAcylation enhances anaplastic thyroid carcinoma malignancy. Oncol. Lett. 2016, 12, 572–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wu, Z.; He, J.; Jin, Y.; Chu, C.; Cao, Y.; Gu, F.; Wang, H.; Hou, C.; Liu, X.; et al. OGT regulated O-GlcNAcylation promotes pa-pillary thyroid cancer malignancy via activating YAP. Oncogene 2021, 40, 4859–4871. [Google Scholar] [CrossRef] [PubMed]
- de Nigris, F.; Cerutti, J.; Morelli, C.; Califano, D.; Chiariotti, L.; Viglietto, G.; Santelli, G.; Fusco, A. Isolation of a SIR-like gene, SIR-T8, that is overexpressed in thyroid carcinoma cell lines and tissues. Br. J. Cancer 2002, 86, 917–923. [Google Scholar] [CrossRef] [Green Version]
- Frye, R. “SIRT8” expressed in thyroid cancer is actually SIRT7. Br. J. Cancer 2002, 87, 1479. [Google Scholar] [CrossRef] [Green Version]
- Petrulea, M.S.; Plantinga, T.; Smit, J.W.; Georgescu, C.E.; Netea-Maier, R.T. PI3K/Akt/mTOR: A promising therapeutic target for non-medullary thyroid carcinoma. Cancer Treat. Rev. 2015, 41, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Prasongsook, N.; Kumar, A.; Chintakuntlawar, A.; Foote, R.L.; Kasperbauer, J.; Molina, J.; Garces, Y.; Ma, D.; Wittich, M.A.N.; Rubin, J.; et al. Survival in Response to Multimodal Therapy in Anaplastic Thyroid Cancer. J. Clin. Endocrinol. Metab. 2017, 102, 4506–4514. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.; Tulla, K.; Maker, A.V.; Burman, K.D.; Prabhakar, B.S. Therapeutic advances in anaplastic thyroid cancer: A current per-spective. Mol. Cancer. 2018, 17, 154–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.; Di Cristofano, F.; Ranieri, M.; De Martino, D.; Di Cristofano, A. PI3K/mTOR inhibition potentiates and extends palbo-ciclib activity in anaplastic thyroid cancer. Endocr. Relat. Cancer 2019, 26, 425–436. [Google Scholar] [CrossRef]
- Sherr, C.J. The Pezcoller lecture: Cancer cell cycles revisited. Cancer Res. 2000, 60, 3689–3695. [Google Scholar] [PubMed]
- Lee, H.J.; Lee, W.K.; Kang, C.W.; Ku, C.R.; Cho, Y.H.; Lee, E.J. A selective cyclin-dependent kinase 4, 6 dual inhibitor, Ribociclib (LEE011) inhibits cell proliferation and induces apoptosis in aggressive thyroid cancer. Cancer Lett. 2018, 417, 131–140. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, B.; Cutts, R.J.; Liu, Y.; Hrebien, S.; Huang, X.; Fenwick, K.; André, F.; Loibl, S.; Loi, S.; Garcia-Murillas, I.; et al. The Genetic Landscape and Clonal Evolution of Breast Cancer Resistance to Palbociclib plus Fulvestrant in the PALOMA-3 Trial. Cancer Discov. 2018, 8, 1390–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waddington, C.H. The epigenotype. 1942. Int. J. Epidemiol. 2012, 41, 10–13. [Google Scholar] [CrossRef] [Green Version]
- Russo, D.; Damante, G.; Puxeddu, E.; Durante, C.; Filetti, S. Epigenetics of thyroid cancer and novel therapeutic targets. J. Mol. Endocrinol. 2011, 46, R73–R81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Rodero, S.; Delgado-Álvarez, E.; Fernández, A.F.; Fernández-Morera, J.L.; Menéndez-Torre, E.; Fraga, M.F. Epigenetic alterations in endocrine-related cancer. Endocr.-Relat. Cancer 2014, 21, R319–R330. [Google Scholar] [CrossRef]
- Kass, S.U.; Pruss, D.; Wolffe, A.P. How does DNA methylation repress transcription? Trends Genet. 1997, 13, 444–449. [Google Scholar] [CrossRef]
