Simple Summary
Triple-negative breast cancer (TNBC) outcomes are improving since the implementation of immunotherapy. However, objective response rates are still limited to a select group of patients. This is partly due to TNBC intrinsic immune evasive mechanisms and the lack of proper tumor microenvironment immune system activation. Dynamic epigenetic modifications contribute to immune surveillance and immune escape in cancer and can be reverted through epigenetic drugs. This review summarizes the epigenetic changes in TNBC cells and their contribution to the cancer cell–immunity cycle. Furthermore, it also describes how epigenetic drugs may provide novel biomarkers for immunotherapy and enhance the immune response. This manuscript lists the current clinical trials using epigenetic drugs alone or combined with either immune checkpoint inhibitors or small molecules.
Abstract
Triple-negative breast cancer (TNBC) is defined by the absence of estrogen receptor and progesterone receptor and human epidermal growth factor receptor 2 (HER2) overexpression. This malignancy, representing 15–20% of breast cancers, is a clinical challenge due to the lack of targeted treatments, higher intrinsic aggressiveness, and worse outcomes than other breast cancer subtypes. Immune checkpoint inhibitors have shown promising efficacy for early-stage and advanced TNBC, but this seems limited to a subgroup of patients. Understanding the underlying mechanisms that determine immunotherapy efficiency is essential to identifying which TNBC patients will respond to immunotherapy-based treatments and help to develop new therapeutic strategies. Emerging evidence supports that epigenetic alterations, including aberrant chromatin architecture conformation and the modulation of gene regulatory elements, are critical mechanisms for immune escape. These alterations are particularly interesting since they can be reverted through the inhibition of epigenetic regulators. For that reason, several recent studies suggest that the combination of epigenetic drugs and immunotherapeutic agents can boost anticancer immune responses. In this review, we focused on the contribution of epigenetics to the crosstalk between immune and cancer cells, its relevance on immunotherapy response in TNBC, and the potential benefits of combined treatments.
Keywords:
epigenetics; TNBC; immunotherapy; cancer; breast cancer; epigenetic drugs; immune system; immune checkpoint 1. Introduction
Triple-negative breast cancer (TNBC), which represents 15–20% of breast cancers (BC), is classified based on the exclusion criteria of lack of estrogen and progesterone receptor expression and absence of human epidermal growth factor receptor 2 (HER2) overexpression. TNBC is particularly aggressive, with a higher probability of metastatic progression and a lack of effective targeted therapies [1]. Given its classification method of BC, TNBC is a heterogeneous malignancy that encloses tumors with different histopathological and molecular features [2]. Thus, several studies focused on the classification of TNBC subtypes. The first classification approach took advantage of the microarray technology to identify six different TNBC subtypes: basal-like 1 (BL1), basal-like 2 (BL2), mesenchymal (M), mesenchymal stem-like (MSL), immunomodulatory (IM), and luminal androgen receptor (LAR) [3]. TNBC subclassification allows for a better understanding of this disease, yet, it has shown limitations in that it fails to predict specific and effective treatments for the TNBC subtype.
This has encouraged the search for new therapeutic alternatives, such as PARP inhibitors and, more recently, immune checkpoint inhibitors (ICI). Thus, different regimens combining immunotherapy and chemotherapy are currently approved by the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for TNBC patients: The combination of nab-paclitaxel and atezolizumab [4] and pembrolizumab in combination with chemotherapy has demonstrated positive results in metastatic TNBC [5]. It was recently shown that neoadjuvant immunotherapy combined with chemotherapy shows increased pathological complete response rates in early-stage TNBC [6,7]. Nevertheless, the iRECIST objective response rate (ORR) for unselected cohorts of TNBC patients remains below 10% [8]. The adaptive phase II TONIC clinical trial revealed that the induction of the tumor and immune system cells with low doses of doxorubicin significantly improves ICI response in advanced TNBC patients (ORR: 35%), independently of the tumor mutational burden [9]. This study highlighted the importance of the dynamic phenotypic adaptation of TNBC and immune cells for immune response.
Unlike other solid tumors, TNBC displays a low frequency of genetic alterations, highlighting the relevance of epigenetic modifications during cancer progression and establishing aggressive phenotypes. Two well-established epigenetic modifications involve DNA methylation and histone modifications. DNA methylation mainly occurs on the 5th position of cytosines, followed by guanosines (also referred to as 5mC). The catalog of histone modifications is far more complex since it involves more than a single chemical group and position. These fine-tuned chemical modifications are controlled by a set of enzymes with rising importance in cancer research: writers are involved in depositing chemical marks, erasers are responsible for the removal, and readers recognize the epigenetic code and recruit other proteins [10]. In general, epigenetic alterations involving DNA or histone modifications are summarized based on the impact on the associated gene(s). Thus, gene promoter hypermethylation or repressive histone marks are usually associated with silencing of the nearby genes (i.e., tumor suppressor genes [11] such as the BRCA1 gene [12]).
Nevertheless, beyond gene promoters, epigenetic alterations can affect gene regulatory elements (GRE) such as enhancer and insulator elements determining activation of cancer-associated gene expression programs and global DNA hypomethylation leading to genomic instability and the potential oncogene reactivation [13]. An overview of epigenetic mechanisms is summarized in Figure 1. However, epigenetic modifications are not only being involved in tumor suppressor gene silencing and oncogene reactivation. Due to the dynamic nature of epigenetic modifications, it is important to consider the epigenetic landscape of the tumor microenvironment (TME), its adaptation to tumor-induced changes, and its contribution to immunotherapy [14]. This will allow for understanding the interplay between TNBC and immune cells and identifying biomarkers to better stratify patients for immunotherapy or complementary therapeutic targets. In addition, chemical removal of aberrant epigenetic marks using small molecule inhibitors may enhance the immune response by expressing immunogenic antigens and reactivating transposable elements [15]. In this context, understanding the epigenetic mechanisms involved in TNBC immune-suppressive pathways and immune cells activation represents an opportunity to improve the selection of TNBC patients for immunotherapy and explore alternative and complementary therapeutic targets. In this review, we cover the importance of epigenetic mechanisms in immunotherapy response in patients with TNBC.
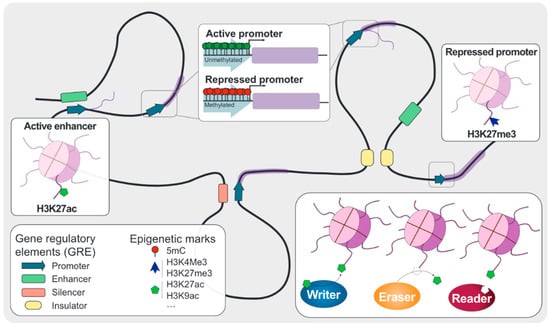
Figure 1.
Overview of epigenetics. Gene regulatory elements (GRE) are defined according to their location and effect on the associated gene expression. Promoters are located close to the transcription start site of their associated genes and facilitate the transcription machinery deposition. This deposition can be aided or blocked by distant GRE called enhancers and silencers, respectively. Chromatin architecture is dynamically regulated by another group of GRE called insulators, which mediate in the topological associating domains (TADs) formation and further contributing to gene expression. DNA and histone modifications are tightly regulated by three different groups of proteins: writers, which are involved in the deposition of these chemical marks; erasers, which are responsible for removing these modifications; and readers, which can recognize the epigenetic code and recruit other proteins.
2. Epigenetic Relevance on Antitumor Immune Response
Both innate and adaptive immunity mediates the cancer immune response. Innate immunity is performed by natural killers (NKs), dendritic cells (DC), eosinophils, and tumor-associated macrophages (TAMs) [16]. The activation of these cells involves a fine-tuned epigenetic modulation of the gene expression program [17,18,19]. Conversely, epigenetic alterations contribute to immunosuppression by innate immune cells. For instance, epigenetic silencing through promoter hypermethylation of NKG2D ligands impairs NK function in acute myeloid leukemia [20]. Focusing on TNBC, TAM polarization into M2 protumorigenic macrophages is mediated by miR-200C [21], and TAM infiltration is associated with a higher risk of distant metastasis in TNBC [22].
In 2013, Chen and Mellman proposed the cancer–immunity cycle (Figure 2), consisting of sequential steps describing adaptive immunity against tumor cells. Briefly, cancer cells express aberrant antigens (step 1) called tumor-associated antigens (TAAs), which are released into the TME after cell death. TAAs are captured by antigen-presenting cells (APCs), such as DCs, that migrate to the lymph nodes, where they present the TAAs through the major histocompatibility complex (MHC) to naïve T-cells (step 2). This presentation drives T-cell priming and activation (step 3). Finally, activated cytotoxic T lymphocytes (CTLs) leave the lymph nodes and enter the bloodstream (step 4), where chemokine detection leads to extravasation into the TME (step 5). There, cancer cells must be recognized by CTLs (step 6) and killed (step 7) [23]. Although epigenetic changes are involved in all the listed steps of the cancer-immunity cycle, our review emphasizes the epigenetic regulation of those steps involving tumor cells (steps 1, 5, 6, 7; Figure 2). Epigenetic changes involving DC maturation and CTL activation have been reviewed elsewhere [24,25].
