
The Fra-1/AP-1 Oncoprotein: From the “Undruggable” Transcription Factor to Therapeutic Targeting




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
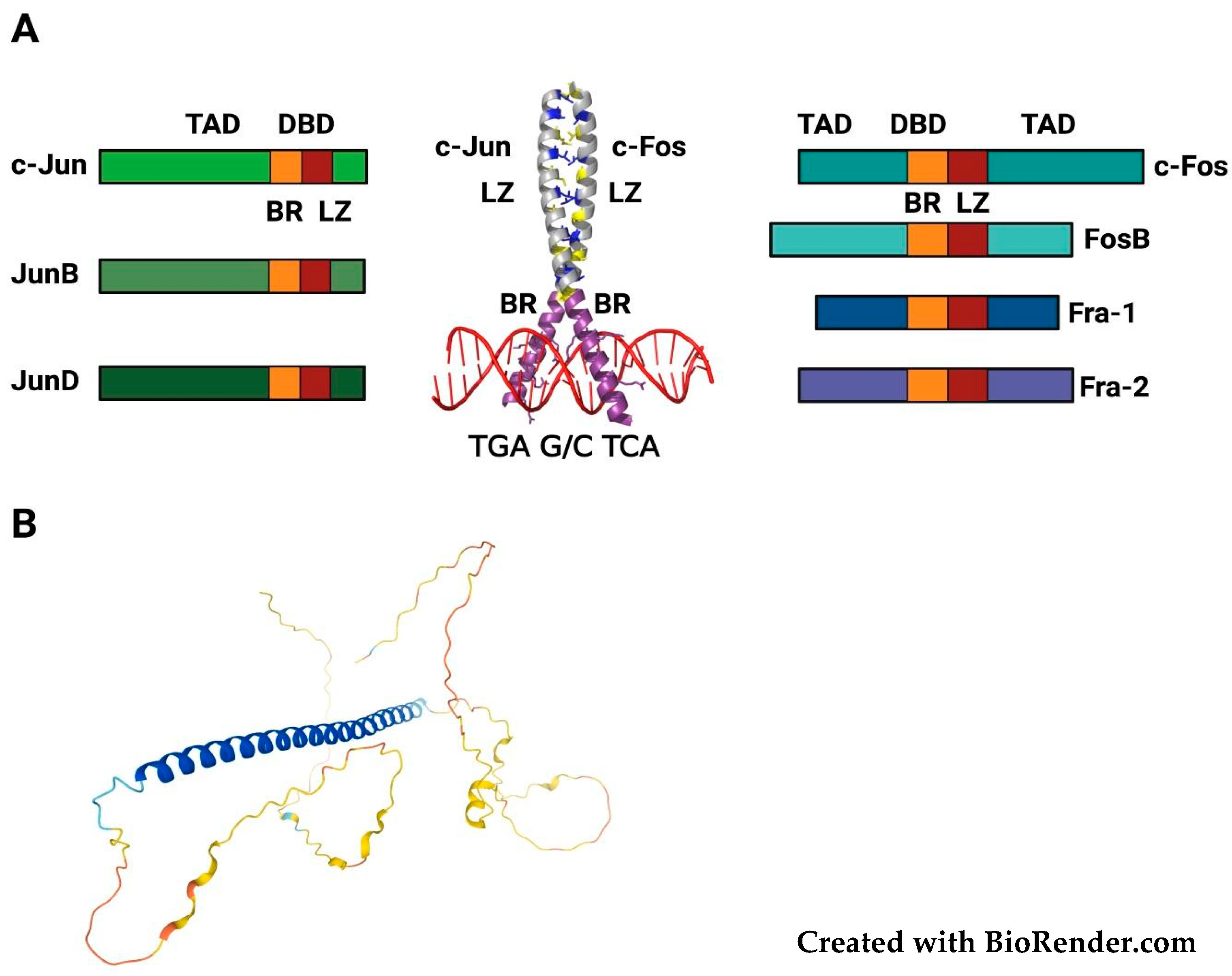
2. Structure, Regulation and Functional Roles of FOSL1/Fra-1
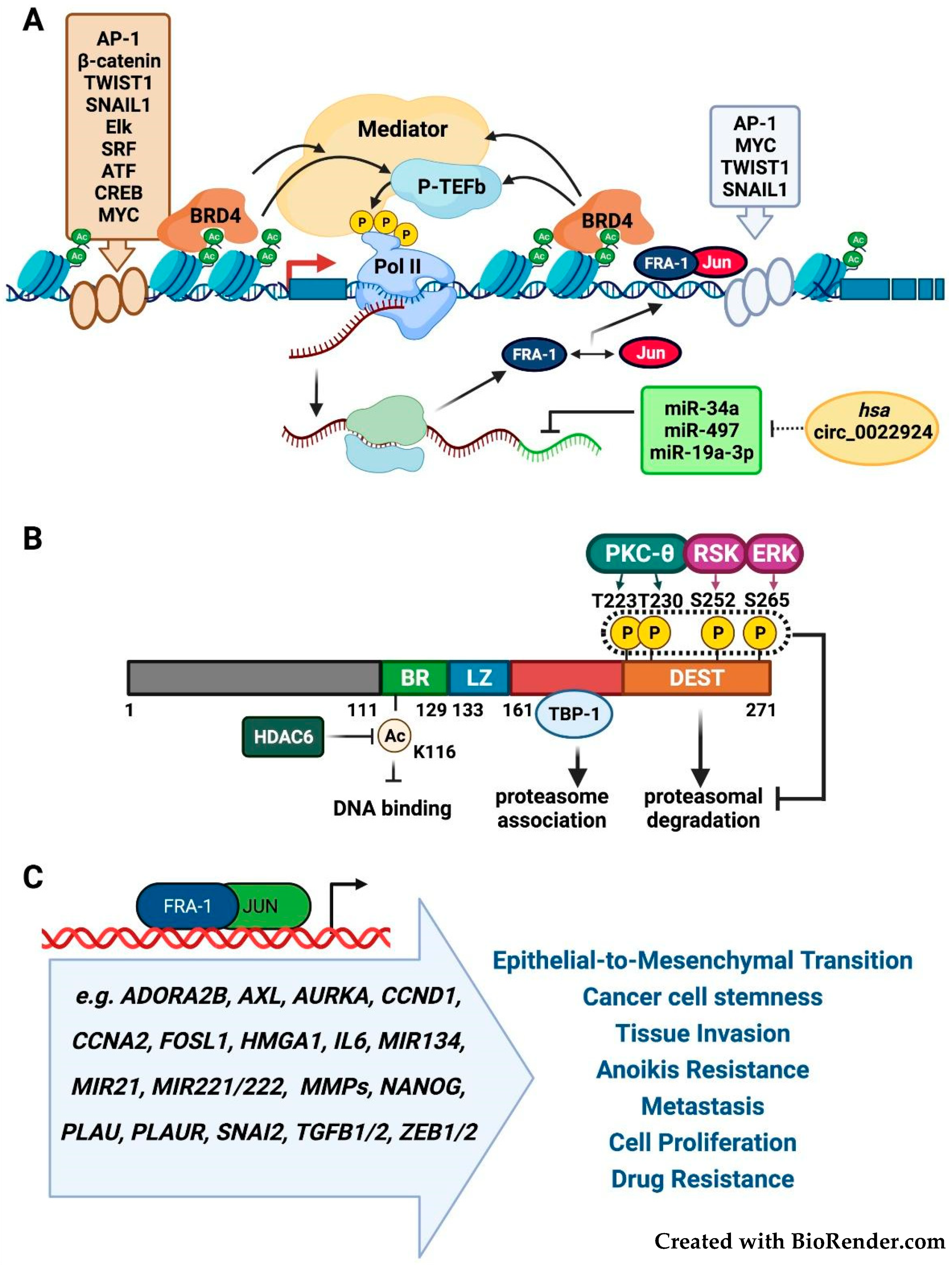
2.1. FOSL1/Fra-1 Structure and Regulation
2.2. Fra-1 in Tumor Growth, Invasion, and Metastasis
2.3. Fra-1 as Prognostic Biomarker and Cancer Cell Addiction to Fra-1 Overexpression
2.4. Fra-1 in Drug Resistance and Drug Addiction Mechanisms
2.5. Fra-1 in Drug Resistance and DNA Repair Mechanisms
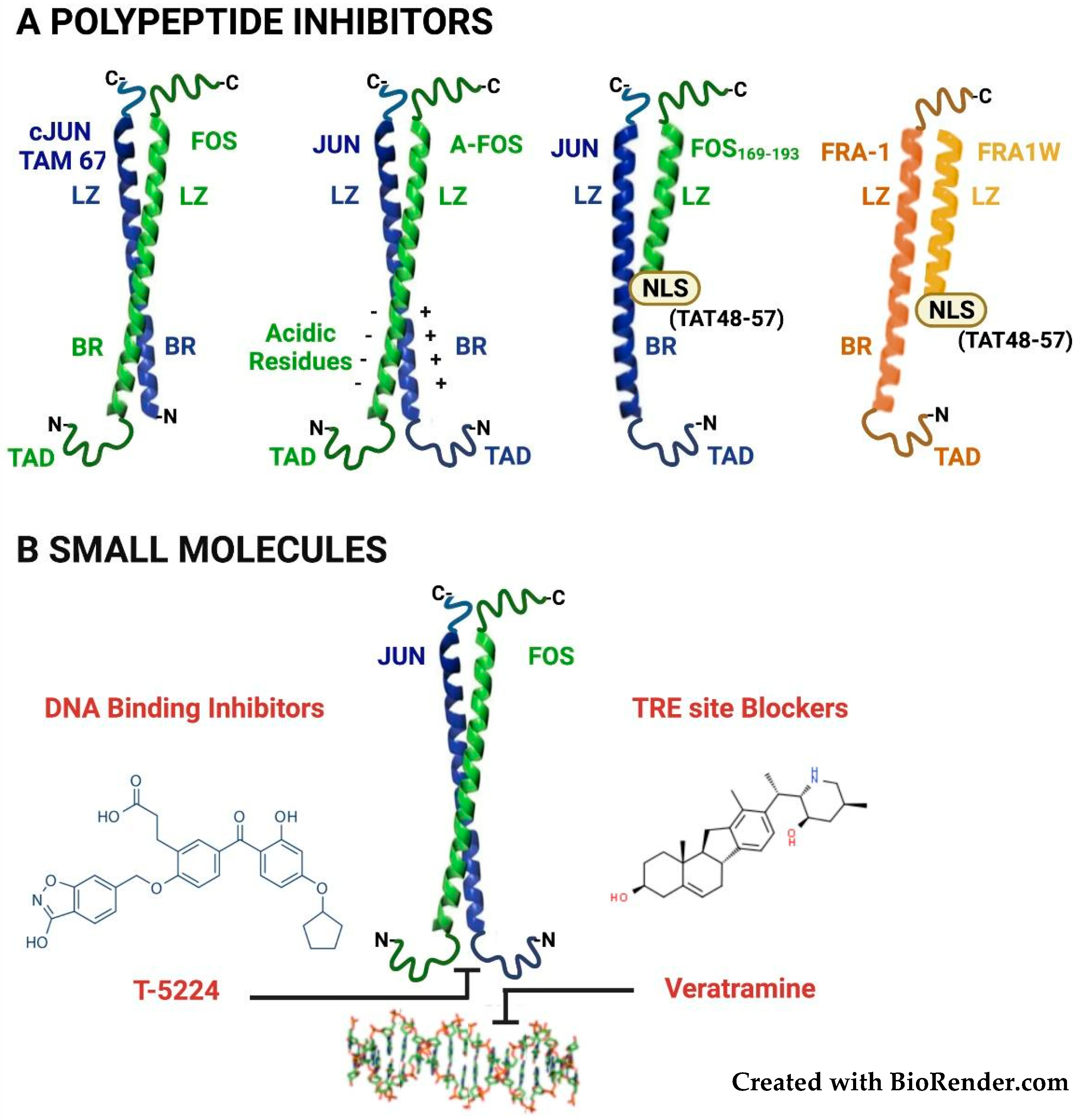
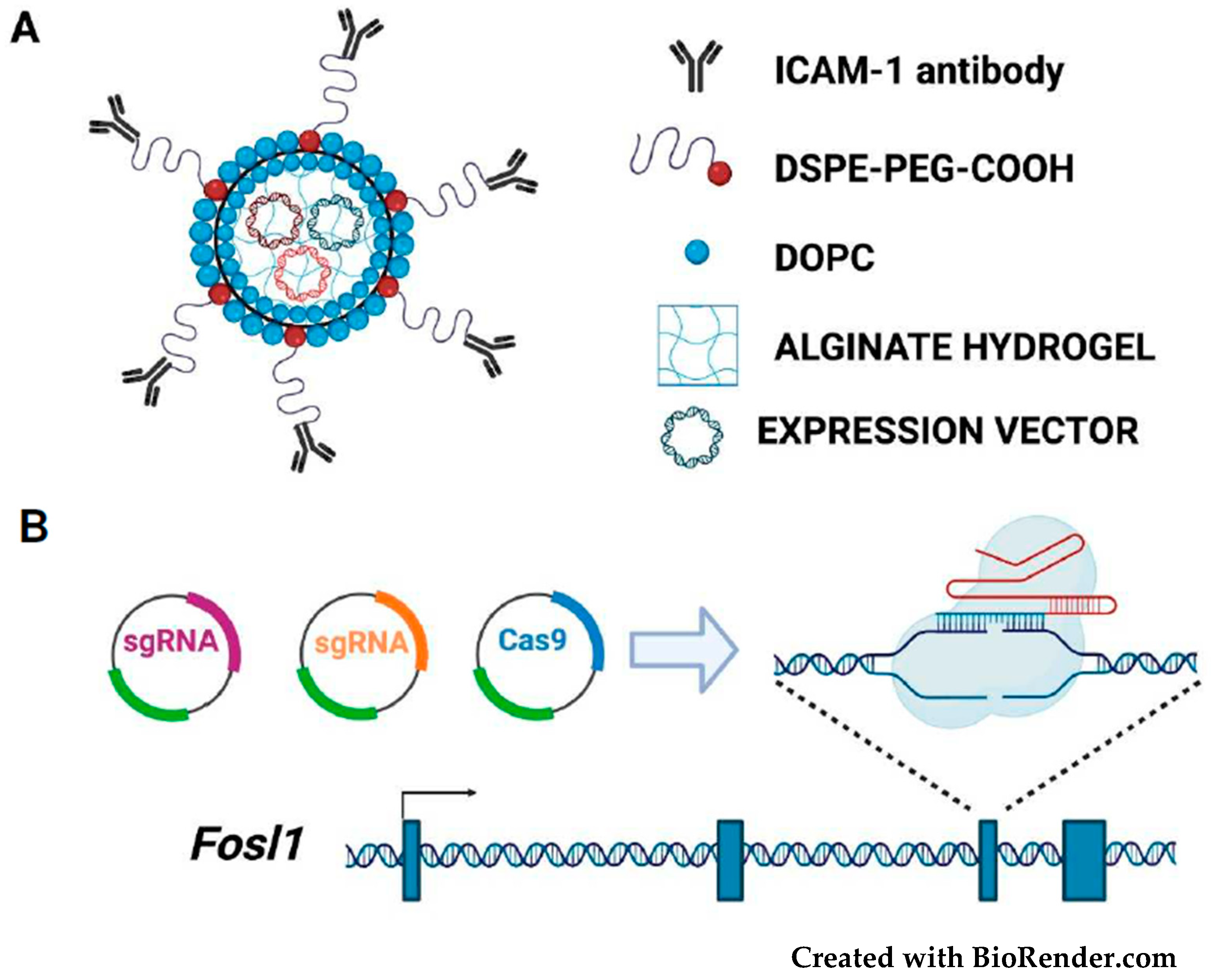
3. Therapeutic Targeting of FOSL1/Fra-1 in Neoplastic Cells
3.1. Targeting the Fra-1/AP-1 DNA-Binding Heterodimers
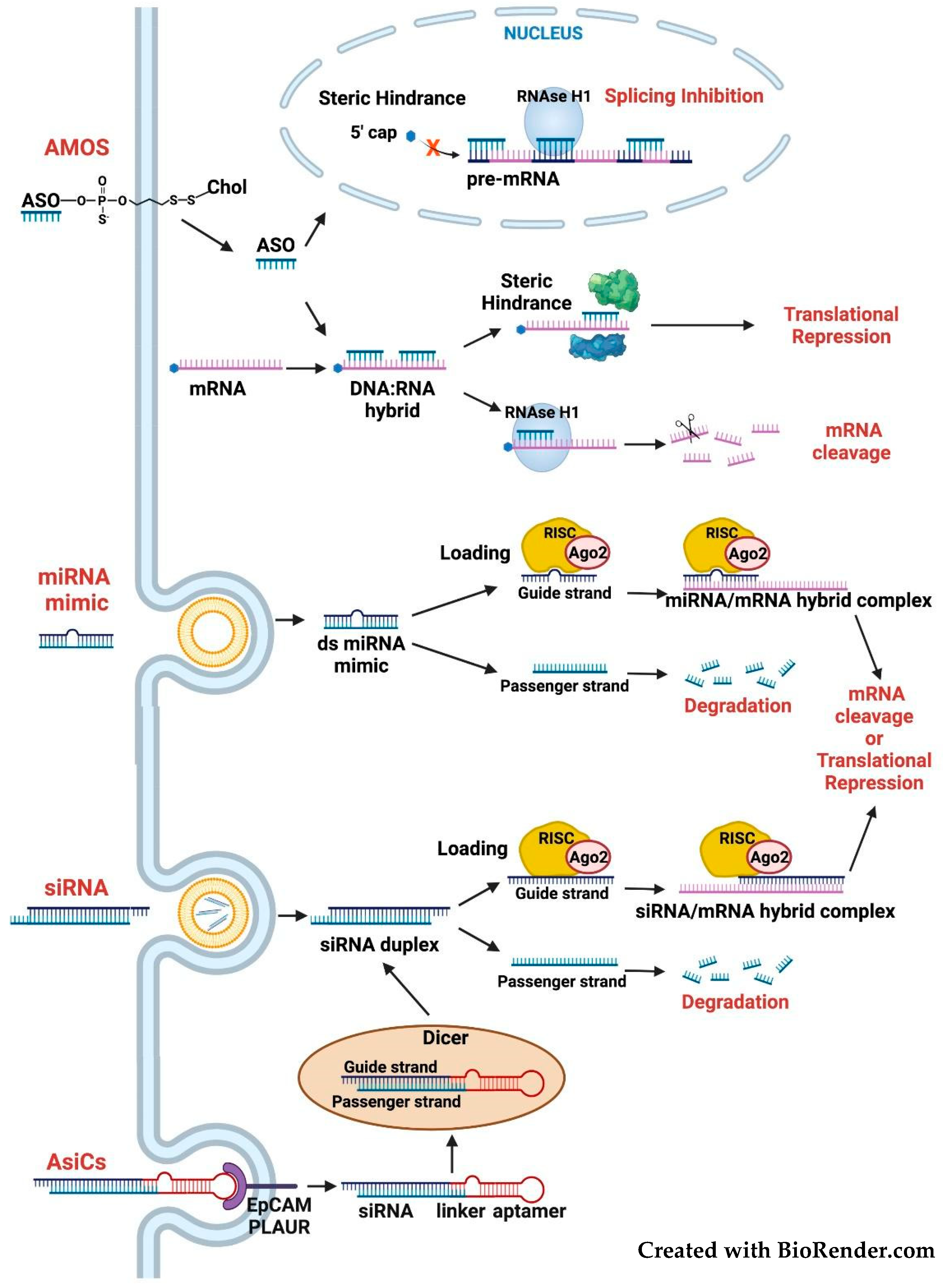
3.2. Targeting the FOSL1 Gene and Fra-1 mRNA
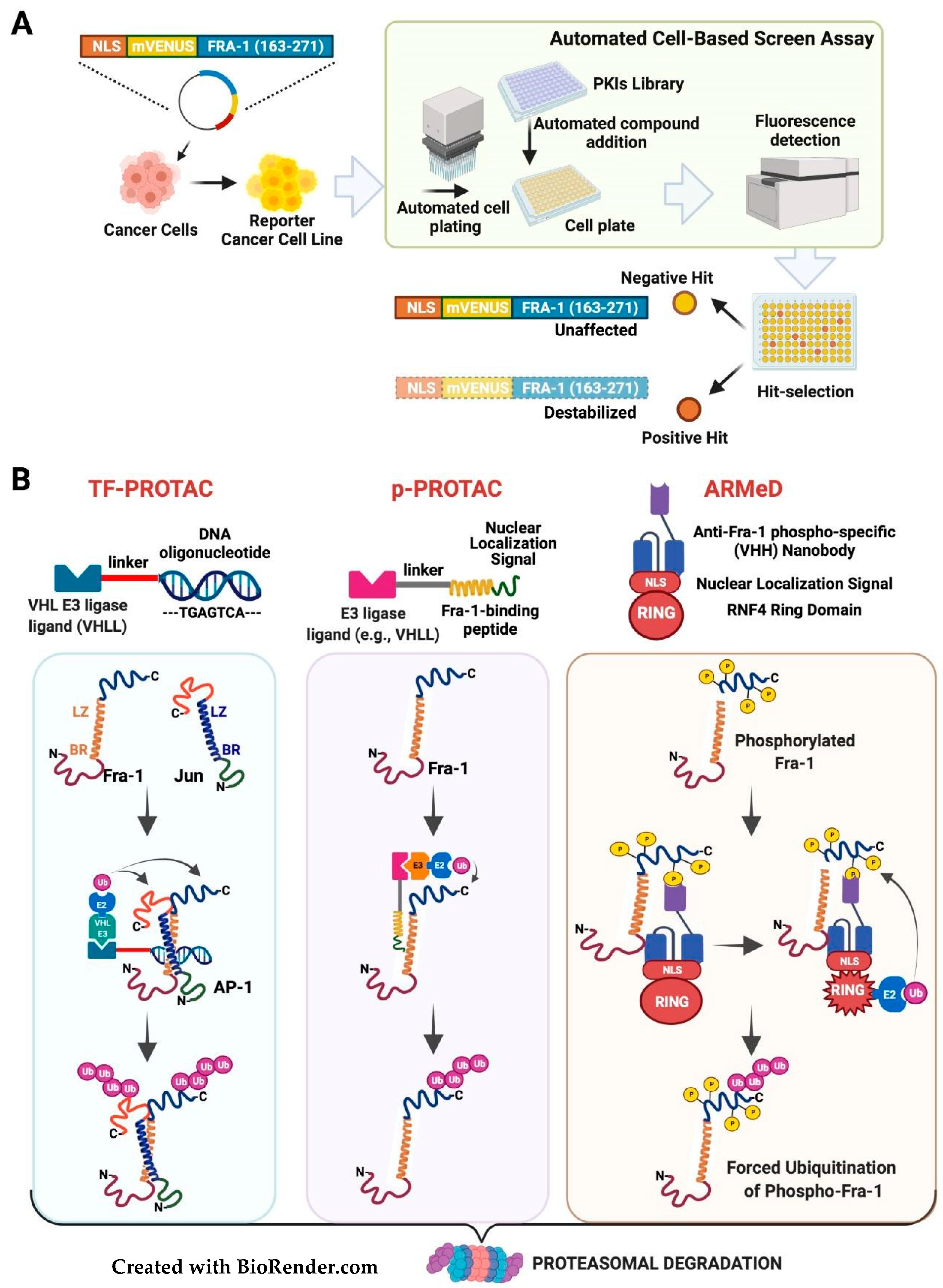
3.3. Targeting the Fra-1 Protein Stability
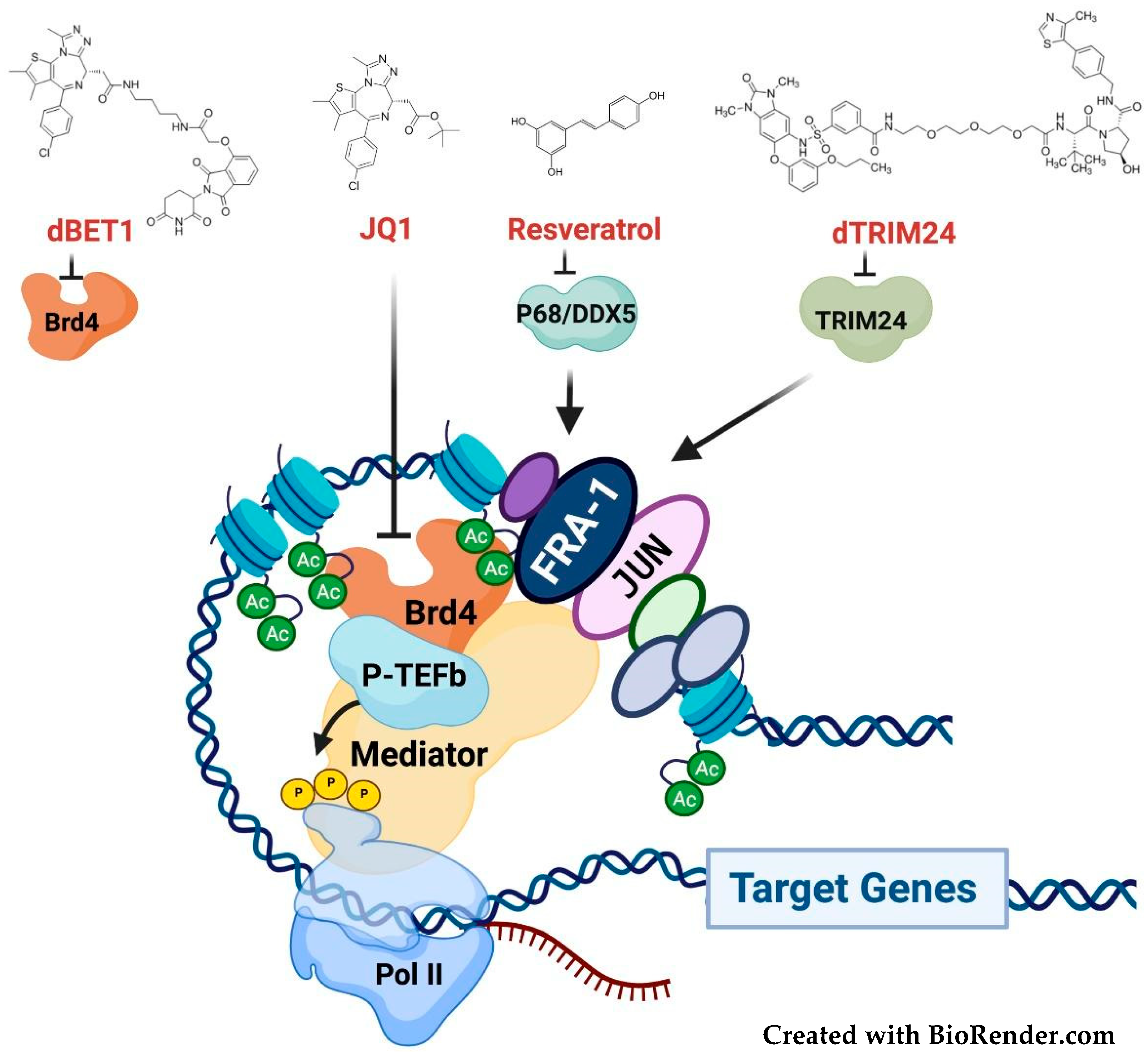
3.4. Targeting the Fra-1/AP-1-Mediated Transactivation
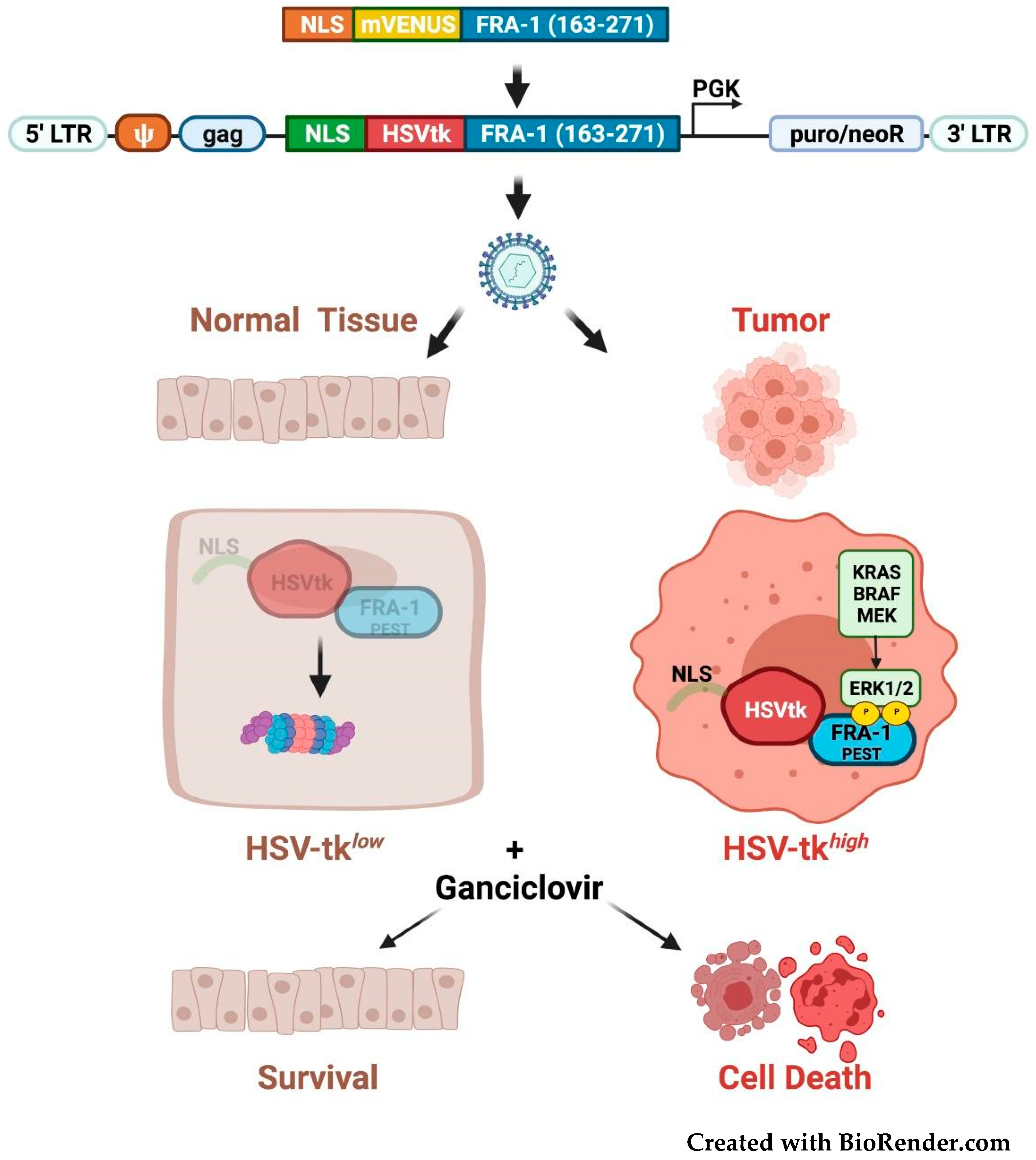
3.5. Fra-1-Based Suicide Gene Therapy Strategies
4. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Darnell, J.E., Jr. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 740–749. [Google Scholar] [CrossRef]
- Hess, J.; Angel, P.; Schorpp-Kistner, M. AP-1 subunits: Quarrel and harmony among siblings. J. Cell Sci. 2004, 117, 5965–5973. [Google Scholar] [CrossRef] [PubMed]
- Bejjani, F.; Evanno, E.; Zibara, K.; Piechaczyk, M.; Jariel-Encontre, I. The AP-1 transcriptional complex: Local switch or remote command? Biochim. Biophys. Acta 2019, 1872, 11–23. [Google Scholar] [CrossRef]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Tulchinsky, E. FRA-1 as a driver of tumour heterogeneity: A nexus between oncogenes and embryonic signalling pathways in cancer. Oncogene 2014, 34, 4421–4428. [Google Scholar] [CrossRef] [PubMed]
- Talotta, F.; Casalino, L.; Verde, P. The nuclear oncoprotein Fra-1: A transcription factor knocking on therapeutic applications door. Oncogene 2020, 39, 4491–4506. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Xie, H.; Dou, Y.; Yuan, J.; Zeng, D.; Xiao, S. Expression and function of FRA1 protein in tumors. Mol. Biol. Rep. 2020, 47, 737–752. [Google Scholar] [CrossRef] [PubMed]
- Rajamohan, S.; Reddy, S. FOSL1 (FOS-like antigen 1). Atlas Genet. Cytogenet. Oncol. Haematol. 2013, 5, 313–318. [Google Scholar] [CrossRef]
- Shu, L.; Chen, A.; Li, L.; Yao, L.; He, Y.; Xu, J.; Gu, W.; Li, Q.; Wang, K.; Zhang, T.; et al. NRG1 regulates Fra-1 transcription and metastasis of triple-negative breast cancer cells via the c-Myc ubiquitination as manipulated by ERK1/2-mediated Fbxw7 phosphorylation. Oncogene 2022, 41, 907–919. [Google Scholar] [CrossRef] [PubMed]
- Zippo, A.; Serafini, R.; Rocchigiani, M.; Pennacchini, S.; Krepelova, A.; Oliviero, S. Histone Crosstalk between H3S10ph and H4K16ac Generates a Histone Code that Mediates Transcription Elongation. Cell 2009, 138, 1122–1136. [Google Scholar] [CrossRef] [PubMed]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Abraham, B.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-Enhancers in the Control of Cell Identity and Disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef]
- Wu, J.; Wu, G.; Lv, L.; Ren, Y.-F.; Zhang, X.-J.; Xue, Y.-F.; Li, G.; Lu, X.; Sun, Z.; Tang, K.-F. MicroRNA-34a inhibits migration and invasion of colon cancer cells via targeting to Fra-1. Carcinogenesis 2011, 33, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Li, Y.; Gao, J.; Zhang, T.; Li, S.; Luo, A.; Chen, H.; Ding, F.; Wang, X.; Liu, Z. MicroRNA-34 suppresses breast cancer invasion and metastasis by directly targeting Fra-1. Oncogene 2013, 32, 4294–4303. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Shen, Q.; Zhang, P. miR-497 suppresses epithelial–mesenchymal transition and metastasis in colorectal cancer cells by targeting fos-related antigen-1. OncoTargets Ther. 2016, 9, 6597–6604. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Z.; Chen, C.; Liu, Y.; Si, Q.; Chuang, T.-H.; Li, N.; Gomezcabrero, A.; Reisfeld, R.A.; Xiang, R.; et al. MicroRNA-19a-3p inhibits breast cancer progression and metastasis by inducing macrophage polarization through downregulated expression of Fra-1 proto-oncogene. Oncogene 2014, 33, 3014–3023. [Google Scholar] [CrossRef] [PubMed]
- Santer, L.; Bär, C.; Thum, T. Circular RNAs: A Novel Class of Functional RNA Molecules with a Therapeutic Perspective. Mol. Ther. 2019, 27, 1350–1363. [Google Scholar] [CrossRef] [PubMed]
- Salzman, J.; Chen, R.E.; Olsen, M.N.; Wang, P.L.; Brown, P.O. Cell-type specific features of circular RNA expression. PLoS Genet. 2013, 9, e1003777. [Google Scholar] [CrossRef]
