1. Introduction
Angiogenesis, the formation of new blood vessels from pre-existing endothelial cells, is regulated by the balance of pro-angiogenic and anti-angiogenic factors present in a tissue [
1]. In solid tumors, including lung cancer, the mechanisms involved in angiogenesis have been considered as therapeutic targets, because angiogenesis is essential for primary tumor growth and for metastasis in advanced cancers [
1,
2]. It is known that hypoxia in the tumor mass, which is a major cause of angiogenesis, leads to the overproduction of pro-angiogenic factors [
1,
3]. The best characterized family of angiogenic factors is the vascular endothelial growth factor (VEGF) family, which consists of VEGF-A through D and placenta growth factor (PlGF, also named phosphatidylinositol glycan anchor biosynthesis class F (PIGF)) [
1,
2]. VEGFs binds to a family of transmembrane receptor tyrosine kinases (RTKs) called VEGF receptors (VEGFR1-3). VEGF-A, mainly responsible for vessel formation in adult tissues, binds to VEGFR-1 with higher affinity; however, it has been reported that VEGFR-2, which is the primary receptor involved in endothelial cell proliferation and migration, mediates VEGF-A-induced angiogenesis in cancers [
2]. In addition to VEGF family members, other factors (such as fibroblast growth factors (FGFs)) and chemokines have also been implicated in the processes of normal and tumoral angiogenesis [
4]. Tumor angiogenesis has unique characteristics of abnormal and poorly organized vessels with increased permeability. These features may result from the overproduction of various angiogenic factors from the tumor and stromal cells [
1,
2]. Targeting tumor angiogenesis has been approached through two methods: monoclonal antibodies that block VEGF–VEGFR binding, and small molecule TKIs that inhibit the downstream VEGFR mediated signaling [
1].
C-type lectin 11A (CLEC11A, also named stem cell growth factor (SCGF)) is originally identified as an autocrine factor secreted by leukemia cell lines [
5]. Full length human CLEC11A protein consists of 323 amino acids with the lectin domain at its carboxyl terminus [
6,
7]. CLEC11A is also a factor promoting endothelial cell differentiation from AC133-positive progenitor cells [
8]. Genetic ablation in mice shows that CLEC11A is also an autocrine factor for osteoblasts, and its abolition causes reduced limb and vertebral bone formation [
9]. In contrast to the earlier findings, CLEC11A-null mice do not have obvious defects in hematopoiesis [
9]. CLEC11A contains potential integrin-binding motifs (RGD and TDF motifs), and it has been shown that Integrin α11 is the receptor for CLEC11A in osteoblasts [
10]. Nevertheless, the RGD motif is not present in the mouse CLEC11A protein [
6,
10].
Our current study showed that CLEC11A expression was increased in LAC cells expressing mutated EGFR. The elevation of CLEC11A was confirmed in clinical LAC tissues, and high CLEC11A expression in LAC was associated with a worse overall survival. CLEC11A was a chemo-attractant for mouse endothelial cells and enhanced angiogenesis induced by VEGF-A or FGF1. Our results from cellular and animal studies supported that CLEC11A might promote LAC progression through enhancing tumor angiogenesis.
2. Materials and Methods
2.1. Cell Cultures
NL20-immortalized human bronchial epithelial cells and SVEC4-10 mouse endothelial cells were obtained from the National Health Research Institutes (NHRI) Cell Bank and cultured as described [
11,
12]. Human lung cancer cell lines (H1299 and its derivatives, CL1-0, H661, H358, H1975, HCC1650, PE089, and HCC827) were cultured as described previously [
13,
14,
15]. NL20, CL1-0, H661, H358, and H1299 cells express wild type (WT) EGFR; H1975 cell line has L858R/T790M double mutations; H1650, PE089, and HCC827 cells have
EGFR exon 19 deletion (Del). H1299 derivatives expressing CLEC11A were established by transfecting the CLEC11A expression vector into H1299 cells using the lipofectamine 2000 reagent (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions, and were subjected to Zeocin (Invitrogen, Waltham, MA, USA) selection for one week to establish permanently transfected cells. HCC827–vector control and HCC827-shCLEC11A cells were established by transfecting an empty pSuper-puromycin vector or a vector encoding a small hairpin RNA against CLEC11A (shCLEC11A) into H1299 cells using the lipofectamine 2000 reagent. The transfected cells were then subjected to puromycin selection (1 μg/mL) for one week to establish permanently transfected cells. The isolation and culture of mouse primary aortic artery endothelial cell (AAE cell) was performed as described previously [
16].