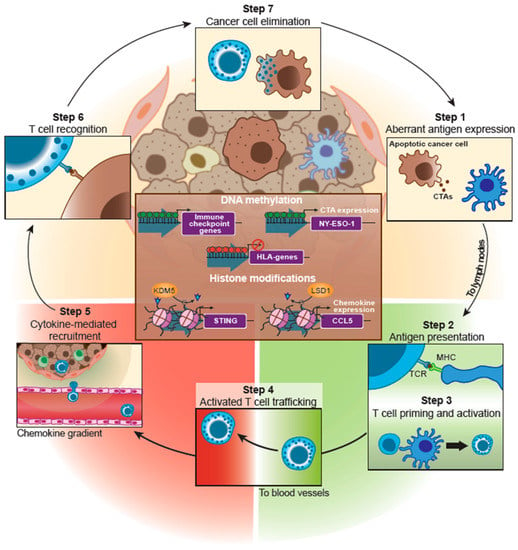
Figure 2.
Epigenetic regulation of cancer–immunity cycle in TNBC. This scheme shows epigenetic mechanisms involved in TNBC immune escape (brown square in the middle). It also includes the different steps of the cancer immunity cycle. Scheme based on Chen and Mellman (2013) cancer-cell immunity cycle [23]. Abbreviations: CTAs (Cancer testis antigens), TCR (T-cell receptor), MHC (Major Histocompatibility complex).
2.1. Aberrant Antigen Expression
The tumor-specific global hypomethylation and chromatin organization promote the reactivation of epigenetically silenced genes in healthy tissues [26]. Some of these genes may trigger an immune response since they act as neoantigens [27] or immunogenic cancer-testis antigens (CTAs), which act as TAAs. CTAs are downregulated in somatic adult tissues but aberrantly expressed in different malignancies. In addition, treatment with demethylating agents promotes CTA re-expression, suggesting that DNA methylation is responsible for silencing in somatic tissues [28]. Similarly, gene promoter hypermethylation has been proposed as a mechanism able to silence neo-antigen expression [27]. In TNBC, specifically, different studies have identified the upregulation of NY-ESO-1, MAGE-1, WT1, and SPANXB1 [29,30,31]. Thus, aberrant CTA expression in TNBC has become a therapeutic opportunity. Clinical trials based on vaccines targeting CTAs are currently being performed in solid tumors [32]. Beyond their potential relevance as therapeutic targets, CTAs also have an impact on TNBC tumor biology. For example, SPANXB1 expression was associated with increased migration and invasion abilities. In addition, its mRNA and protein levels were negatively correlated with the metastasis suppressor gene SH3GL2 [31].
Global hypomethylation also disturbs the immune response through the abnormal expression of endogenous retroviruses (ERV) [33]. Although ERV activation drives the expression of oncogenes [34], this alteration also produces double-stranded RNA molecules, which promote an IFN-mediated viral mimicry response [35], activating the innate immune response. Nevertheless, cancer cells may overcome this setback. For example, taxane-resistant TNBC cell lines display global DNA hypomethylation, but an epigenetic switch prevents ERV activation. EZH2, a histone writer whose upregulation is mainly observed in TNBC [36], represses ERV sequences through H3K27me3 histone mark deposition, avoiding viral mimicry and eluding the immune system [37]. Viral mimicry and its further antiviral signaling have been observed after spliceosome-targeted therapies in TNBC, promoting innate and adaptive immune responses and providing new therapeutic strategies [38]. Taken together, the modulation of the TNBC epigenome may promote viral mimicry responses or tumor-associated antigen expression, which in turn may trigger an immune response.
2.2. Chemokine-Mediated Recruitment
After antigen presentation and T-cell activation in the lymph nodes, CD8+ cytotoxic lymphocytes (CTLs) migrate through blood vessels. There, CTLs may detect a chemokine gradient and extravasate into the TME. The number of CTLs recruited at the tumor site is a predictor of immunotherapy response across different cancers [39], mediated by chemokine production on the tumor site [40]. Furthermore, high levels of the C-X-C motif chemokine ligands 9 and 10 (CXCL9 and CXCL10), C-C motif chemokine ligand 5 (CCL5), and IFN-y are associated with enhanced levels of CTLs in the TME [41]. This recruitment correlates with increased survival and lower levels of cancer metastasis in cancer patients [42]. Using TNBC cell lines, Qin et al. elucidated the possible mechanisms that underlie epigenetic dysregulation in the activity of chemokines and how they blocked antitumor immune cells’ circulation. They concluded that an epigenetic modifier, Lysine-Specific Demethylase 1 (LSD1), altered the cell landscape in TNBC through chemokine silencing, especially CCL5 and CXCL10 expression [43].
2.3. T-Cell Recognition & Antigen Presentation
Inhibition of the immune system is closely interconnected with tumor progression and development. The downregulation of antigen processing and presentation, especially the lack of MHC class I expression, allows tumor cells to evade immune surveillance [44]. Downregulation of MHC class I occurs more commonly than full elimination since full depletion makes cancer cells sensitive to the effect of NK cells via non-classical MHC molecules [45]. Moreover, epigenetic dysregulation of antigen processing and the presentation of machinery-related genes has been observed in cancer cells. These alterations include the depletion of the MHC class I transactivator NLRC5 and the HLA class II-chaperone CD74 through DNA methylation in different types of cancer [46,47].
MHC class I impairment has been observed as a mechanism of immunotherapeutic resistance, particularly in the TNBC apocrine subtype [48] and metastatic TNBC [49]. In metastatic TNBC, high expression of the transmembrane protein MAL2 diminishes the level and stability of the antigen-loaded MHC class I on the cell membrane, promoting an ineffective antigen presentation and consequently limited recognition by CD8+ T-cells [50].
In TNBC, aberrant overexpression of the MHC class II pathway is associated with increased T-cell infiltration and prolonged progression-free survival (PFS) [51]. In addition, the expression of MHC class II molecules, such as HLA-DR, in tumor tissue has been linked to the presence of tumor-infiltrating lymphocytes (TILs) and high expression of CD4, CD3D, and CD8A [52]. MHC class I and MHC class II expression are regulated through promoter DNA methylation of their coding HLA genes. An inverse correlation exists between the mRNA expression levels and DNA methylation on the surrounding region of transcription start sites of HLA genes in BC. Moreover, hypermethylation of HLA promoters also correlates with decreased CD8A mRNA levels [53].
Epigenetic modulation using the DNA Methyltransferase inhibitor (DNMTi) guadecitabine, a next-generation hypomethylating agent, promotes effective CD8 T-cell responses via increased MHC class I expression, increased IFN-y secretion, and recruitment of CTLs into the TME. Thus, DNMTi treatment might have countless effects on the interplay between tumor and immune systems, such as immune response activation and demethylation of MHC class I [53,54]. Regarding MHC class I, studies have identified the melanoma-associated antigen-A11 (MAGE-A11) peptides presented by HLA class I molecules [55,56,57]. MAGE-A11 is a CTA usually expressed in BC and related to poor prognosis. Therefore, its induction is relevant for the recognition and killing of BC cells by CTLs. Furthermore, MAGE-A11 antigens induced cytotoxicity on MAGE-A11-positive TNBC cells by effector CTLs [58].
Different alterations may lead to the release of double-stranded DNA into the cytosol. These events activate the cGAS-STING pathway, which provokes a signaling cascade that enhances the transcriptional expression of type I interferons and other immune-stimulatory genes, promoting an immune response [59]. Given its role as an immune-stimulatory pathway, cancer cells tend to downregulate STING expression levels. For instance, KDM5-mediated histone demethylation and transcription repression was observed in BC cell lines. However, this epigenetic signaling has not been studied in TNBC cell lines or in vivo models. [60]. Given this relevance, the development of human STING agonists has arisen. In fact, the administration of STING agonists resensitized TNBC immunocompetent mice against ICI [61,62].