- Basbous, J.; Chalbos, D.; Hipskind, R.; Jariel-Encontre, I.; Piechaczyk, M. Ubiquitin-Independent Proteasomal Degradation of Fra-1 Is Antagonized by Erk1/2 Pathway-Mediated Phosphorylation of a Unique C-Terminal Destabilizer. Mol. Cell. Biol. 2007, 27, 3936–3950. [Google Scholar] [CrossRef]
- Belguise, K.; Milord, S.; Galtier, F.; Moquet-Torcy, G.; Piechaczyk, M.; Chalbos, D. The PKCθ pathway participates in the aberrant accumulation of Fra-1 protein in invasive ER-negative breast cancer cells. Oncogene 2012, 31, 4889–4897. [Google Scholar] [CrossRef] [PubMed]
- Talotta, F.; Mega, T.; Bossis, G.; Casalino, L.; Basbous, J.; Jariel-Encontre, I.; Piechaczyk, M.; Verde, P. Heterodimerization with Fra-1 cooperates with the ERK pathway to stabilize c-Jun in response to the RAS oncoprotein. Oncogene 2010, 29, 4732–4740. [Google Scholar] [CrossRef] [PubMed]
- Casalino, L.; Cesare, D.D.; Verde, P. Accumulation of Fra-1 in Ras-Transformed Cells Depends on Both Transcriptional Autoregulation and MEK-Dependent Posttranslational Stabilization. Mol. Cell. Biol. 2003, 23, 4401–4415. [Google Scholar] [CrossRef] [PubMed]
- Pakay, J.L.; Diesch, J.; Gilan, O.; Yip, Y.-Y.; Sayan, E.; Kolch, W.; Mariadason, J.M.; Hannan, R.D.; Tulchinsky, E.; Dhillon, A.S. A 19S proteasomal subunit cooperates with an ERK MAPK-regulated degron to regulate accumulation of Fra-1 in tumour cells. Oncogene 2011, 31, 1817–1824. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ni, H.; Lan, L.; Wei, X.; Xiang, R.; Wang, Y. Fra-1 protooncogene regulates IL-6 expression in macrophages and promotes the generation of M2d macrophages. Cell Res. 2010, 20, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Dimitri, C.A.; Yoon, S.-O.; Dowdle, W.; Blenis, J. ERK2 but Not ERK1 Induces Epithelial-to-Mesenchymal Transformation via DEF Motif-Dependent Signaling Events. Mol. Cell 2010, 38, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Sayan, A.E.; Stanford, R.; Vickery, R.; Grigorenko, E.; Diesch, J.; Kulbicki, K.; Edwards, R.; Pal, R.; Greaves, P.; Jariel-Encontre, I.; et al. Fra-1 controls motility of bladder cancer cells via transcriptional upregulation of the receptor tyrosine kinase AXL. Oncogene 2012, 31, 1493–1503. [Google Scholar] [CrossRef]
- Bakiri, L.; Macho-Maschler, S.; Custic, I.; Niemiec, J.; Guío-Carrión, A.; Hasenfuss, S.C.; Eger, A.; Müller, M.; Beug, H.; Wagner, E.F. Fra-1/AP-1 induces EMT in mammary epithelial cells by modulating Zeb1/2 and TGFβ expression. Cell Death Differ. 2014, 22, 336–350. [Google Scholar] [CrossRef]
- Diesch, J.; Sanij, E.; Gilan, O.; Love, C.; Tran, H.; Fleming, N.I.; Ellul, J.; Amalia, M.; Haviv, I.; Pearson, R.B.; et al. Widespread FRA1-Dependent Control of Mesenchymal Transdifferentiation Programs in Colorectal Cancer Cells. PLoS ONE 2014, 9, e88950. [Google Scholar] [CrossRef] [PubMed]
- Tolza, C.; Bejjani, F.; Evanno, E.; Mahfoud, S.; Moquet-Torcy, G.; Gostan, T.; Maqbool, M.A.; Kirsh, O.; Piechaczyk, M.; Jariel-Encontre, I. AP-1 Signaling by Fra-1 Directly Regulates HMGA1 Oncogene Transcription in Triple-Negative Breast Cancers. Mol. Cancer Res. 2019, 17, 1999–2014. [Google Scholar] [CrossRef] [PubMed]
- Adiseshaiah, P.; Lindner, D.J.; Kalvakolanu, D.V.; Reddy, S.P. FRA-1 Proto-Oncogene Induces Lung Epithelial Cell Invasion and Anchorage-Independent Growth In vitro, but Is Insufficient to Promote Tumor Growth In vivo. Cancer Res. 2007, 67, 6204–6211. [Google Scholar] [CrossRef] [PubMed]
- Adiseshaiah, P.; Vaz, M.; Machireddy, N.; Kalvakolanu, D.V.; Reddy, S.P. A Fra-1-dependent, matrix metalloproteinase driven EGFR activation promotes human lung epithelial cell motility and invasion. J. Cell. Physiol. 2008, 216, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Desmet, C.J.; Gallenne, T.; Prieur, A.; Reyal, F.; Visser, N.L.; Wittner, B.S.; Smit, M.A.; Geiger, T.R.; Laoukili, J.; Iskit, S.; et al. Identification of a pharmacologically tractable Fra-1/ADORA2B axis promoting breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2013, 110, 5139–5144. [Google Scholar] [CrossRef]
- Iskit, S.; Schlicker, A.; Wessels, L.; Peeper, D.S. Fra-1 is a key driver of colon cancer metastasis and a Fra-1 classifier predicts disease-free survival. Oncotarget 2015, 6, 43146–43161. [Google Scholar] [CrossRef] [PubMed]
- Gallenne, T.; Ross, K.N.; Visser, N.L.; Salony, S.; Desmet, C.J.; Wittner, B.S.; Wessels, L.F.A.; Ramaswamy, S.; Peeper, D.S. Systematic functional perturbations uncover a prognostic genetic network driving human breast cancer. Oncotarget 2017, 8, 20572–20587. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, A.; Perurena, N.; Guruceaga, E.; Mazur, P.K.; Martinez-Canarias, S.; Zandueta, C.; Valencia, K.; Arricibita, A.; Gwinn, D.; Sayles, L.C.; et al. An integrative approach unveils FOSL1 as an oncogene vulnerability in KRAS-driven lung and pancreatic cancer. Nat. Commun. 2017, 8, 14294. [Google Scholar] [CrossRef]