- Baylin, S.B.; Herman, J.G. DNA hypermethylation in tumorigenesis: Epigenetics joins genetics. Trends Genet. 2000, 16, 168–174. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Wen, H.; Shi, X. Lysine methylation: Beyond histones. Acta Biochim. Biophys Sin. 2012, 44, 14–27. [Google Scholar] [CrossRef] [Green Version]
- Xing, M. Gene Methylation in Thyroid Tumorigenesis. Endocrinology 2007, 148, 948–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, M.; Usadel, H.; Cohen, Y.; Tokumaru, Y.; Guo, Z.; Westra, W.B.; Tong, B.C.; Tallini, G.; Udelsman, R.; Califano, J.A.; et al. Methylation of the thyroid-stimulating hormone receptor gene in epithelial thyroid tumors: A marker of malig-nancy and a cause of gene silencing. Cancer Res. 2003, 63, 2316–2321. [Google Scholar] [PubMed]
- Hu, S.; Liu, D.; Tufano, R.P.; Carson, K.A.; Rosenbaum, E.; Cohen, Y.; Holt, E.H.; Kiseljak-Vassiliades, K.; Rhoden, K.J.; Tolaney, S.; et al. Association of aberrant meth-ylation of tumor suppressor genes with tumor aggressiveness and BRAF mutation in papillary thyroid cancer. Int. J. Cancer 2006, 119, 2322–2329. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Nakazawa, T.; Ma, D.; Niu, D.; Mochizuki, K.; Kawasaki, T.; Nakamura, N.; Yamane, T.; Kobayashi, M.; Katoh, R. Epige-netic silencing of TTF-1/NKX2-1 through DNA hypermethylation and histone H3 modulation in thyroid carcinomas. Lab-Vest 2009, 89, 791–799. [Google Scholar]
- Zuo, H.; Gandhi, M.; Edreira, M.M.; Hochbaum, D.; Nimgaonkar, V.L.; Zhang, P.; Dipaola, J.; Evdokimova, V.; Altschuler, D.L.; Nikifo-rov, Y.E. Downregulation of Rap1GAP through epigenetic silencing and loss of heterozygosity promotes invasion and progression of thyroid tumors. Cancer Res. 2010, 70, 1389–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tell, G.; Pines, A.; Arturi, F.; Cesaratto, L.; Adamson, E.; Puppin, C.; Presta, I.; Russo, D.; Filetti, S.; Damante, G. Control of Phosphatase and Tensin Homolog (PTEN) Gene Expression in Normal and Neoplastic Thyroid Cells. Endocrinology 2004, 145, 4660–4666. [Google Scholar] [CrossRef] [PubMed]
- Schlumberger, M.; Lacroix, L.; Russo, D.; Filetti, S.; Bidart, J.-M. Defects in iodide metabolism in thyroid cancer and implications for the follow-up and treatment of patients. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, G.M.; Yatin, M.; Marcinek, R.; Ain, K.B. Restoration of iodide uptake in dedifferentiated thyroid carcinoma: Rela-tionship to human Na+/I-symporter gene methylation status. J. Clin. Endocrinol. Metab. 1999, 84, 2449–2457. [Google Scholar]
- Haugen, B.R. Redifferentiation therapy in advanced thyroid cancer. Curr. Drug Targets Immune Endocr Metab. Disord 2004, 4, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.P.; Matthijs, G. The evolving genetic landscape of congenital disorders of glycosylation. Biochim. Et. Biophys. Acta (BBA)-Gen. Subj. 2021, 1865, 129976. [Google Scholar] [CrossRef]
- Fuster, M.M.; Esko, J.D. The Sweet and Sour of Cancer: Glycans as Novel Therapeutic Targets. Nat. Rev. Cancer 2005, 5, 526–542. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Stowell, S.R.; Ju, T.; Cummings, R.D. Protein glycosylation in cancer. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 473–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoyagi, Y.; Suzuki, Y.; Igarashi, K. The usefulness of simultaneous determinations of glucosaminylation and fucosylation indices of alpha‚ Äêfetoprotein in the differential diagnosis of neoplastic diseases of the liver. Cancer 1991, 67, 2390–2394. [Google Scholar] [CrossRef]