2.4. Cancer Cell Elimination
Once recognized, CTLs can eliminate cancer cells, whereas immune checkpoints (ICs) can compromise this immune response. ICs include different inhibitory pathways that control the intensity and duration of immune responses [63]. It is well known that in the TME, cancer cells can escape the cytotoxic effect of T-cells by activating IC pathways [64]. The best-described strategies to de-activate T-cells are the binding of PD-1 on T-cells to programmed death-ligand 1 (PD-L1) on cancer cells and APCs [65] and CTLA-4 on T-cells to CD80/86 on APCs [66]. Different IC proteins, such as TIM-3, CTLA-4, and LAG-3, are upregulated in primary BC through epigenetic mechanisms, including DNA hypomethylation and decreased repressive histone marks H3K27me3 and H3K9me3, in the promoter regions [67]. Regarding PD-L1 regulation, it is well established that DNA methylation affects its expression in different cancers, including melanoma and gastric cancer [68,69]. However, PD-L1 seems to be completely demethylated in BC, and its regulation is based on histone modifications [67,70]. The upregulation of different immune checkpoint proteins is supported by aberrant epigenetics events that may be corrected using epigenetic drugs. Thus, histone deacetylase inhibitors (HDACi) and DNMTi can revert the aberrant expression and restore antitumor immunity [71].
In this context, Terranova-Barberio et al. evaluated the effect of HDACi combined with ICI to enhance immunotherapy responses in TNBC in vivo. They observed that HDACi upregulates PD-L1 and HLA-DR expression in TNBC cells and improves the response to PD-1/CTLA-4 blockade in a TNBC mouse model, decreasing tumor growth and improving survival. This was associated with increased T-cell tumor infiltration and downregulation of CD4+ and FOXP3+ T-cells [72].
Beyond CTLA-4 and PD-1/PD-L1, there is an increasing interest in inhibitory receptors as potential targets in immunotherapy. CD155 and CD112 are expressed on the tumor cell surface. These markers interact with the T-cell immunoreceptor with Ig, and ITIM domains (TIGIT) found on NK, CD8+, and CD4+ T-cell membranes [73]. TIGIT is poorly expressed on naïve T-cells, but it is overexpressed after promoter hypomethylation and FOXP3 binding [74]. The overexpression of TIGIT ligand CD155, encoded by the PVR gene, has been reported in metastatic BC compared with normal tissue [75].
Furthermore, TNBC displays higher CD155 expression than other BC subtypes [75,76]. Interestingly, an active enhancer has been found close to the PVR promoter, which may explain this phenotype (Figure 3). This overexpression was observed in all BC subtypes, being more prevalent in TNBC. CD155 expression correlated with a worse prognosis in BC, pointing out its relevance on new treatments and the outcome prediction [75]. B7-H3 is another promising target in TNBC. Its enhanced expression on TAMs and cancer cells in TNBC patients promoted a pro-angiogenic state and correlated with a worse prognosis [77]. The antibody-mediated inhibition of this immune receptor enhanced the therapeutic effect of PD-1 blockade in TNBC murine models [78]. An in vitro study performed on a TNBC cell line revealed that B7-H3 knockdown reduced glycolytic activity and sensitized cells against AKT/mTOR inhibitors [79]. Different studies point out different epigenetic mechanisms disrupted cancer progression, promoting increased B7-H3 expression levels [80,81,82].
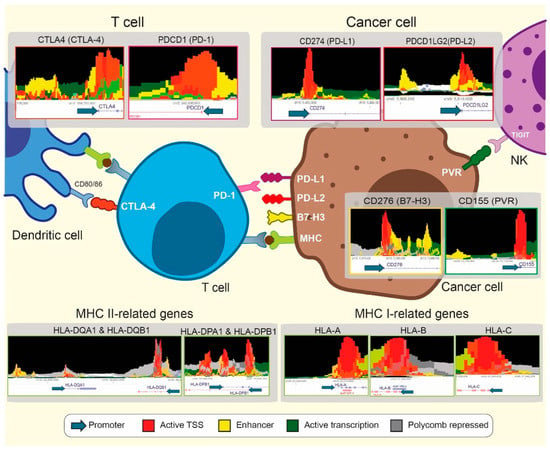
Figure 3.
Therapeutic opportunities combining epigenetics and immunotherapy. Different GRE, including promoters and enhancers, have been identified close to the transcription start site (TSS) of IC-related genes using the Epilogos visualization tool (https://epilogos.altius.org/, accessed on 29 April 2021). A total of 127 human samples from the Roadmap Consortium were considered when cancer cell-expressing genes were interrogated. In contrast, the blood & T cells category was selected from T cell and NK expressing genes.
3. Other Connections between Epigenetics and Immunotherapy in TNBC
3.1. Tumor Microenvironment
The TME composition also impacts immunotherapy response. Tumors can be classified as ‘hot’ when they exert T-cell infiltration and inflammation or ‘cold’ when enriched in immunosuppressive cells, such as TAMs, myeloid-derived suppressor cells (MDSCs), and regulatory T-cells (Tregs). Hot tumors show a better response against IC therapies. For that reason, several strategies—including epigenetic reprogramming—aim to turn ‘cold’ tumors into ‘hot’ [71] (Figure 4).
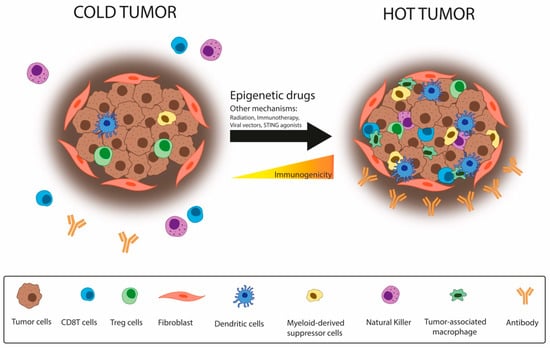
Figure 4.
Epigenetic drugs contribute to turning cold tumors into hot. Cancer cells modulate their microenvironment to promote immune escape, increasing the presence of immunosuppressive cells on the tumor site and becoming a “cold tumor”, which displays a worse response against immunotherapy. Epigenetic drugs may switch this state, enhancing the immune response through the recruitment of CTLs, becoming a “hot tumor”. Hot tumors respond better to immunotherapy.
Consequently, the presence of CD8+ infiltrating T-cells is associated with a good prognosis and a better response against anti-PD-L1 treatments in TNBC and other cancer subtypes [83,84,85]. EZH2 overexpression correlates with decreased infiltration of CD8+ T-cells, translating into a ‘cold’ phenotype. Persistent antigen stimulation of CD8+ T-cells and an immunosuppressive TME contribute to CD8+ T-cell ‘exhaustion’, which is translated into the activation of Pdcd1 (encoding PD-1) and Il-10 signaling pathways [86]. This alteration has been observed in melanoma murine cell lines [87]. As far as we know, no study has covered the epigenetic role in this state of cell dysfunction, focusing on TNBC.
MDSCs also generate an immunosuppressive environment through many mechanisms, including nutrient depletion and Tregs recruitment [88]. In addition, this population contributes to the development of premetastatic niches [89]. Interestingly, low doses of epigenetic treatments impaired this population in TNBC murine models [90] and disrupted the metastatic niche formation in BC [89]. Eosinophils display pleiotropic effects on tumor sites since they can produce and secrete cytotoxic proteins and angiogenic and matrix-remodeling factors [91]. In TNBC, relative eosinophil count has been associated with a lower relapse rate, suggesting tumor-inhibiting phenotype in this particular malignancy [92]. Cancer cells can reprogram the TAMs expression patterns through different mechanisms. TNBC cell lines displayed paracrine signaling that epigenetically activated the ID4 promoter region in cancer cells but also in TAMs. This crosstalk promoted the activation of an angiogenic program, partially sustained by the downregulation of miR-15b/107 in TAMs [93,94]. The connections between TAMs and anti-PD-1/PD-L1 therapies are comprehensively reviewed by Santoni et al. [95].
3.2. Metabolic Rewiring
Metabolic reprogramming is another hallmark of cancer. Despite its intrinsic heterogeneity, TNBC displays a higher glucose dependence when compared with other BC subtypes, a feature partially sustained by epigenetic alterations. These alterations include promoter hypermethylation of the gluconeogenic enzyme FBP1 [96] and HIF-1α stabilization by the lncRNA LINK-A, among others [97]. This glucose dependence is translated into an increase in lactate production even in normoxic conditions, a phenomenon known as the “Warburg effect” [98]. The acidification of the TME and the lactate release inhibit the immune response [99]. Aerobic glycolysis also promotes MDSC recruitment, which contributes to immune suppression [100]. In addition, lactate was recently identified as a novel histone posttranslational modification [101]. Further research may highlight the relevance of epigenetic mechanisms in the pH regulation in TME and its potential role in immune response [102].
The kynurenine pathway (KP) also couples epigenetic alterations and immune response. During inflammation, IFN-gamma induces indoleamine 2,3-dioxygenase (IDO) expression in cancer cells and MDSCs, the first enzyme of the KP [103]. IDO overexpression occurs preferentially on TNBC with basal-like subtypes [104,105]. It seems to be negatively regulated by promoter hypermethylation on ER+ BC [106]. IDO enzymatic activity may promote tryptophan depletion and immunosuppressive metabolites synthesis, inhibiting T-cell activity and inducing immune tolerance [107,108].