- Elangovan, I.M.; Vaz, M.; Tamatam, C.R.; Potteti, H.R.; Reddy, N.M.; Reddy, S.P. FOSL1 Promotes Kras-induced Lung Cancer through Amphiregulin and Cell Survival Gene Regulation. Am. J. Respir. Cell Mol. Biol. 2018, 58, 625–635. [Google Scholar] [CrossRef]
- Luo, Y.; Zhou, H.; Krueger, J.; Kaplan, C.; Liao, D.; Markowitz, D.; Liu, C.; Chen, T.; Chuang, T.-H.; Xiang, R.; et al. The role of proto-oncogene Fra-1 in remodeling the tumor microenvironment in support of breast tumor cell invasion and progression. Oncogene 2010, 29, 662–673. [Google Scholar] [CrossRef]
- Reisfeld, R.A. The Tumor Microenvironment: A Target for Combination therapy of Breast Cancer. Crit. Rev. Oncog. 2013, 18, 115–133. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ren, G.; Wang, T.; Chen, Y.; Gong, C.; Bai, Y.; Wang, B.; Qi, H.; Shen, J.; Zhu, L.; et al. Aberrantly expressed Fra-1 by IL-6/STAT3 transactivation promotes colorectal cancer aggressiveness through epithelial–mesenchymal transition. Carcinogenesis 2015, 36, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Talotta, F.; Cimmino, A.; Matarazzo, M.R.; Casalino, L.; De Vita, G.; D’Esposito, M.; Di Lauro, R.; Verde, P. An autoregulatory loop mediated by miR-21 and PDCD4 controls the AP-1 activity in RAS transformation. Oncogene 2009, 28, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Hatley, M.; Patrick, D.; Garcia, M.R.; Richardson, J.A.; Bassel-Duby, R.; Van Rooij, E.; Olson, E.N. Modulation of K-Ras-Dependent Lung Tumorigenesis by MicroRNA-21. Cancer Cell 2010, 18, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Sánchez, D.; Arriaga-Canon, C.; Pedroza-Torres, A.; Rosa-Velázquez, I.A.D.L.; González-Barrios, R.; Contre-ras-Espinosa, L.; Montiel-Manríquez, R.; Castro-Hernández, C.; Fragoso-Ontiveros, V.; Álvarez-Gómez, R.M.; et al. The Promising Role of MiR-21 as a Cancer Biomarker and Its Importance in RNA-Based Therapeutics. Mol. Ther. Nucleic Acids 2020, 20, 409–420. [Google Scholar] [CrossRef]
- Wu, J.; Sun, Y.; Zhang, P.-Y.; Qian, M.; Zhang, H.; Chen, X.; Ma, D.; Xu, Y.; Chen, X.; Tang, K.-F. The Fra-1–miR-134–SDS22 feedback loop amplifies ERK/JNK signaling and reduces chemosensitivity in ovarian cancer cells. Cell Death Dis. 2016, 7, e2384. [Google Scholar] [CrossRef]
- Stinson, S.; Lackner, M.R.; Adai, A.T.; Yu, N.; Kim, H.-J.; O’Brien, C.; Spoerke, J.; Jhunjhunwala, S.; Boyd, Z.; Januario, T.; et al. TRPS1 Targeting by miR-221/222 Promotes the Epithelial-to-Mesenchymal Transition in Breast Cancer. Sci. Signal. 2011, 4, ra41. [Google Scholar] [CrossRef]
- Tam, W.L.; Lu, H.; Buikhuisen, J.; Soh, B.S.; Lim, E.; Reinhardt, F.; Wu, Z.J.; Krall, J.A.; Bierie, B.; Guo, W.; et al. Protein Kinase C α Is a Central Signaling Node and Therapeutic Target for Breast Cancer Stem Cells. Cancer Cell 2013, 24, 347–364. [Google Scholar] [CrossRef]
- Wang, T.; Song, P.; Zhong, T.; Wang, X.; Xiang, X.; Liu, Q.; Chen, H.; Xia, T.; Liu, H.; Niu, Y.; et al. The inflammatory cytokine IL-6 induces FRA1 deacetylation promoting colorectal cancer stem-like properties. Oncogene 2019, 38, 4932–4947. [Google Scholar] [CrossRef]
- Marques, C.; Unterkircher, T.; Kroon, P.; Oldrini, B.; Izzo, A.; Dramaretska, Y.; Ferrarese, R.; Kling, E.; Schnell, O.; Nelander, S.; et al. NF1 regulates mesenchymal glioblastoma plasticity and aggressiveness through the AP-1 transcription factor FOSL1. eLife 2021, 10, e64846. [Google Scholar] [CrossRef]
- Zhao, C.; Qiao, Y.; Jonsson, P.; Wang, J.; Xu, L.; Rouhi, P.; Sinha, I.; Cao, Y.; Williams, C.; Dahlman-Wright, K. Genome-wide Profiling of AP-1–Regulated Transcription Provides Insights into the Invasiveness of Triple-Negative Breast Cancer. Cancer Res. 2014, 74, 3983–3994. [Google Scholar] [CrossRef]
- Franco, H.L.; Nagari, A.; Malladi, V.S.; Li, W.; Xi, Y.; Richardson, D.; Allton, K.L.; Tanaka, K.; Li, J.; Murakami, S.; et al. Enhancer transcription reveals subtype-specific gene expression programs controlling breast cancer pathogenesis. Genome Res. 2018, 28, 159–170. [Google Scholar] [CrossRef]
- Vallejo, A.; Erice, O.; Entrialgo-Cadierno, R.; Feliu, I.; Guruceaga, E.; Perugorria, M.J.; Olaizola, P.; Muggli, A.; Macaya, I.; O’Dell, M.; et al. FOSL1 promotes cholangiocarcinoma via transcriptional effectors that could be therapeutically targeted. J. Hepatol. 2021, 75, 363–376. [Google Scholar] [CrossRef]
- Zhang, M.; Hoyle, R.G.; Ma, Z.; Sun, B.; Cai, W.; Cai, H.; Xie, N.; Zhang, Y.; Hou, J.; Liu, X.; et al. FOSL1 promotes metastasis of head and neck squamous cell carcinoma through super-enhancer-driven transcription program. Mol. Ther. 2021, 29, 2583–2600. [Google Scholar] [CrossRef]