- Taketa, K.; Endo, Y.; Sekiya, C. A Collaborative Study for the Evaluation of Lectin-Reactive α-Fetoproteins in Early Detec-tion of Hepatocellular Carcinoma. Cancer Res. 1993, 53, 5419–5423. [Google Scholar]
- Cummings, R.D.; Pierce, J.M. The Challenge and Promise of Glycomics. Chem. Biol. 2014, 21, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nozawa, Y.; Ami, H.; Suzuki, S.; Tuchiya, A.; Abe, R.; Abe, M. Distribution of sialic acid-dependent carbohydrate epitope in thy-roid tumors: Immunoreactivity of FB21 in paraffin-embedded tissue sections. Pathol. Int. 1999, 49, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Miyauchi, A.; Yoshida, H.; Uruno, T.; Nakano, K.; Takamura, Y.; Miya, A.; Kobayashi, K.; Yokozawa, T.; Matsuzuka, F.; et al. Expression of α1,6-fucosyltransferase (FUT8) in papillary carcinoma of the thyroid: Its linkage to biological aggressiveness and anaplastic transformation. Cancer Lett. 2003, 200, 167–172. [Google Scholar] [CrossRef]
- Noda, K.; Miyoshi, E.; Uozumi, N.; Yanagidani, S.; Ikeda, Y.; Gao, C.-X.; Suzuki, K.; Yoshihara, H.; Yoshikawa, M.; Kawano, K.; et al. Gene expression of fucosyltransferase in human hepatoma tissues: A possible implication for increased fucosylation of fetoprotein. Hepatology 1998, 28, 944–952. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Ikeda, Y.; Miyoshi, E.; Yagiimura, Y.; Ishikawa, M.; Taniguchi, N. a1, 6 fucosyltransferase is highly and specifi-cally expressed in human ovarian serous adenocarcinomas. Int. J. Cancer 2000, 88, 914–919. [Google Scholar] [CrossRef]
- Qin, H.; Liu, J.; Yu, M.; Wang, H.; Thomas, A.M.; Li, S.; Yan, Q.; Wang, L. FUT7 promotes the malignant transformation of follicular thyroid carcinoma through α-1,3-fucosylation of EGF receptor. Exp. Cell Res. 2020, 393, 112095. [Google Scholar] [CrossRef] [PubMed]
- Martin-Satue, M.; de Castellarnau, C.; Blanco, J. Overexpression of alpha(1,3)-fucosyltransferase VII is sufficient for the ac-quisition of lung colonization phenotype in human lung adenocarcinoma HAL-24Luc cells. Br. J. Canc. 1999, 80, 1169–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Qi, H.L.; Zhang, Y.; Zhang, X.Y.; Chen, H.L. Transfection of the c-erbB2/neu gene upregulates the expression of sialyl Lewis X, alpha1,3-fucosyltransferase VII, and metastatic potential in a human hepatocarcinoma cell line. Eur. J. Biochem 2001, 268, 3501–3512. [Google Scholar] [CrossRef]
- Ito, Y.; Akinaga, A.; Yamanaka, K.; Nakagawa, T.; Kondo, A.; Dickson, R.B.; Lin, C.-Y.; Miyauchi, A.; Taniguchi, N.; Miyoshi, E. Co-expression of matriptase and N-acetylglucosaminyltransferase V in thyroid cancer tissues—its possible role in prolonged stability in vivo by aberrant glycosylation. Glycobiology 2006, 16, 368–374. [Google Scholar] [CrossRef]
- Citterio, C.E.; Targovnik, H.M.; Arvan, P. The role of thyroglobulin in thyroid hormonogenesis. Nat. Rev. Endocrinol. 2019, 15, 323–338. [Google Scholar] [CrossRef]