3.3. Epithelial-to-Mesenchymal Transition
Many epigenetic alterations are associated with epithelial-to-mesenchymal transition (EMT) in TNBC, which have been deeply discussed by Khaled et al. [109]. The authors summarize different epigenetic mechanisms—including DNA methylation, histone modification, and long non-coding RNA (lncRNA) interactions that promote EMT and metastasis. Interestingly, EMT has also been identified as a resistance mechanism to immunotherapy in TNBC [110]. Among other mechanisms, the axis miR-200/ZEB1, which controls the metastasis program, promotes PD-L1 expression [111].
4. Better Together, the Combination of Epigenetic Inhibitors and Immunotherapy
Since their approval, epigenetic drugs have demonstrated efficacy in cancer treatment, especially in hematological malignancies [112]. Clinical adoption of epigenetic drugs may increase during the following years, fueled by their potential role in preventing metastasis [89] and potential combination with immunotherapy [113]. Epigenetic modulation of molecular pathways involved in the cancer cell immunity cycle (Figure 2) through inhibitors against writers, readers, and erasers (Figure 1) promote a decrease in immune evasion and sensitize cancer cells against ICIs [114]. DNMTi and HDACi promote upregulation of CTAs, PDL-1/PD-L2 and MHC-I-related genes expression, the reactivation of repetitive elements, and an increase in the chemokine expression and release [15].
In addition to the effect on cancer cells, low doses of epigenetic drugs impact different immune cell populations: HDAC6 inhibition promotes the activation of naïve T-cells [115], whereas Class I HDACi increase the response of T CD8+ and NK cells [116]. Treg lymphocytes activity can also be modulated through epigenetic drugs. Tregs display an immune-suppressive program, which is orchestrated by the transcription factor FOXP3. Thus, the inhibitor-mediated decrease of FOXP3 instability through the inhibition of the histone acetyltransferase EP300 may contribute to reestablishing the immune response [117,118].
DNMT1, whose upregulation on all BC subtypes correlates with a worse prognosis [119], could be modulated with decitabine, a DNMTi [120]. Decitabine efficacy is being tested with carboplatin and in combination with pembrolizumab, an anti-PD-1 antibody (NCT03295552 and NCT02957968, respectively). Furthermore, the reversibility of estrogen receptor depletion is being tested by combining decitabine and tamoxifen (NCT01194908). 5’-azacytidine, one of the first DNMTi, is being tested alone or combined with entinostat, an HDAC I inhibitor (NCT01292083 and NCT01349959, respectively). Clinical trials involving epigenetic drugs alone or combined with either immunotherapy or chemotherapy are listed in Table 1.

Table 1.
Ongoing clinical trials of epigenetic drugs in TNBC.
Entinostat has been tested in combination with first-line treatments such as doxorubicin [121] and immunotherapy. Entinostat enhanced the effect of an IL-15 agonist and a vaccine against TAAs in TNBC murine models [122] and displayed synergistic effects when combined with a PD-L1 inhibitor through its effect on MDSCs [123]. Clinical trials involving this epigenetic drug alone (NCT03361800) or in combination with atezolizumab (a PD-L1 inhibitor; NCT02708680) are being conducted. In addition, another HDAC I inhibitor called vorinostat is being clinically tested (NCT01695057). Beyond the first generation of epigenetic drugs targeting DNMTs and HDACs, more recently, inhibitors for bromodomains have been developed. BRD4 was identified as a potential target in TNBC [124]. Since then, preclinical and clinical studies have highlighted that chemical inhibition of this BET bromodomain is an efficient treatment in TNBC patients [125,126,127]. Moreover, novel approaches using BET-proteolysis targeting chimeric compounds (BET-PROTACs), which allosterically inhibit BET bromodomains and bind a ubiquitin ligase, have been tested in TNBC with promising results, even in BET-resistant tumors [128]. Infiltration of TAMs in the TME correlates to BET inhibitor resistance in TNBC [129]. Furthermore, a recent study showed that BRD4 blockade impairs PD-L1 expression in TNBC [130].
5. Future Perspectives
The presence of epigenetic alterations modulates the immune response in TNBC, impairing antigen expression, decreasing chemokine production, developing an immunosuppressive TME, and promoting tumor survival through promoting immune-evasive mechanisms. Epigenome signatures involved in immune escape may represent an opportunity to identify complementary therapies and patients that may not respond to ICIs, as tested in other cancer types, such as non-small cell lung cancer [131]. In addition, these new approaches may complement the current biomarkers involved in response to immunotherapy by building integrative nomograms including epigenetic features and additional biomarkers, such as tumor neoantigens, intratumor heterogeneity, tumor metabolism, tumor-immune microenvironment gut microbiome, and checkpoints targets. These include PD-L1 protein expression and a fraction of high PD-1 mRNA [84]. Altogether, the interaction of these parameters may be depicted in individual patients as ‘cancer immunograms’ that will allow a more personalized stratification to guide immunotherapy decision-making [132].
Furthermore, the reported benefits of epigenetic drugs combined with immunotherapy might lead to strategies to further improve immunotherapy in TNBC patients. We also believe that the research focusing on cancer epigenetics will move to key GRE beyond promoters recently reported in TNBC [133]. In fact, key IC-related genes display enhancer elements close to the promoter region with potential involvement in immune response, as described in Figure 3. The characterization of these elements will provide a better understanding of cancer biology behind immune response and may provide new therapeutic targets. Taken together, growing evidence supports that epigenetic modulation is relevant for immunotherapy, and its clinical implementation may be a turning point to improve the outcome of patients with the most aggressive type of BC.
Author Contributions
All authors listed a relevant, direct, and intellectual contribution to the work and approved the final version of the manuscript for publication. P.L.-A. and S.Í.-M. wrote the original draft and designed the figures. K.M., L.V., J.I.J.O. and M.L.D. provided significant feed-back for the whole manuscript, especially contributing to the translational sections. M.E.-M. and B.S. actively participated in draft editing and review. D.M.M. provided the main guidelines and supervised the whole manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Instituto de la Salud Carlos III Miguel Servet Project (#CP17/00188 to D.M.M.) and AES2019 (#I19/01514 to D.M.M.), the Institut d’Investigació Sanitària Illes Balears (IdISBa) FUTURMed FOLIUM program (to B.S.), the Associates for Breast and Prostate Cancer Studies (ABCs to J.I.J.O.) Foundation, the Fashion Footwear Association of New York (FFANY to J.I.J.O.) Foundation, the Asociación Española Contra el Cancer (AECC to M.E.M.) Foundation, the Balearic Islands Government Margalida Comas program (to P.L.-A., the Fundación Francisco Cobos, the Hendrika Roet fund (to L.V.), and the UCLA Breast Cancer Epigenetics Research Program (to M.L.D.).