- Chen, S.; Youhong, T.; Tan, Y.; He, Y.; Ban, Y.; Cai, J.; Li, X.; Xiong, W.; Zeng, Z.; Li, G.; et al. EGFR-PKM2 signaling promotes the metastatic potential of nasopharyngeal carcinoma through induction of FOSL1 and ANTXR 2. Carcinogenesis 2019, 41, 723–733. [Google Scholar] [CrossRef]
- Usui, A.; Hoshino, I.; Akutsu, Y.; Sakata, H.; Nishimori, T.; Murakami, K.; Kano, M.; Shuto, K.; Matsubara, H. The molecular role of Fra-1 and its prognostic significance in human esophageal squamous cell carcinoma. Cancer 2011, 118, 3387–3396. [Google Scholar] [CrossRef] [PubMed]
- Toyozumi, T.; Hoshino, I.; Takahashi, M.; Usui, A.; Akutsu, Y.; Hanari, N.; Murakami, K.; Kano, M.; Akanuma, N.; Suitoh, H.; et al. Fra-1 Regulates the Expression of HMGA1, which is Associated with a Poor Prognosis in Human Esophageal Squamous Cell Carcinoma. Ann. Surg. Oncol. 2016, 24, 3446–3455. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Jin, X.; Yuan, Y.; Deng, P.; Jiang, L.; Zeng, X.; Li, X.-S.; Wang, Z.-Y.; Chen, Q.-M. Prognostic value from integrative analysis of transcription factors c-Jun and Fra-1 in oral squamous cell carcinoma: A multicenter cohort study. Sci. Rep. 2017, 7, 7522. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, H.; Mu, X.; Cui, J.; Peng, Z. Dysregulation of Fra1 expression by Wnt/β-catenin signalling promotes glioma aggressiveness through epithelial–mesenchymal transition. Biosci. Rep. 2017, 37, 20160643. [Google Scholar] [CrossRef] [PubMed]
- Antony, J.; Tan, T.Z.; Kelly, Z.; Low, J.; Choolani, M.; Recchi, C.; Gabra, H.; Thiery, J.P.; Huang, R.Y.-J. The GAS6-AXL signaling network is a mesenchymal (Mes) molecular subtype–specific therapeutic target for ovarian cancer. Sci. Signal. 2016, 9, ra97. [Google Scholar] [CrossRef]
- Leupold, J.H.; Asangani, I.; Maurer, G.D.; Lengyel, E.; Post, S.; Allgayer, H. Src Induces Urokinase Receptor Gene Expression and Invasion/Intravasation via Activator Protein-1/p-c-Jun in Colorectal Cancer. Mol. Cancer Res. 2007, 5, 485–496. [Google Scholar] [CrossRef]
- Casalino, L.; Bakiri, L.; Talotta, F.; Weitzman, J.; Fusco, A.; Yaniv, M.; Verde, P. Fra-1 promotes growth and survival in RAS-transformed thyroid cells by controlling cyclin A transcription. EMBO J. 2007, 26, 1878–1890. [Google Scholar] [CrossRef]
- Román, M.; López, I.; Guruceaga, E.; Baraibar, I.; Ecay, M.; Collantes, M.; Nadal, E.; Vallejo, A.; Cadenas, S.; Miguel, M.E.; et al. Inhibitor of Differentiation-1 Sustains Mutant KRAS-Driven Progression, Maintenance, and Metastasis of Lung Adenocarcinoma via Regulation of a FOSL1 Network. Cancer Res. 2019, 79, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Rennhack, J.P.; Arnoff, T.E.; Thaker, M.; Younger, S.T.; Doench, J.G.; Huang, A.Y.; Yang, A.; Aguirre, A.J.; Wang, B.; et al. SMAD4 represses FOSL1 expression and pancreatic cancer metastatic colonization. Cell Rep. 2021, 36, 109443. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wang, E.; Milazzo, J.P.; Wang, Z.; Kinney, J.B.; Vakoc, C.R. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat. Biotechnol. 2015, 33, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Wilson, M.M.; Weinberg, R.A.; Lees, J.A.; Guen, V.J. Emerging Mechanisms by which EMT Programs Control Stemness. Trends Cancer 2020, 6, 775–780. [Google Scholar] [CrossRef]
- van Staalduinen, J.; Baker, D.; ten Dijke, P.; van Dam, H. Epithelial–mesenchymal-transition-inducing transcription factors: New targets for tackling chemoresistance in cancer? Oncogene 2018, 37, 6195–6211. [Google Scholar] [CrossRef]
- Stathis, A.; Bertoni, F. BET Proteins as Targets for Anticancer Treatment. Cancer Discov. 2018, 8, 24–36. [Google Scholar] [CrossRef]
- Bejjani, F.; Tolza, C.; Boulanger, M.; Downes, D.; Romero, R.; Maqbool, M.A.; El Aabidine, A.Z.; Andrau, J.-C.; Lebre, S.; Brehelin, L.; et al. Fra-1 regulates its target genes via binding to remote enhancers without exerting major control on chromatin architecture in triple negative breast cancers. Nucleic Acids Res. 2021, 49, 2488–2508. [Google Scholar] [CrossRef] [PubMed]
- Shu, S.; Wu, H.-J.; Ge, J.Y.; Zeid, R.; Harris, I.S.; Jovanović, B.; Murphy, K.; Wang, B.; Qiu, X.; Endress, J.E.; et al. Synthetic Lethal and Resistance Interactions with BET Bromodomain Inhibitors in Triple-Negative Breast Cancer. Mol. Cell 2020, 78, 1096–1113.e8. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef]