- Yang, S.-X.; Pollock, H.; Rawitch, A.B. Glycosylation in Human Thyroglobulin: Location of the N-Linked Oligosaccharide Units and Comparison with Bovine Thyroglobulin. Arch. Biochem. Biophys. 1996, 327, 61–70. [Google Scholar] [CrossRef]
- Shimizu, K.; Nakamura, K.; Kobatake, S.; Satomura, S.; Maruyama, M.; Kameko, F.; Tajiri, J.; Kato, R. The clinical utility of Lens culinaris agglutinin-reactive thyroglobulin ratio in serum for distinguishing benign from malignant conditions of the thyroid. Clin. Chim. Acta 2007, 379, 101–104. [Google Scholar] [CrossRef] [Green Version]
- Kanai, T.; Amakawa, M.; Kato, R.; Shimizu, K.; Nakamura, K.; Ito, K.-I.; Hama, Y.; Fujimori, M.; Amano, J. Evaluation of a new method for the diagnosis of alterations of Lens culinaris agglutinin binding of thyroglobulin molecules in thyroid carcinoma. Clin. Chem. Lab. Med. (CCLM) 2009, 47, 1285–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Tan, M.; Xie, Z.; Dai, L.; Chen, Y.; Zhao, T. Identification of lysine succinylation as a new post-translational modifica-tion. Nat. Chem. Biol. 2011, 7, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Gibson, G.E. Succinylation Links Metabolism to Protein Functions. Neurochem. Res. 2019, 44, 2346–2359. [Google Scholar] [CrossRef]
- Xie, Z.; Dai, J.; Dai, L.; Tan, M.; Cheng, Z.; Wu, Y.; Boeke, J.; Zhao, Y. Lysine Succinylation and Lysine Malonylation in Histones. Mol. Cell. Proteom. 2012, 11, 100–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simithy, J.; Sidoli, S.; Yuan, Z.-F.; Coradin, M.; Bhanu, N.V.; Marchione, D.; Klein, B.J.; Bazilevsky, G.A.; McCullough, C.E.; Magin, R.; et al. Characterization of histone acylations links chromatin modifications with metabolism. Nat. Commun. 2017, 8, 1141. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.R.; Bhatt, D.P.; O’Connell, T.M.; Thompson, J.W.; Dubois, L.G.; Backos, D.S.; Yang, H.; Mitchell, G.A.; Ilkayeva, O.R.; Stevens, R.D.; et al. A class of reactive acyl-CoA species reveals the non-enzymatic origins of protein acylation. Cell Metab. 2017, 25, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Guo, Y.R.; Liu, K.; Yin, Z.; Liu, R.; Xia, Y.; Tan, L.; Yang, P.; Lee, J.H.; Li, X.J.; et al. KAT2A coupled with the α-KGDH complex acts as a histone H3 succinyltransferase. Nature 2017, 552, 273–277. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Yuan, Y.; Yuan, H.; Wang, J.; Yun, H.; Geng, Y.; Zhao, M.; Li, L.; Weng, Y.; Liu, Z.; et al. Histone acetyltransferase 1 is a suc-cinyltransferase for histones and non-histones and promotes tumorigenesis. EMBO Rep. 2021, 22, e50967. [Google Scholar] [CrossRef]
- Mu, R.; Ma, Z.; Lu, C.; Wang, H.; Cheng, X.; Tuo, B.; Fan, Y.; Liu, X.; Taolang, L. Role of succinylation modification in thyroid cancer and breast cancer. Am. J. Cancer Res. 2021, 11, 4683–4699. [Google Scholar]
- Dunn, J.; Kim, P.; Dunn, A. Favored sites for thyroid hormone formation on the peptide chains of human thyroglobulin. J. Biol. Chem. 1982, 257, 88–94. [Google Scholar] [CrossRef]
- Shifrin, S.; Kohn, L.D. Binding of thyroglobulin to bovine thyroid membranes. Role of specific amino acids in receptor recogni-tion. J. Biol. Chem. 1981, 256, 10600–10605. [Google Scholar] [CrossRef]