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Hudis, C.A.; Gianni, L. Triple-negative breast cancer: An unmet medical need. Oncologist 2011, 16, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ensenyat-Mendez, M.; Llinàs-Arias, P.; Orozco, J.I.J.; Íñiguez-Muñoz, S.; Salomon, M.P.; Sesé, B.; DiNome, M.L.; Marzese, D.M. Current Triple-Negative Breast Cancer Subtypes: Dissecting the Most Aggressive Form of Breast Cancer. Front. Oncol. 2021, 11, 2311. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emens, L.A.; Molinero, L.; Loi, S.; Rugo, H.S.; Schneeweiss, A.; Diéras, V.; Iwata, H.; Barrios, C.H.; Nechaeva, M.; Duc, A.N.; et al. Atezolizumab and nab-Paclitaxel in Advanced Triple-Negative Breast Cancer: Biomarker Evaluation of the IMpassion130 Study. J. Natl. Cancer Instig. 2021, 113, 1005–1016. [Google Scholar] [CrossRef]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.-A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. KEYNOTE-355 Investigators Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Zhang, H.; Barrios, C.H.; Saji, S.; Jung, K.H.; Hegg, R.; Koehler, A.; Sohn, J.; Iwata, H.; Telli, M.L.; et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): A randomised, double-blind, phase 3 trial. Lancet 2020, 396, 1090–1100. [Google Scholar] [CrossRef]
- Adams, S.; Schmid, P.; Rugo, H.S.; Winer, E.P.; Loirat, D.; Awada, A.; Cescon, D.W.; Iwata, H.; Campone, M.; Nanda, R.; et al. Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: Cohort A of the phase II KEYNOTE-086 study. Ann. Oncol. 2019, 30, 397–404. [Google Scholar] [CrossRef] [Green Version]
- McGrail, D.J.; Pilié, P.G.; Rashid, N.U.; Voorwerk, L.; Slagter, M.; Kok, M.; Jonasch, E.; Khasraw, M.; Heimberger, A.B.; Lim, B.; et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann. Oncol. 2021, 32, 661–672. [Google Scholar] [CrossRef]
- Simó-Riudalbas, L.; Esteller, M. Targeting the histone orthography of cancer: Drugs for writers, erasers and readers. Br. J. Pharmacol. 2015, 172, 2716–2732. [Google Scholar] [CrossRef] [Green Version]
- Llinàs-Arias, P.; Esteller, M. Epigenetic inactivation of tumour suppressor coding and non-coding genes in human cancer: An update. Open Biol. 2017, 7, 170152. [Google Scholar] [CrossRef] [Green Version]
- Tutt, A.; Tovey, H.; Cheang, M.C.U.; Kernaghan, S.; Kilburn, L.; Gazinska, P.; Owen, J.; Abraham, J.; Barrett, S.; Barrett-Lee, P.; et al. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: The TNT Trial. Nat. Med. 2018, 24, 628–637. [Google Scholar] [CrossRef] [Green Version]
- Skvortsova, K.; Stirzaker, C.; Taberlay, P. The DNA methylation landscape in cancer. Essays Biochem. 2019, 63, 797–811. [Google Scholar]
- Mokhtari, R.B.; Sambi, M.; Qorri, B.; Baluch, N.; Ashayeri, N.; Kumar, S.; Cheng, H.-L.M.; Yeger, H.; Das, B.; Szewczuk, M.R. The Next-Generation of Combination Cancer Immunotherapy: Epigenetic Immunomodulators Transmogrify Immune Training to Enhance Immunotherapy. Cancers 2021, 13, 3596. [Google Scholar] [CrossRef] [PubMed]
- Barrero, M.J. Epigenetic Regulation of the Non-Coding Genome: Opportunities for Immuno-Oncology. Epigenomes 2020, 4, 22. [Google Scholar] [CrossRef]
- Kather, J.N.; Halama, N. Harnessing the innate immune system and local immunological microenvironment to treat colorectal cancer. Br. J. Cancer 2019, 120, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Leavenworth, J.W.; Li, Y.; Luo, Q.; Xie, H.; Liu, X.; Huang, S.; Yan, H.; Fu, Z.; Zhang, L.Y.; et al. Ezh2 regulates differentiation and function of natural killer cells through histone methyltransferase activity. Proc. Natl. Acad. Sci. USA 2015, 112, 15988–15993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez-Errico, D.; Vento-Tormo, R.; Sieweke, M.; Ballestar, E. Epigenetic control of myeloid cell differentiation, identity and function. Nat. Rev. Immunol. 2015, 15, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Baragaño Raneros, A.; Martín-Palanco, V.; Fernandez, A.F.; Rodriguez, R.M.; Fraga, M.F.; Lopez-Larrea, C.; Suarez-Alvarez, B. Methylation of NKG2D ligands contributes to immune system evasion in acute myeloid leukemia. Genes Immun. 2015, 16, 71–82. [Google Scholar] [CrossRef]
- Meng, Z.; Zhang, R.; Wang, Y.; Zhu, G.; Jin, T.; Li, C.; Zhang, S. miR-200c/PAI-2 promotes the progression of triple negative breast cancer via M1/M2 polarization induction of macrophage. Int. Immunopharmacol. 2020, 81, 106028. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.-Y.; Luo, R.-Z.; Peng, R.-J.; Wang, S.-S.; Xue, C. High infiltration of tumor-associated macrophages in triple-negative breast cancer is associated with a higher risk of distant metastasis. OncoTargets Ther. 2014, 7, 1475–1480. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Ubreva, J.; Garcia-Gomez, A.; Ballestar, E. Epigenetic mechanisms of myeloid differentiation in the tumor microenvironment. Curr. Opin. Pharmacol. 2017, 35, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gomez, A.; Rodríguez-Ubreva, J.; Ballestar, E. Epigenetic interplay between immune, stromal and cancer cells in the tumor microenvironment. Clin. Immunol. 2018, 196, 64–71. [Google Scholar] [CrossRef]
- Kim, R.; Kulkarni, P.; Hannenhalli, S. Derepression of Cancer/testis antigens in cancer is associated with distinct patterns of DNA hypomethylation. BMC Cancer 2013, 13, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Bakir, M.A.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanić, M.; Reading, J.L.; Joshi, K.; et al. TRACERx consortium Neoantigen-directed immune escape in lung cancer evolution. Nature 2019, 567, 479–485. [Google Scholar] [CrossRef]
- Barrero, M.J. Epigenetic Strategies to Boost Cancer Immunotherapies. Int. J. Mol. Sci. 2017, 18, 1108. [Google Scholar] [CrossRef] [Green Version]
- Raghavendra, A.; Kalita-de Croft, P.; Vargas, A.C.; Smart, C.E.; Simpson, P.T.; Saunus, J.M.; Lakhani, S.R. Expression of MAGE-A and NY-ESO-1 cancer/testis antigens is enriched in triple-negative invasive breast cancers. Histopathology 2018, 73, 68–80. [Google Scholar] [CrossRef] [Green Version]
- Curigliano, G.; Bagnardi, V.; Ghioni, M.; Louahed, J.; Brichard, V.; Lehmann, F.F.; Marra, A.; Trapani, D.; Criscitiello, C.; Viale, G. Expression of tumor-associated antigens in breast cancer subtypes. Breast 2020, 49, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Kannan, A.; Philley, J.V.; Hertweck, K.L.; Ndetan, H.; Singh, K.P.; Sivakumar, S.; Wells, R.B.; Vadlamudi, R.K.; Dasgupta, S. Cancer Testis Antigen Promotes Triple Negative Breast Cancer Metastasis and is Traceable in the Circulating Extracellular Vesicles. Sci. Rep. 2019, 9, 11632. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, M.; Tono, Y.; Miyahara, Y.; Muraoka, D.; Harada, N.; Kageyama, S.; Sasaki, T.; Hori, Y.; Soga, N.; Uchida, K.; et al. First-in-human phase I clinical trial of the NY-ESO-1 protein cancer vaccine with NOD2 and TLR9 stimulants in patients with NY-ESO-1-expressing refractory solid tumors. Cancer Immunol. Immunother. 2020, 69, 663–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.A.; Ohtani, H.; Chakravarthy, A.; De Carvalho, D.D. Epigenetic therapy in immune-oncology. Nat. Rev. Cancer 2019, 19, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.S.; Shah, N.M.; Du, A.Y.; Dailey, Z.Z.; Pehrsson, E.C.; Godoy, P.M.; Zhang, D.; Li, D.; Xing, X.; Kim, S.; et al. Transposable elements drive widespread expression of oncogenes in human cancers. Nat. Genet. 2019, 51, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [Green Version]
- Holm, K.; Grabau, D.; Lövgren, K.; Aradottir, S.; Gruvberger-Saal, S.; Howlin, J.; Saal, L.H.; Ethier, S.P.; Bendahl, P.-O.; Stål, O.; et al. Global H3K27 trimethylation and EZH2 abundance in breast tumor subtypes. Mol. Oncol. 2012, 6, 494–506. [Google Scholar] [CrossRef] [Green Version]