- Liu, X.; Li, H.; Rajurkar, M.; Li, Q.; Cotton, J.L.; Ou, J.; Zhu, L.; Goel, H.L.; Mercurio, A.M.; Park, J.-S.; et al. Tead and AP1 Coordinate Transcription and Motility. Cell Rep. 2016, 14, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Feldker, N.; Ferrazzi, F.; Schuhwerk, H.; Widholz, S.A.; Guenther, K.; Frisch, I.; Jakob, K.; Kleemann, J.; Riegel, D.; Bönisch, U.; et al. Genome-wide cooperation of EMT transcription factor ZEB 1 with YAP and AP-1 in breast cancer. EMBO J. 2020, 39, e103209. [Google Scholar] [CrossRef]
- Maurus, K.; Hufnagel, A.; Geiger, F.; Graf, S.; Berking, C.; Heinemann, A.; Paschen, A.; Kneitz, S.; Stigloher, C.; Geissinger, E.; et al. The AP-1 transcription factor FOSL1 causes melanocyte reprogramming and transformation. Oncogene 2017, 36, 5110–5121. [Google Scholar] [CrossRef] [PubMed]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Osborne, J.; Hutchinson, P.; Tse, G.; et al. A Switch in the Expression of Embryonic EMT-Inducers Drives the Development of Malignant Melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef]
- Richard, G.; Dalle, S.; Monet, M.; Ligier, M.; Boespflug, A.; Pommier, R.; de la Fouchardiere, A.; Perier-Muzet, M.; Depaepe, L.; Barnault, R.; et al. ZEB1-mediated melanoma cell plasticity enhances resistance to MAPK inhibitors. EMBO Mol. Med. 2016, 8, 1143–1161. [Google Scholar] [CrossRef]
- Suda, K.; Tomizawa, K.; Osada, H.; Maehara, Y.; Yatabe, Y.; Sekido, Y.; Mitsudomi, T. Conversion from the “oncogene addiction” to “drug addiction” by intensive inhibition of the EGFR and MET in lung cancer with activating EGFR mutation. Lung Cancer 2012, 76, 292–299. [Google Scholar] [CrossRef]
- Das Thakur, M.; Salangsang, F.; Landman, A.S.; Sellers, W.R.; Pryer, N.K.; Levesque, M.P.; Dummer, R.; McMahon, M.; Stuart, D.D. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013, 494, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Roux, J.; Pages, C.; Malouf, D.; Seguin, N.B.; Madjlessi, N.; Baccard, M.; Comte, C.; Archimbaud, A.; Battistella, M.; Viguier, M.; et al. BRAF inhibitor rechallenge in patients with advanced BRAF V600-mutant melanoma. Melanoma Res. 2015, 25, 559–563. [Google Scholar] [CrossRef]
- Kong, X.; Kuilman, T.; Shahrabi, A.; Boshuizen, J.; Kemper, K.; Song, J.-Y.; Niessen, H.W.M.; Rozeman, E.A.; Foppen, M.H.G.; Blank, C.U.; et al. Cancer drug addiction is relayed by an ERK2-dependent phenotype switch. Nature 2017, 550, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Hong, A.; Moriceau, G.; Sun, L.; Lomeli, S.; Piva, M.; Damoiseaux, R.; Holmen, S.L.; Sharpless, N.E.; Hugo, W.; Lo, R.S. Exploiting Drug Addiction Mechanisms to Select against MAPKi-Resistant Melanoma. Cancer Discov. 2018, 8, 74–93. [Google Scholar] [CrossRef]
- Rao, M.; Shi, B.; Yuan, Y.; Wang, Y.; Chen, Y.; Liu, X.; Li, X.; Zhang, M.; Liu, X.; Sun, X. The positive correlation between drug addiction and drug dosage in vemurafenib-resistant melanoma cells is underpinned by activation of ERK1/2-FRA-1 pathway. Anti-Cancer Drugs 2020, 31, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Rashidian, M.; Reinhardt, F.; Bagnato, A.; Keckesova, Z.; Ploegh, H.L.; Weinberg, R.A. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res. 2017, 77, 3982–3989. [Google Scholar] [CrossRef]
- Lee, M.-H.; Yanagawa, J.; Tran, L.; Walser, T.C.; Bisht, B.; Fung, E.; Park, S.J.; Zeng, G.; Krysan, K.; Wallace, W.D.; et al. FRA1 contributes to MEK-ERK pathway-dependent PD-L1 upregulation by KRAS mutation in premalignant human bronchial epithelial cells. Am. J. Transl. Res. 2020, 12, 409–427. [Google Scholar]
- He, H.; Song, D.; Sinha, I.; Hessling, B.; Li, X.; Haldosen, L.-A.; Zhao, C. Endogenous interaction profiling identifies DDX5 as an oncogenic coactivator of transcription factor Fra-1. Oncogene 2019, 38, 5725–5738. [Google Scholar] [CrossRef]
- Song, D.; He, H.; Sinha, I.; Hases, L.; Yan, F.; Archer, A.; Haldosen, L.-A.; Zhao, C.; Williams, C. Blocking Fra-1 sensitizes triple-negative breast cancer to PARP inhibitor. Cancer Lett. 2021, 506, 23–34. [Google Scholar] [CrossRef]
- Brown, P.H.; Alani, R.; Preis, L.H.; Szabo, E.; Birrer, M.J. Suppression of oncogene-induced transformation by a deletion mutant of c-jun. Oncogene 1993, 8, 877–886. [Google Scholar] [PubMed]
- Young, M.R.; Li, J.-J.; Rincón, M.; Flavell, R.A.; Sathyanarayana, B.K.; Hunziker, R.; Colburn, N. Transgenic mice demonstrate AP-1 (activator protein-1) transactivation is required for tumor promotion. Proc. Natl. Acad. Sci. USA 1999, 96, 9827–9832. [Google Scholar] [CrossRef] [PubMed]
- Olive, M.; Krylov, D.; Echlin, D.R.; Gardner, K.; Taparowsky, E.J.; Vinson, C. A Dominant Negative to Activation Protein-1 (AP1) That Abolishes DNA Binding and Inhibits Oncogenesis. J. Biol. Chem. 1997, 272, 18586–18594. [Google Scholar] [CrossRef]