2.2. Antibodies and Reagents
The anti-CLEC11A monoclonal antibody (Cat# MAB1904) was purchased from R&D System (Minneapolis, MN, USA). The anti-FAK (Cat# sc-1688), anti-integrins α2 (Cat# sc-6586), α5 (Cat# sc-10729), and β1 (Cat# sc-6622), p130-Cas (Cat# sc-20029), and peroxidase-conjugated secondary antibodies were purchased from Santa Cruz (Santa Cruz, CA, USA). The anti-active integrin β1 antibody (Cat# 550531) was obtained from BD Pharmingen (San Diego, CA, USA). The anti-EGFR (Cat#2232), anti-pEGFR (Y1068, Cat# 2234), anti-pFAK (Y576/577, Cat# 3281), and anti-pp130-Cas (Y165, Cat# 4015) were purchased from Cell Signaling Technology (Beverly, MA, USA). The anti-pFAK (Y397; Cat# ab81298) antibody was purchased from Abcam (Cambridge, UK). The anti-actin (Cat# A5441) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Anti-TGFBI (Cat# GTX100744) and anti-tubulin (Cat# GTX62882) antibodies were obtained from GeneTex (Hsinchu, Taiwan). G418 and Zeocin were purchased from Sigma-Aldrich (St. Louis, MO, USA) and Invitrogen (Carlsbad, CA, USA), respectively. VEGF-A 165 (Cat# 292-VE) and basic FGF (Cat# 234-FSE) were purchased from R&D System (Minneapolis, MN, USA).
2.3. CLEC11A Expression and Knockdown Vectors
The coding sequence of human CLEC11A was PCR-amplified using a cDNA pool from H1299-EGFR-Del cells as the template. The forward primer sequence is 5′-GCGGGATCCACCATGCAGGCAGCCTGGCTTTTGGGG and the reverse primer is 5′-GCGGTCGACATGAAGGGGAACTCGCAGACGTAG. The amplified DNA fragment was digested by restriction enzymes (Bam HI and Sal I) and was inserted between the Bam HI and Xho I sites of the pcDNA4-His-Myc B vector (Invitrogen). The D63E, D133E, and D63E/D133E mutations of CLEC11A were generated using the site-directed mutagenesis method. The plasmid-expressing small hairpin RNA against human CLEC11A (shCLEC11A) was constructed using the pSuper-retro-puro vector (OligoEngine, Seattle, WA, USA) as a backbone. A 19-nucleotide sequence (5′-TCCTGCCGGAACTGTTGAG) corresponding to the nucleotides 195–213 of human CLEC11A coding region was used for designing the inverted-repeat insertion in the vector according to the manufacturer’s instructions.
2.4. Cell Extracts Preparation
Whole cell extract was prepared by suspending 2 × 106 cells in 200 μL of lysis buffer (50 mM Tris (pH 8.0), 150 mM NaCl, 1% Triton X-100, 0.5% deoxycholate, 0.1% SDS, 2 μg/mL leupeptin, 5 μg/mL aprotinin, 1 mM PMSF, 1 mM DTT, and 1 mM Na3VO4). For His-tagged CLEC11A pull-down assays, the cell extracts were prepared using lysis buffer without deoxycholate and SDS. The cell extracts were kept on ice and vigorously mixed four times with 5 min intervals. The extracts were cleared by centrifugation at 14,000× g for 10 min, and the supernatants were collected for further analyses or stored at −80 °C.
2.5. Immuno-Blots, Immuno-Fluorescence (IF) Staining, and His-Tagged Protein Pull-Down Assays
Immuno-blotting and IF staining assays were performed as described previously [
17]. For pull-down assays, His-tagged CLEC11A and mutant proteins were immobilized on TALON metal affinity resin beads (BD Biosciences, San Jose, CA, USA) and were incubated with 2 mg of SVEC4-10 cell extracts for 2 h at 4 °C with continuous rotation. The complexes were then washed with the lysis buffer (without deoxycholate and SDS) twice, and affinity-purified proteins were boiled in SDS sample buffer and examined by immuno-blotting.
2.6. Cell Migration and Statistical Analyses
Transwell migration assays were performed as described previously with modifications [
17]. Forty-thousand SVEC4-10 cells were seeded in the top wells (DMEM with 1% FCS) and allowed for Transwell migration for 8 h toward the bottom chambers (DMEM with 1% FCS) with specified attractants. The Transwell membranes were subjected to crystal violet staining, and the data presented were the results of the quantitative data from 15 observed fields of represented experiments. Statistical analyses were performed by two-tailed Student’s
t-tests.
2.7. Matrigel Angiogenesis Assay
C57BL/6 mice were injected subcutaneously in both flanks with 300 μL of growth factor-reduced Matrigel (Corning, Corning, NY, USA) supplemented with basic FGF or VEGF-A (100 ng/mL) in the presence or absence of CLEC11A (1 μg/mL). The control (without CLEC11A) and experimental group (with CLEC11A) were injected into the same mouse in the right and left flank, respectively. Two weeks later, the Matrigel plugs were carefully harvested, weighed, and minced in ice-cold deionized water. The mixtures were kept at 4 °C overnight for hemoglobin extraction. Then, the mixtures were centrifugated at 13,000 rpm for 20 min at 4 °C and the supernatants were collected for hemoglobin measurement by a colorimetric assay using Drabkin’s reagent (Sigma-Aldrich). A total of 250 µL of the reaction mixture was placed in a well of a 96-well plate and the absorbance was measured at 540 nm.