- Deblois, G.; Tonekaboni, S.A.M.; Grillo, G.; Martinez, C.; Kao, Y.I.; Tai, F.; Ettayebi, I.; Fortier, A.-M.; Savage, P.; Fedor, A.N.; et al. Epigenetic Switch-Induced Viral Mimicry Evasion in Chemotherapy-Resistant Breast Cancer. Cancer Discov. 2020, 10, 1312–1329. [Google Scholar] [CrossRef] [PubMed]
- Bowling, E.A.; Wang, J.H.; Gong, F.; Wu, W.; Neill, N.J.; Kim, I.S.; Tyagi, S.; Orellana, M.; Kurley, S.J.; Dominguez-Vidaña, R.; et al. Spliceosome-targeted therapies trigger an antiviral immune response in triple-negative breast cancer. Cell 2021, 184, 384–403.e21. [Google Scholar] [CrossRef]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; van de Vijver, K.K.; de Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef] [Green Version]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [Green Version]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Vasilatos, S.N.; Chen, L.; Wu, H.; Cao, Z.; Fu, Y.; Huang, M.; Vlad, A.M.; Lu, B.; Oesterreich, S.; et al. Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances antitumor efficacy of immune checkpoint blockade. Oncogene 2019, 38, 390–405. [Google Scholar] [CrossRef]
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 2018, 18, 139–147. [Google Scholar] [CrossRef]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [Green Version]
- Shukla, A.; Cloutier, M.; Appiya Santharam, M.; Ramanathan, S.; Ilangumaran, S. The MHC Class-I Transactivator NLRC5: Implications to Cancer Immunology and Potential Applications to Cancer Immunotherapy. Int. J. Mol. Sci. 2021, 22, 1964. [Google Scholar] [CrossRef]
- Zeiner, P.S.; Zinke, J.; Kowalewski, D.J.; Bernatz, S.; Tichy, J.; Ronellenfitsch, M.W.; Thorsen, F.; Berger, A.; Forster, M.T.; Muller, A.; et al. CD74 regulates complexity of tumor cell HLA class II peptidome in brain metastasis and is a positive prognostic marker for patient survival. Acta Neuropathol. Commun. 2018, 6, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dusenbery, A.C.; Maniaci, J.L.; Hillerson, N.D.; Dill, E.A.; Bullock, T.N.; Mills, A.M. MHC Class I Loss in Triple-negative Breast Cancer: A Potential Barrier to PD-1/PD-L1 Checkpoint Inhibitors. Am. J. Surg. Pathol. 2021, 45, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.H.; Hood, B.L.; Beck, H.C.; Conrads, T.P.; Ditzel, H.J.; Leth-Larsen, R. Downregulation of antigen presentation-associated pathway proteins is linked to poor outcome in triple-negative breast cancer patient tumors. Oncoimmunology 2017, 6, e1305531. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Wang, L.; Wan, C.; Sun, Y.; Van der Jeught, K.; Zhou, Z.; Dong, T.; So, K.M.; Yu, T.; Li, Y.; et al. MAL2 drives immune evasion in breast cancer by suppressing tumor antigen presentation. J. Clin. Invest. 2021, 131. [Google Scholar] [CrossRef]
- Forero, A.; Li, Y.; Chen, D.; Grizzle, W.E.; Updike, K.L.; Merz, N.D.; Downs-Kelly, E.; Burwell, T.C.; Vaklavas, C.; Buchsbaum, D.J.; et al. Expression of the MHC Class II Pathway in Triple-Negative Breast Cancer Tumor Cells Is Associated with a Good Prognosis and Infiltrating Lymphocytes. Cancer Immunol. Res. 2016, 4, 390–399. [Google Scholar] [CrossRef] [Green Version]
- Stewart, R.L.; Matynia, A.P.; Factor, R.E.; Varley, K.E. Spatially-resolved quantification of proteins in triple negative breast cancers reveals differences in the immune microenvironment associated with prognosis. Sci. Rep. 2020, 10, 6598. [Google Scholar] [CrossRef] [Green Version]
- Luo, N.; Nixon, M.J.; Gonzalez-Ericsson, P.I.; Sanchez, V.; Opalenik, S.R.; Li, H.; Zahnow, C.A.; Nickels, M.L.; Liu, F.; Tantawy, M.N.; et al. DNA methyltransferase inhibition upregulates MHC-I to potentiate cytotoxic T lymphocyte responses in breast cancer. Nat. Commun. 2018, 9, 248. [Google Scholar] [CrossRef]
- Luo, N.; Sugiura, A.; Balko, J.M. Therapeutic potential of DNA methyltransferase inhibitors with immune checkpoint inhibitor therapy in breast cancer. Cell Stress 2018, 2, 69–71. [Google Scholar] [CrossRef] [Green Version]
- Roch, N.; Kutup, A.; Vashist, Y.; Yekebas, E.; Kalinin, V.; Izbicki, J.R. Coexpression of MAGE-A peptides and HLA class I molecules in hepatocellular carcinoma. Anticancer. Res. 2010, 30, 1617–1623. [Google Scholar]
- Guo, L.; Sang, M.; Liu, Q.; Fan, X.; Zhang, X.; Shan, B. The expression and clinical significance of melanoma-associated antigen-A1, -A3 and -A11 in glioma. Oncol. Lett. 2013, 6, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Park, J.-W.; Chun, Y.-S. Jumonji histone demethylases as emerging therapeutic targets. Pharmacol. Res. 2016, 105, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sang, M.; Gu, L.; Liu, F.; Li, W.; Yin, D.; Wu, Y.; Liu, S.; Huang, W.; Shan, B. Zebularine Treatment Induces MAGE-A11 Expression and Improves CTL Cytotoxicity Using a Novel Identified HLA-A2-restricted MAGE-A11 Peptide. J. Immunother. 2017, 40, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Cao, J.; Cai, W.L.; Lang, S.M.; Horton, J.R.; Jansen, D.J.; Liu, Z.Z.; Chen, J.F.; Zhang, M.; Mott, B.T.; et al. KDM5 histone demethylases repress immune response via suppression of STING. PLoS Biol. 2018, 16, e2006134. [Google Scholar] [CrossRef] [Green Version]
- Cheng, N.; Watkins-Schulz, R.; Junkins, R.D.; David, C.N.; Johnson, B.M.; Montgomery, S.A.; Peine, K.J.; Darr, D.B.; Yuan, H.; McKinnon, K.P.; et al. A nanoparticle-incorporated STING activator enhances antitumor immunity in PD-L1-insensitive models of triple-negative breast cancer. JCI Insight 2018, 3, e120638. [Google Scholar] [CrossRef]
- Sceneay, J.; Goreczny, G.J.; Wilson, K.; Morrow, S.; DeCristo, M.J.; Ubellacker, J.M.; Qin, Y.; Laszewski, T.; Stover, D.G.; Barrera, V.; et al. Interferon Signaling Is Diminished with Age and Is Associated with Immune Checkpoint Blockade Efficacy in Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 1208–1227. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakimi, K.; Karasaki, T.; Matsushita, H.; Sugie, T. Advances in personalized cancer immunotherapy. Breast Cancer 2017, 24, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [Green Version]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasidharan Nair, V.; Salhat, H.E.; Taha, R.Z.; John, A.; Ali, B.R.; Elkord, E. DNA methylation and repressive H3K9 and H3K27 trimethylation in the promoter regions of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, and PD-L1 genes in human primary breast cancer. Clin. Epigenetics 2018, 10, 78. [Google Scholar] [CrossRef] [Green Version]
- Micevic, G.; Thakral, D.; McGeary, M.; Bosenberg, M.W. PD-L1 methylation regulates PD-L1 expression and is associated with melanoma survival. Pigment. Cell Melanoma Res. 2019, 32, 435–440. [Google Scholar] [CrossRef]
- Lv, D.; Xing, C.; Cao, L.; Zhuo, Y.; Wu, T.; Gao, N. PD-L1 gene promoter methylation represents a potential diagnostic marker in advanced gastric cancer. Oncol. Lett. 2020, 19, 1223–1234. [Google Scholar] [CrossRef] [Green Version]
- Darvin, P.; Sasidharan Nair, V.; Elkord, E. PD-L1 Expression in Human Breast Cancer Stem Cells Is Epigenetically Regulated through Posttranslational Histone Modifications. J. Oncol. 2019, 2019, 3958908. [Google Scholar] [CrossRef] [Green Version]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef]
- Terranova-Barberio, M.; Thomas, S.; Ali, N.; Pawlowska, N.; Park, J.; Krings, G.; Rosenblum, M.D.; Budillon, A.; Munster, P.N. HDAC inhibition potentiates immunotherapy in triple negative breast cancer. Oncotarget 2017, 8, 114156–114172. [Google Scholar] [CrossRef] [Green Version]