- Bonovich, M.; Olive, M.; Reed, E.; O’Connell, B.; Vinson, C. Adenoviral delivery of A-FOS, an AP-1 dominant negative, selectively inhibits drug resistance in two human cancer cell lines. Cancer Gene Ther. 2002, 9, 62–70. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gerdes, M.J.; Myakishev, M.; Frost, N.A.; Rishi, V.; Moitra, J.; Acharya, A.; Levy, M.R.; Park, S.; Glick, A.; Yuspa, S.H.; et al. Activator Protein-1 Activity Regulates Epithelial Tumor Cell Identity. Cancer Res. 2006, 66, 7578–7588. [Google Scholar] [CrossRef]
- Baxter, D.; Perry, S.R.; Hill, T.A.; Kok, W.M.; Zaccai, N.R.; Brady, R.L.; Fairlie, D.P.; Mason, J.M. Downsizing Proto-oncogene cFos to Short Helix-Constrained Peptides That Bind Jun. ACS Chem. Biol. 2017, 12, 2051–2061. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Ghamsari, L.; Rotolo, J.A.; Kappel, B.J.; Mason, J.M. Combined computational and intracellular peptide library screening: Towards a potent and selective Fra1 inhibitor. RSC Chem. Biol. 2021, 2, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, K.; Chaki, H.; Takakura, T.; Yokotani, J.; Aikawa, Y.; Shiozawa, S.; Gouda, H.; Hirono, S. Design, Synthesis, and Biological Evaluation of New Cyclic Disulfide Decapeptides That Inhibit the Binding of AP-1 to DNA. J. Med. Chem. 2004, 47, 4239–4246. [Google Scholar] [CrossRef] [PubMed]
- Ye, N.; Ding, Y.; Wild, C.; Shen, Q.; Zhou, J. Small Molecule Inhibitors Targeting Activator Protein 1 (AP-1). J. Med. Chem. 2014, 57, 6930–6948. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, Y.; Morimoto, K.; Yamamoto, T.; Chaki, H.; Hashiramoto, A.; Narita, H.; Hirono, S.; Shiozawa, S. Treatment of arthritis with a selective inhibitor of c-Fos/activator protein-1. Nat. Biotechnol. 2008, 26, 817–823. [Google Scholar] [CrossRef]
- Makino, H.; Seki, S.; Yahara, Y.; Shiozawa, S.; Aikawa, Y.; Motomura, H.; Nogami, M.; Watanabe, K.; Sainoh, T.; Ito, H.; et al. A selective inhibition of c-Fos/activator protein-1 as a potential therapeutic target for intervertebral disc degeneration and associated pain. Sci. Rep. 2017, 7, 16983. [Google Scholar] [CrossRef]
- Ucero, A.C.; Bakiri, L.; Roediger, B.; Suzuki, M.; Jimenez, M.; Mandal, P.; Braghetta, P.; Bonaldo, P.; Paz-Ares, L.; Fustero-Torre, C.; et al. Fra-2–expressing macrophages promote lung fibrosis. J. Clin. Investig. 2019, 129, 3293–3309. [Google Scholar] [CrossRef]
- Rajasekaran, S.; Vaz, M.; Reddy, S.P. Fra-1/AP-1 Transcription Factor Negatively Regulates Pulmonary Fibrosis In Vivo. PLoS ONE 2012, 7, e41611. [Google Scholar] [CrossRef]
- Kamide, D.; Yamashita, T.; Araki, K.; Tomifuji, M.; Tanaka, Y.; Tanaka, S.; Shiozawa, S.; Shiotani, A. Selective activator protein-1 inhibitor T-5224 prevents lymph node metastasis in an oral cancer model. Cancer Sci. 2016, 107, 666–673. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhang, K.; Li, Y.; Ren, Y.; Shi, J.; Gu, Y.; Qiu, S.; Liu, S.; Cheng, Y.; Qiao, Y.; et al. OLFML2A is necessary for anti-triple negative breast cancer effect of selective activator protein-1 inhibitor T-5224. Transl. Oncol. 2021, 14, 101100. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Liu, K.; Li, H.; Wang, J.; Zhu, J.; Hao, P.; Zhu, L.; Zhang, S.; Shan, L.; Ma, W.; et al. Veratramine modulates AP-1-dependent gene transcription by directly binding to programmable DNA. Nucleic Acids Res. 2017, 46, 546–557. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Yang, J.; Huang, J.; Auguste, D.T.; Moses, M.A. Therapeutic genome editing of triple-negative breast tumors using a noncationic and deformable nanolipogel. Proc. Natl. Acad. Sci. USA 2019, 116, 18295–18303. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef]
- Annis, M.G.; Ouellet, V.; Rennhack, J.P.; L’Espérance, S.; Rancourt, C.; Mes-Masson, A.-M.; Andrechek, E.R.; Siegel, P.M. Integrin-uPAR signaling leads to FRA-1 phosphorylation and enhanced breast cancer invasion. Breast Cancer Res. 2018, 20, 9. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Kim, W.; Lee, S.; Kim, H.S.; Song, M.; Cha, Y.H.; Kim, Y.-H.; Shin, J.; Lee, E.-S.; Joo, Y.; Song, J.J.; et al. Targeting mutant KRAS with CRISPR-Cas9 controls tumor growth. Genome Res. 2018, 28, 374–382. [Google Scholar] [CrossRef]
- Rossi, J.J.; Rossi, D.J. siRNA Drugs: Here to Stay. Mol. Ther. 2021, 29, 431–432. [Google Scholar] [CrossRef]
- Xiong, H.; Veedu, R.N.; Diermeier, S.D. Recent Advances in Oligonucleotide Therapeutics in Oncology. Int. J. Mol. Sci. 2021, 22, 3295. [Google Scholar] [CrossRef]
- Khvalevsky, E.Z.; Gabai, R.; Rachmut, I.H.; Horwitz, E.; Brunschwig, Z.; Orbach, A.; Shemi, A.; Golan, T.; Domb, A.J.; Yavin, E.; et al. Mutant KRAS is a druggable target for pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20723–20728. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Kang, Y.-K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.-L.; Kim, T.-Y.; et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer 2020, 122, 1630–1637. [Google Scholar] [CrossRef] [PubMed]