- Lai, X.; Umbricht, C.B.; Fisher, K.; Bishop, J.; Shi, Q.; Chen, S. Identification of novel biomarker and therapeutic target candidates for diagnosis and treatment of follicular carcinoma. J. Proteom. 2017, 166, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Smestad, J.; Erber, L.; Chen, Y.; Maher, L.J. Chromatin Succinylation Correlates with Active Gene Expression and Is Perturbed by Defective TCA Cycle Metabolism. iScience 2018, 2, 63–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Posttranslational Modification | Proposed Role in Thyroid Cancer |
|---|---|
| Ubiquitination | UBE4A-regulated ubiquitination and degradation of PCBP1 may be involved in the tumorigenesis of thyroid cancer. Thyroid cancer may be regulated by Smurf1-induced ubiquitination and the degradation of the tumor suppressor Kisspeptin-1. |
| Sumoylation | Reduced levels of sumoylated PDGF-C may be involved in tumorigenesis in thyroid cancer. Sumoylation of the tumor repressor CCDC6 may be a factor involved in thyroid cancer, providing an additional mechanism of sustained neoplastic growth. Sumoylation of TFAP2A results in altered patterns of gene expression associated with anaplastic thyroid cancer. |
| Acetylation | Histone acetylation varies by thyroid cancer subtype. Both histone acetyltransferase inhibitors and histone deacetylase inhibitors may have a role in the treatment of thyroid cancer. |
| Phosphorylation | Increased Akt phosphorylation and activity in thyroid cancer most often reflects stimulated PI3K-Akt signaling. CDK4/6-dependent phosphorylation of tumor suppressor RB may be important in the progression of anaplastic thyroid cancer. |
| Methylation | The PTEN tumor-suppressor gene is silenced by promotor methylation. PTEN gene promoter methylation induces a tumorigenic effect in the thyroid via dephosphorylation of PI3K. |
| Glycosylation | Sialic acid epitopes on glycoproteins are found in most follicular thyroid cancers (93%), but less than half (45%) of papillary thyroid cancers The stabilization of matriptase via glycosylation by GNT-V may be involved in the early tumorigenesis of PTC. |
| Succinylation | Succinyl-CoA ligase subunit beta may be a useful biomarker for follicular thyroid cancer. Mutations in succinate dehydrogenase in Cowden Syndrome may represent a role of succinylation in thyroid cancer. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Broekhuis, J.M.; James, B.C.; Cummings, R.D.; Hasselgren, P.-O. Posttranslational Modifications in Thyroid Cancer: Implications for Pathogenesis, Diagnosis, Classification, and Treatment. Cancers 2022, 14, 1610. https://doi.org/10.3390/cancers14071610
Broekhuis JM, James BC, Cummings RD, Hasselgren P-O. Posttranslational Modifications in Thyroid Cancer: Implications for Pathogenesis, Diagnosis, Classification, and Treatment. Cancers. 2022; 14(7):1610. https://doi.org/10.3390/cancers14071610
Chicago/Turabian StyleBroekhuis, Jordan M., Benjamin C. James, Richard D. Cummings, and Per-Olof Hasselgren. 2022. "Posttranslational Modifications in Thyroid Cancer: Implications for Pathogenesis, Diagnosis, Classification, and Treatment" Cancers 14, no. 7: 1610. https://doi.org/10.3390/cancers14071610
APA StyleBroekhuis, J. M., James, B. C., Cummings, R. D., & Hasselgren, P.-O. (2022). Posttranslational Modifications in Thyroid Cancer: Implications for Pathogenesis, Diagnosis, Classification, and Treatment. Cancers, 14(7), 1610. https://doi.org/10.3390/cancers14071610