- Chauvin, J.-M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Maksimovic, J.; Naselli, G.; Qian, J.; Chopin, M.; Blewitt, M.E.; Oshlack, A.; Harrison, L.C. Genome-wide DNA methylation analysis identifies hypomethylated genes regulated by FOXP3 in human regulatory T cells. Blood 2013, 122, 2823–2836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.-C.; Zhou, Q.; Song, Q.-K.; Wang, R.-B.; Lyu, S.; Guan, X.; Zhao, Y.-J.; Wu, J.-P. Overexpression of an Immune Checkpoint (CD155) in Breast Cancer Associated with Prognostic Significance and Exhausted Tumor-Infiltrating Lymphocytes: A Cohort Study. J. Immunol. Res. 2020, 2020, 3948928. [Google Scholar] [CrossRef] [PubMed]
- Stamm, H.; Oliveira-Ferrer, L.; Grossjohann, E.-M.; Muschhammer, J.; Thaden, V.; Brauneck, F.; Kischel, R.; Müller, V.; Bokemeyer, C.; Fiedler, W.; et al. Targeting the TIGIT-PVR immune checkpoint axis as novel therapeutic option in breast cancer. Oncoimmunology 2019, 8, e1674605. [Google Scholar] [CrossRef]
- Cheng, N.; Bei, Y.; Song, Y.; Zhang, W.; Xu, L.; Zhang, W.; Yang, N.; Bai, X.; Shu, Y.; Shen, P. B7-H3 augments the pro-angiogenic function of tumor-associated macrophages and acts as a novel adjuvant target for triple-negative breast cancer therapy. Biochem. Pharmacol. 2021, 183, 114298. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-H.; Martin-Orozco, N.; Zheng, P.; Li, J.; Zhang, P.; Tan, H.; Park, H.J.; Jeong, M.; Chang, S.H.; Kim, B.-S.; et al. Inhibition of the B7-H3 immune checkpoint limits tumor growth by enhancing cytotoxic lymphocyte function. Cell Res. 2017, 27, 1034–1045. [Google Scholar] [CrossRef]
- Nunes-Xavier, C.E.; Karlsen, K.F.; Tekle, C.; Pedersen, C.; Øyjord, T.; Hongisto, V.; Nesland, J.M.; Tan, M.; Sahlberg, K.K.; Fodstad, Ø. Decreased expression of B7-H3 reduces the glycolytic capacity and sensitizes breast cancer cells to AKT/mTOR inhibitors. Oncotarget 2016, 7, 6891–6901. [Google Scholar] [CrossRef] [Green Version]
- Kanchan, R.K.; Perumal, N.; Atri, P.; Chirravuri Venkata, R.; Thapa, I.; Klinkebiel, D.L.; Donson, A.M.; Perry, D.; Punsoni, M.; Talmon, G.A.; et al. MiR-1253 exerts tumor-suppressive effects in medulloblastoma via inhibition of CDK6 and CD276 (B7-H3). Brain Pathol. 2020, 30, 732–745. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Z.; Zhang, C.; Liu, X.; Li, G.; Liu, S.; Sun, L.; Liang, J.; Hu, H.; Liu, Y.; et al. Genetic and clinical characterization of B7-H3 (CD276) expression and epigenetic regulation in diffuse brain glioma. Cancer Sci. 2018, 109, 2697–2705. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-Y.; Qi, B.; Wang, F.; Lin, Z.-R.; Li, M.-Y.; Yin, W.-J.; Zhu, Y.-Y.; He, L.; Yu, Y.; Yang, F.; et al. PBK phosphorylates MSL1 to elicit epigenetic modulation of CD276 in nasopharyngeal carcinoma. Oncogenesis 2021, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Vihervuori, H.; Autere, T.A.; Repo, H.; Kurki, S.; Kallio, L.; Lintunen, M.M.; Talvinen, K.; Kronqvist, P. Tumor-infiltrating lymphocytes and CD8+ T cells predict survival of triple-negative breast cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 3105–3114. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Ruppin, E. Multiomics Prediction of Response Rates to Therapies to Inhibit Programmed Cell Death 1 and Programmed Cell Death 1 Ligand. JAMA Oncol. 2019, 5, 1614–1618. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Thike, A.A.; Li, H.; Yeong, J.; Koo, S.-L.; Dent, R.A.; Tan, P.H.; Iqbal, J. Increased CD4 and CD8-positive T cell infiltrate signifies good prognosis in a subset of triple-negative breast cancer. Breast Cancer Res. Treat. 2016, 156, 237–247. [Google Scholar] [CrossRef]
- Villanueva, L.; Álvarez-Errico, D.; Esteller, M. The Contribution of Epigenetics to Cancer Immunotherapy. Trends Immunol. 2020, 41, 676–691. [Google Scholar] [CrossRef]
- Miller, B.C.; Sen, D.R.; Abosy, R.A.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef]
- Gomez, S.; Tabernacki, T.; Kobyra, J.; Roberts, P.; Chiappinelli, K.B. Combining epigenetic and immune therapy to overcome cancer resistance. Semin. Cancer Biol. 2020, 65, 99–113. [Google Scholar] [CrossRef]
- Lu, Z.; Zou, J.; Li, S.; Topper, M.J.; Tao, Y.; Zhang, H.; Jiao, X.; Xie, W.; Kong, X.; Vaz, M.; et al. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature 2020, 579, 284–290. [Google Scholar] [CrossRef]
- Kim, K.; Skora, A.D.; Li, Z.; Liu, Q.; Tam, A.J.; Blosser, R.L.; Diaz, L.A.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11774–11779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grisaru-Tal, S.; Itan, M.; Klion, A.D.; Munitz, A. A new dawn for eosinophils in the tumour microenvironment. Nat. Rev. Cancer 2020, 20, 594–607. [Google Scholar] [CrossRef] [PubMed]
- Onesti, C.E.; Josse, C.; Boulet, D.; Thiry, J.; Beaumecker, B.; Bours, V.; Jerusalem, G. Blood eosinophilic relative count is prognostic for breast cancer and associated with the presence of tumor at diagnosis and at time of relapse. Oncoimmunology 2020, 9, 1761176. [Google Scholar] [CrossRef] [PubMed]
- Donzelli, S.; Milano, E.; Pruszko, M.; Sacconi, A.; Masciarelli, S.; Iosue, I.; Melucci, E.; Gallo, E.; Terrenato, I.; Mottolese, M.; et al. Expression of ID4 protein in breast cancer cells induces reprogramming of tumour-associated macrophages. Breast Cancer Res. BCR 2018, 20, 59. [Google Scholar] [CrossRef] [PubMed]
- Donzelli, S.; Sacconi, A.; Turco, C.; Gallo, E.; Milano, E.; Iosue, I.; Blandino, G.; Fazi, F.; Fontemaggi, G. Paracrine Signaling from Breast Cancer Cells Causes Activation of ID4 Expression in Tumor-Associated Macrophages. Cells 2020, 9, 418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoni, M.; Romagnoli, E.; Saladino, T.; Foghini, L.; Guarino, S.; Capponi, M.; Giannini, M.; Cognigni, P.D.; Ferrara, G.; Battelli, N. Triple negative breast cancer: Key role of Tumor-Associated Macrophages in regulating the activity of anti-PD-1/PD-L1 agents. Biochim. Et Biophys. Acta. Rev. Cancer 2018, 1869, 78–84. [Google Scholar] [CrossRef]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.M.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.; Li, C.; Xing, Z.; Hu, Q.; Liang, K.; Han, L.; Wang, C.; Hawke, D.H.; Wang, S.; Zhang, Y.; et al. The LINK-A lncRNA activates normoxic HIF1α signalling in triple-negative breast cancer. Nat. Cell Biol. 2016, 18, 213–224. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.X.; Choi, S.Y.C.; Niu, X.; Kang, N.; Xue, H.; Killam, J.; Wang, Y. Lactic Acid and an Acidic Tumor Microenvironment suppress Anticancer Immunity. Int. J. Mol. Sci. 2020, 21, 8363. [Google Scholar] [CrossRef]
- Li, W.; Tanikawa, T.; Kryczek, I.; Xia, H.; Li, G.; Wu, K.; Wei, S.; Zhao, L.; Vatan, L.; Wen, B.; et al. Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab 2018, 28, 87–103.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Lee, T.-Y. Lactate: A multifunctional signaling molecule. Yeungnam Univ. J. Med. 2021, 38, 183–193. [Google Scholar] [CrossRef]
- Mowat, C.G. Role of Kynurenine Pathway in Cancer Biology. In Targeting the Broadly Pathogenic Kynurenine Pathway; Springer: Cham, Switzerland, 2015; pp. 273–286. [Google Scholar]
- Jacquemier, J.; Bertucci, F.; Finetti, P.; Esterni, B.; Charafe-Jauffret, E.; Thibult, M.-L.; Houvenaeghel, G.; Van den Eynde, B.; Birnbaum, D.; Olive, D.; et al. High expression of indoleamine 2,3-dioxygenase in the tumour is associated with medullary features and favourable outcome in basal-like breast carcinoma. Int. J. Cancer 2012, 130, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Dill, E.A.; Dillon, P.M.; Bullock, T.N.; Mills, A.M. IDO expression in breast cancer: An assessment of 281 primary and metastatic cases with comparison to PD-L. Mod. Pathol. 2018, 31, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Dewi, D.L.; Mohapatra, S.R.; Blanco Cabañes, S.; Adam, I.; Somarribas Patterson, L.F.; Berdel, B.; Kahloon, M.; Thürmann, L.; Loth, S.; Heilmann, K.; et al. Suppression of indoleamine-2,3-dioxygenase 1 expression by promoter hypermethylation in ER-positive breast cancer. Oncoimmunology 2017, 6, e1274477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Z.; Yue, L.; Shi, J.; Shao, M.; Wu, T. Role of IDO and TDO in Cancers and Related Diseases and the Therapeutic Implications. J. Cancer 2019, 10, 2771. [Google Scholar] [CrossRef] [Green Version]