- Kruspe, S.; Giangrande, P.H. Aptamer-siRNA Chimeras: Discovery, Progress, and Future Prospects. Biomedicines 2017, 5, 45. [Google Scholar] [CrossRef]
- Gilboa-Geffen, A.; Hamar, P.; LE, T.N.M.; Wheeler, L.A.; Trifonova, R.; Petrocca, F.; Wittrup, A.; Lieberman, J. Gene Knockdown by EpCAM Aptamer–siRNA Chimeras Suppresses Epithelial Breast Cancers and Their Tumor-Initiating Cells. Mol. Cancer Ther. 2015, 14, 2279–2291. [Google Scholar] [CrossRef]
- Zhang, Y.; Xie, X.; Yeganeh, P.N.; Lee, D.-J.; Valle-Garcia, D.; Meza-Sosa, K.F.; Junqueira, C.; Su, J.; Luo, H.R.; Hide, W.; et al. Immunotherapy for breast cancer using EpCAM aptamer tumor-targeted gene knockdown. Proc. Natl. Acad. Sci. USA 2021, 118, e2022830118. [Google Scholar] [CrossRef] [PubMed]
- Vaseva, A.V.; Blake, D.R.; Gilbert, T.; Ng, S.; Hostetter, G.; Azam, S.H.; Ozkan-Dagliyan, I.; Gautam, P.; Bryant, K.L.; Pearce, K.H.; et al. KRAS Suppression-Induced Degradation of MYC Is Antagonized by a MEK5-ERK5 Compensatory Mechanism. Cancer Cell 2018, 34, 807–822.e7. [Google Scholar] [CrossRef]
- Albeck, J.G.; Mills, G.B.; Brugge, J.S. Frequency-Modulated Pulses of ERK Activity Transmit Quantitative Proliferation Signals. Mol. Cell 2012, 49, 249–261. [Google Scholar] [CrossRef]
- Basbous, J.; Jariel-Encontre, I.; Gomard, T.; Bossis, G.; Piechaczyk, M. Ubiquitin-independent versus ubiquitin-dependent proteasomal degradation of the c-Fos and Fra-1 transcription factors: Is there a unique answer? Biochimie 2008, 90, 296–305. [Google Scholar] [CrossRef]
- Hoffmann, E.; Thiefes, A.; Buhrow, D.; Dittrich-Breiholz, O.; Schneider, H.; Resch, K.; Kracht, M. MEK1-dependent Delayed Expression of Fos-related Antigen-1 Counteracts c-Fos and p65 NF-κB-mediated Interleukin-8 Transcription in Response to Cytokines or Growth Factors. J. Biol. Chem. 2005, 280, 9706–9718. [Google Scholar] [CrossRef]
- Yun, S.-I.; Hong, H.K.; Yeo, S.-Y.; Kim, S.-H.; Cho, Y.B.; Kim, K.K. Ubiquitin-Specific Protease 21 Promotes Colorectal Cancer Metastasis by Acting as a Fra-1 Deubiquitinase. Cancers 2020, 12, 207. [Google Scholar] [CrossRef]
- Gao, F.; Li, M.; Zhou, L.; Liu, W.; Zuo, H.; Li, W. Xanthohumol targets the ERK1/2-Fra1 signaling axis to reduce cyclin D1 expression and inhibit non-small cell lung cancer. Oncol. Rep. 2020, 44, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Coleman, K.G.; Crews, C.M. Proteolysis-Targeting Chimeras: Harnessing the Ubiquitin-Proteasome System to Induce Degradation of Specific Target Proteins. Annu. Rev. Cancer Biol. 2018, 2, 41–58. [Google Scholar] [CrossRef]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Zhou, H.; Xu, R.; Zhao, Y.; Chinnaswamy, K.; McEachern, D.; Chen, J.; Yang, C.-Y.; Liu, Z.; Wang, M.; et al. A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo. Cancer Cell 2019, 36, 498–511.e17. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, H.; Kaniskan, H.Ü.; Xie, L.; Chen, X.; Jin, J.; Wei, W. TF-PROTACs Enable Targeted Degradation of Transcription Factors. J. Am. Chem. Soc. 2021, 143, 8902–8910. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Wu, Y.; Chen, J.; Shen, Y.; Zhang, L.; Zhang, H.; Chen, L.; Yuan, H.; Chen, H.; Zhang, W.; et al. The peptide PROTAC modality: A novel strategy for targeted protein ubiquitination. Theranostics 2020, 10, 10141–10153. [Google Scholar] [CrossRef]
- Ma, D.; Zou, Y.; Chu, Y.; Liu, Z.; Liu, G.; Chu, J.; Li, M.; Wang, J.; Sun, S.-Y.; Chang, Z. A cell-permeable peptide-based PROTAC against the oncoprotein CREPT proficiently inhibits pancreatic cancer. Theranostics 2020, 10, 3708–3721. [Google Scholar] [CrossRef] [PubMed]
- Moutel, S.; Bery, N.; Bernard, V.; Keller, L.; Lemesre, E.; De Marco, A.; Ligat, L.; Rain, J.-C.; Favre, G.; Olichon, A.; et al. NaLi-H1: A universal synthetic library of humanized nanobodies providing highly functional antibodies and intrabodies. eLife 2016, 5, e16228. [Google Scholar] [CrossRef]
- Ibrahim, A.F.M.; Shen, L.; Tatham, M.H.; Dickerson, D.; Prescott, A.R.; Abidi, N.; Xirodimas, D.P.; Hay, R.T. Antibody RING-Mediated Destruction of Endogenous Proteins. Mol. Cell 2020, 79, 155–166.e9. [Google Scholar] [CrossRef]
- Huang, C.; Ma, W.-Y.; Dawson, M.I.; Rincon, M.; Flavell, R.A.; Dong, Z. Blocking activator protein-1 activity, but not activating retinoic acid response element, is required for the antitumor promotion effect of retinoic acid. Proc. Natl. Acad. Sci. USA. 1997, 94, 5826–5830. [Google Scholar] [CrossRef]
- Sun, Y.; Lin, Z.; Liu, C.-H.; Gong, Y.; Liegl, R.; Fredrick, T.W.; Meng, S.S.; Burnim, S.B.; Wang, Z.; Akula, J.D.; et al. Inflammatory signals from photoreceptor modulate pathological retinal angiogenesis via c-Fos. J. Exp. Med. 2017, 214, 1753–1767. [Google Scholar] [CrossRef] [PubMed]
- Bishop, T.R.; Zhang, Y.; Erb, M.A. Pharmacological Modulation of Transcriptional Coregulators in Cancer. Trends Pharmacol. Sci. 2019, 40, 388–402. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Lockwood, W.W.; Zejnullahu, K.; Bradner, J.E.; Varmus, H. Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 19408–19413. [Google Scholar] [CrossRef] [PubMed]
- Baker, E.K.; Taylor, S.; Gupte, A.; Sharp, P.P.; Walia, M.; Walsh, N.C.; Zannettino, A.C.; Chalk, A.M.; Burns, C.J.; Walkley, C.R. BET inhibitors induce apoptosis through a MYC independent mechanism and synergise with CDK inhibitors to kill osteosarcoma cells. Sci. Rep. 2015, 5, 10120. [Google Scholar] [CrossRef]
- Bid, H.K.; Phelps, D.A.; Xaio, L.; Guttridge, D.C.; Lin, J.; London, C.; Baker, L.H.; Mo, X.; Houghton, P.J. The Bromodomain BET Inhibitor JQ1 Suppresses Tumor Angiogenesis in Models of Childhood Sarcoma. Mol. Cancer Ther. 2016, 15, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Hoyle, R.G.; Xie, N.; Wang, W.; Cai, H.; Zhang, M.; Ma, Z.; Xiong, G.; Xu, X.; Huang, Z.; et al. A Super-Enhancer Driven by FOSL1 Controls miR-21-5p Expression in Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2021, 11, 656628. [Google Scholar] [CrossRef] [PubMed]
- Ravasi, T.; Suzuki, H.; Cannistraci, C.V.; Katayama, S.; Bajic, V.B.; Tan, K.; Akalin, A.; Schmeier, S.; Kanamori-Katayama, M.; Bertin, N.; et al. An Atlas of Combinatorial Transcriptional Regulation in Mouse and Man. Cell 2010, 140, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.-W.; Wang, Z.; Yiu, T.T.; Akdemir, K.C.; Xia, W.; Winter, S.; Tsai, C.-Y.; Shi, X.; Schwarzer, D.; Plunkett, W.; et al. TRIM24 links a non-canonical histone signature to breast cancer. Nature 2010, 468, 927–932. [Google Scholar] [CrossRef]
- Groner, A.C.; Cato, L.; de Tribolet-Hardy, J.; Bernasocchi, T.; Janouskova, H.; Melchers, D.; Houtman, R.; Cato, A.; Tschopp, P.; Gu, L.; et al. TRIM24 Is an Oncogenic Transcriptional Activator in Prostate Cancer. Cancer Cell 2016, 29, 846–858. [Google Scholar] [CrossRef]
- Lv, D.; Li, Y.; Zhang, W.; Alvarez, A.A.; Song, L.; Tang, J.; Gao, W.-Q.; Hu, B.; Cheng, S.-Y.; Feng, H. TRIM24 is an oncogenic transcriptional co-activator of STAT3 in glioblastoma. Nat. Commun. 2017, 8, 1454. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.; Fedorov, O.; Tallant, C.; Monteiro, O.; Meier, J.; Gamble, V.; Savitsky, P.; Nunez-Alonso, G.A.; Haendler, B.; Rogers, C.; et al. Discovery of a Chemical Tool Inhibitor Targeting the Bromodomains of TRIM24 and BRPF. J. Med. Chem. 2016, 59, 1642–1647. [Google Scholar] [CrossRef] [PubMed]
- Gechijian, L.N.; Buckley, D.L.; Lawlor, M.; Reyes, J.; Paulk, J.; Ott, C.J.; Winter, G.E.; Erb, M.A.; Scott, T.; Xu, M.; et al. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nat. Chem. Biol. 2018, 14, 405–412. [Google Scholar] [CrossRef]
- Taniguchi, T.; Iizumi, Y.; Watanabe, M.; Masuda, M.; Morita, M.; Aono, Y.; Toriyama, S.; Oishi, M.; Goi, W.; Sakai, T. Resveratrol directly targets DDX5 resulting in suppression of the mTORC1 pathway in prostate cancer. Cell Death Dis. 2016, 7, e2211. [Google Scholar] [CrossRef]
- Kraft, T.E.; Parisotto, D.; Schempp, C.; Efferth, T. Fighting Cancer with Red Wine? Molecular Mechanisms of Resveratrol. Crit. Rev. Food Sci. Nutr. 2009, 49, 782–799. [Google Scholar] [CrossRef] [PubMed]
- Day, E.K.; Campbell, A.; Pandolf, A.; Rogerson, T.; Zhong, Q.; Xiao, A.; Purow, B.; Lazzara, M.J. ERK-dependent suicide gene therapy for selective targeting of RTK/RAS-driven cancers. Mol. Ther. 2021, 29, 1585–1601. [Google Scholar] [CrossRef] [PubMed]
- Bushweller, J.H. Targeting transcription factors in cancer—From undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casalino, L.; Talotta, F.; Cimmino, A.; Verde, P. The Fra-1/AP-1 Oncoprotein: From the “Undruggable” Transcription Factor to Therapeutic Targeting. Cancers 2022, 14, 1480. https://doi.org/10.3390/cancers14061480
Casalino L, Talotta F, Cimmino A, Verde P. The Fra-1/AP-1 Oncoprotein: From the “Undruggable” Transcription Factor to Therapeutic Targeting. Cancers. 2022; 14(6):1480. https://doi.org/10.3390/cancers14061480
Chicago/Turabian StyleCasalino, Laura, Francesco Talotta, Amelia Cimmino, and Pasquale Verde. 2022. "The Fra-1/AP-1 Oncoprotein: From the “Undruggable” Transcription Factor to Therapeutic Targeting" Cancers 14, no. 6: 1480. https://doi.org/10.3390/cancers14061480
APA StyleCasalino, L., Talotta, F., Cimmino, A., & Verde, P. (2022). The Fra-1/AP-1 Oncoprotein: From the “Undruggable” Transcription Factor to Therapeutic Targeting. Cancers, 14(6), 1480. https://doi.org/10.3390/cancers14061480