- Heng, B.; Bilgin, A.A.; Lovejoy, D.B.; Tan, V.X.; Milioli, H.H.; Gluch, L.; Bustamante, S.; Sabaretnam, T.; Moscato, P.; Lim, C.K.; et al. Differential kynurenine pathway metabolism in highly metastatic aggressive breast cancer subtypes: Beyond IDO1-induced immunosuppression. Breast Cancer Res. BCR 2020, 22, 113. [Google Scholar] [CrossRef]
- Khaled, N.; Bidet, Y. New Insights into the Implication of Epigenetic Alterations in the EMT of Triple Negative Breast Cancer. Cancers 2019, 11, 559. [Google Scholar] [CrossRef] [Green Version]
- Soundararajan, R.; Fradette, J.J.; Konen, J.M.; Moulder, S.; Zhang, X.; Gibbons, D.L.; Varadarajan, N.; Wistuba, I.I.; Tripathy, D.; Bernatchez, C.; et al. Targeting the Interplay between Epithelial-to-Mesenchymal-Transition and the Immune System for Effective Immunotherapy. Cancers 2019, 11, 714. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.-H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Morel, D.; Jeffery, D.; Aspeslagh, S.; Almouzni, G.; Postel-Vinay, S. Combining epigenetic drugs with other therapies for solid tumours—past lessons and future promise. Nat. Rev. Clin. Oncol. 2020, 17, 91–107. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Yan, Q. Cancer Epigenetics, Tumor Immunity, and Immunotherapy. Trends Cancer 2020, 6, 580–592. [Google Scholar] [CrossRef]
- Chiappinelli, K.B.; Zahnow, C.A.; Ahuja, N.; Baylin, S.B. Combining Epigenetic and Immunotherapy to Combat Cancer. Cancer Res. 2016, 76, 1683–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woan, K.V.; Lienlaf, M.; Perez-Villaroel, P.; Lee, C.; Cheng, F.; Knox, T.; Woods, D.M.; Barrios, K.; Powers, J.; Sahakian, E.; et al. Targeting histone deacetylase 6 mediates a dual anti-melanoma effect: Enhanced antitumor immunity and impaired cell proliferation. Mol. Oncol. 2015, 9, 1447–1457. [Google Scholar] [CrossRef] [Green Version]
- Kroesen, M.; Gielen, P.; Brok, I.C.; Armandari, I.; Hoogerbrugge, P.M.; Adema, G.J. HDAC inhibitors and immunotherapy; a double edged sword? Oncotarget 2014, 5, 6558–6572. [Google Scholar] [CrossRef] [Green Version]
- Hammitzsch, A.; Tallant, C.; Fedorov, O.; O’Mahony, A.; Brennan, P.E.; Hay, D.A.; Martinez, F.O.; Al-Mossawi, M.H.; de Wit, J.; Vecellio, M.; et al. CBP30, a selective CBP/p300 bromodomain inhibitor, suppresses human Th17 responses. Proc. Natl. Acad. Sci. USA 2015, 112, 10768–10773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Wang, L.; Predina, J.; Han, R.; Beier, U.H.; Wang, L.-C.S.; Kapoor, V.; Bhatti, T.R.; Akimova, T.; Singhal, S.; et al. Inhibition of p300 impairs Foxp3⁺ T regulatory cell function and promotes antitumor immunity. Nat. Med. 2013, 19, 1173–1177. [Google Scholar] [CrossRef]
- Wong, K.K. DNMT1: A key drug target in triple-negative breast cancer. Semin. Cancer Biol. 2021, 72, 198–213. [Google Scholar] [CrossRef]
- Yu, J.; Qin, B.; Moyer, A.M.; Nowsheen, S.; Liu, T.; Qin, S.; Zhuang, Y.; Liu, D.; Lu, S.W.; Kalari, K.R.; et al. DNA methyltransferase expression in triple-negative breast cancer predicts sensitivity to decitabine. J. Clin. Investig. 2018, 128, 2376–2388. [Google Scholar] [CrossRef] [Green Version]
- Merino, V.F.; Cho, S.; Nguyen, N.; Sadik, H.; Narayan, A.; Talbot, C.; Cope, L.; Zhou, X.C.; Zhang, Z.; Győrffy, B.; et al. Induction of cell cycle arrest and inflammatory genes by combined treatment with epigenetic, differentiating, and chemotherapeutic agents in triple-negative breast cancer. Breast Cancer Res. BCR 2018, 20, 145. [Google Scholar] [CrossRef]
- Hicks, K.C.; Knudson, K.M.; Lee, K.L.; Hamilton, D.H.; Hodge, J.W.; Figg, W.D.; Ordentlich, P.; Jones, F.R.; Rabizadeh, S.; Soon-Shiong, P.; et al. Cooperative Immune-Mediated Mechanisms of the HDAC Inhibitor Entinostat, an IL15 Superagonist, and a Cancer Vaccine Effectively Synergize as a Novel Cancer Therapy. Clin. Cancer Res. 2020, 26, 704–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christmas, B.J.; Rafie, C.I.; Hopkins, A.C.; Scott, B.A.; Ma, H.S.; Cruz, K.A.; Woolman, S.; Armstrong, T.D.; Connolly, R.M.; Azad, N.A.; et al. Entinostat Converts Immune-Resistant Breast and Pancreatic Cancers into Checkpoint-Responsive Tumors by Reprogramming Tumor-Infiltrating MDSCs. Cancer Immunol. Res. 2018, 6, 1561–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, S.; Lin, C.Y.; He, H.H.; Witwicki, R.M.; Tabassum, D.P.; Roberts, J.M.; Janiszewska, M.; Huh, S.J.; Liang, Y.; Ryan, J.; et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 2016, 529, 413–417. [Google Scholar] [CrossRef] [Green Version]
- Vázquez, R.; Riveiro, M.E.; Astorgues-Xerri, L.; Odore, E.; Rezai, K.; Erba, E.; Panini, N.; Rinaldi, A.; Kwee, I.; Beltrame, L.; et al. The bromodomain inhibitor OTX015 (MK-8628) exerts anti-tumor activity in triple-negative breast cancer models as single agent and in combination with everolimus. Oncotarget 2017, 8, 7598–7613. [Google Scholar] [CrossRef] [Green Version]
- Lewin, J.; Soria, J.-C.; Stathis, A.; Delord, J.-P.; Peters, S.; Awada, A.; Aftimos, P.G.; Bekradda, M.; Rezai, K.; Zeng, Z.; et al. Phase Ib Trial With Birabresib, a Small-Molecule Inhibitor of Bromodomain and Extraterminal Proteins, in Patients With Selected Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 3007–3014. [Google Scholar] [CrossRef]
- Shu, S.; Wu, H.-J.; Ge, J.Y.; Zeid, R.; Harris, I.S.; Jovanović, B.; Murphy, K.; Wang, B.; Qiu, X.; Endress, J.E.; et al. Synthetic Lethal and Resistance Interactions with BET Bromodomain Inhibitors in Triple-Negative Breast Cancer. Mol. Cell 2020, 78, 1096–1113.e8. [Google Scholar] [CrossRef] [PubMed]
- Noblejas-López, M.D.M.; Nieto-Jimenez, C.; Burgos, M.; Gómez-Juárez, M.; Montero, J.C.; Esparís-Ogando, A.; Pandiella, A.; Galán-Moya, E.M.; Ocaña, A. Activity of BET-proteolysis targeting chimeric (PROTAC) compounds in triple negative breast cancer. J. Exp. Clin. Cancer Res. 2019, 38, 383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, J.; Chen, Y.; Mi, Y.; Jin, H.; Wang, L.; Huang, T.; Li, H.; Song, Y.; Cao, J.; Wu, B.; et al. Macrophages confer resistance to BET inhibition in triple-negative breast cancer by upregulating IKBKE. Biochem. Pharmacol. 2020, 180, 114126. [Google Scholar] [CrossRef]
- Jing, X.; Shao, S.; Zhang, Y.; Luo, A.; Zhao, L.; Zhang, L.; Gu, S.; Zhao, X. BRD4 inhibition suppresses PD-L1 expression in triple-negative breast cancer. Exp. Cell Res. 2020, 392, 112034. [Google Scholar] [CrossRef]
- Duruisseaux, M.; Martinez-Cardus, A.; Calleja-Cervantes, M.E.; Morán, S.; Castro de Moura, M.; Davalos, V.; Piñeyro, D.; Sanchez-Cespedes, M.; Girard, N.; Brevet, M.; et al. Epigenetic prediction of response to anti-PD-1 treatment in non-small-cell lung cancer: A multicentre, retrospective analysis. Lancet. Respir. Med. 2018, 6, 771–781. [Google Scholar] [CrossRef]
- Blank, C.U.; Haanen, J.B.; Ribas, A.; Schumacher, T.N. CANCER IMMUNOLOGY. The “cancer immunogram”. Science 2016, 352, 658–660. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Hu, J.; Maryam, A.; Huang, Q.; Zhang, Y.; Ramakrishnan, S.; Li, J.; Ma, H.; Ma, V.W.S.; Cheuk, W.; et al. Defining super-enhancer landscape in triple-negative breast cancer by multiomic profiling. Nat. Commun. 2021, 12, 2242. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).