2.8. RNA Extraction and Real-Time PCR Quantification of mRNA
H1299 derivative (Vec, EGFR, L858R, Del) cells were maintained in regular culture medium or in serum-free medium for 24 h. Total cellular RNA were extracted using the Trizol reagent (Invitrogen). The quantification of mRNA was conducted by real-time PCR on the LightCycler instrument (Roche Molecular Biochemicals, Indianapolis, IN, USA). The PCR specificity was further verified by checking the amplification products using agarose gel electrophoresis. Real-time PCR was performed using a kit of LightCycler FastStart DNA Masterplus SYBR Green I (Roche, Indianapolis, IN, USA) in a total volume of 20 μL in the LightCycler glass capillaries. The reaction program was: initial heating to 95 °C for 10 min, followed by 45 PCR cycles of heating to 95 °C for 10 s, incubation for 10 s at the annealing temperature specific for the use of primer, and incubation for 25 s at 72 °C. We conducted a melting curve analysis after every real-time PCR to identify PCR product and to detect the possible presence of contaminating products. We also quantified the transcripts of the β-Actin gene, and each sample was normalized on the basis of its β-actin mRNA level. The relative quantification was determined using the comparative CT method.
2.9. Experimental Animals
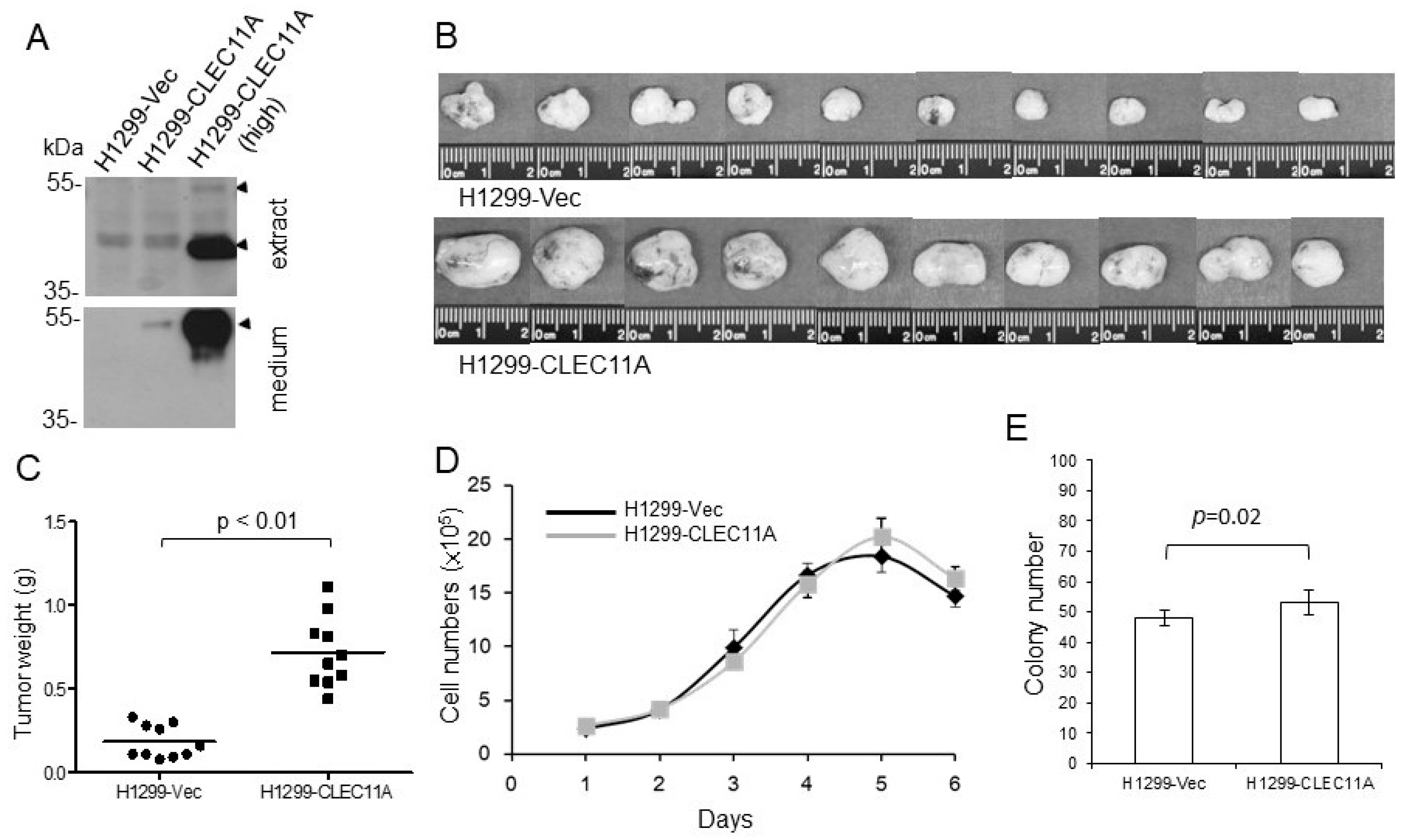
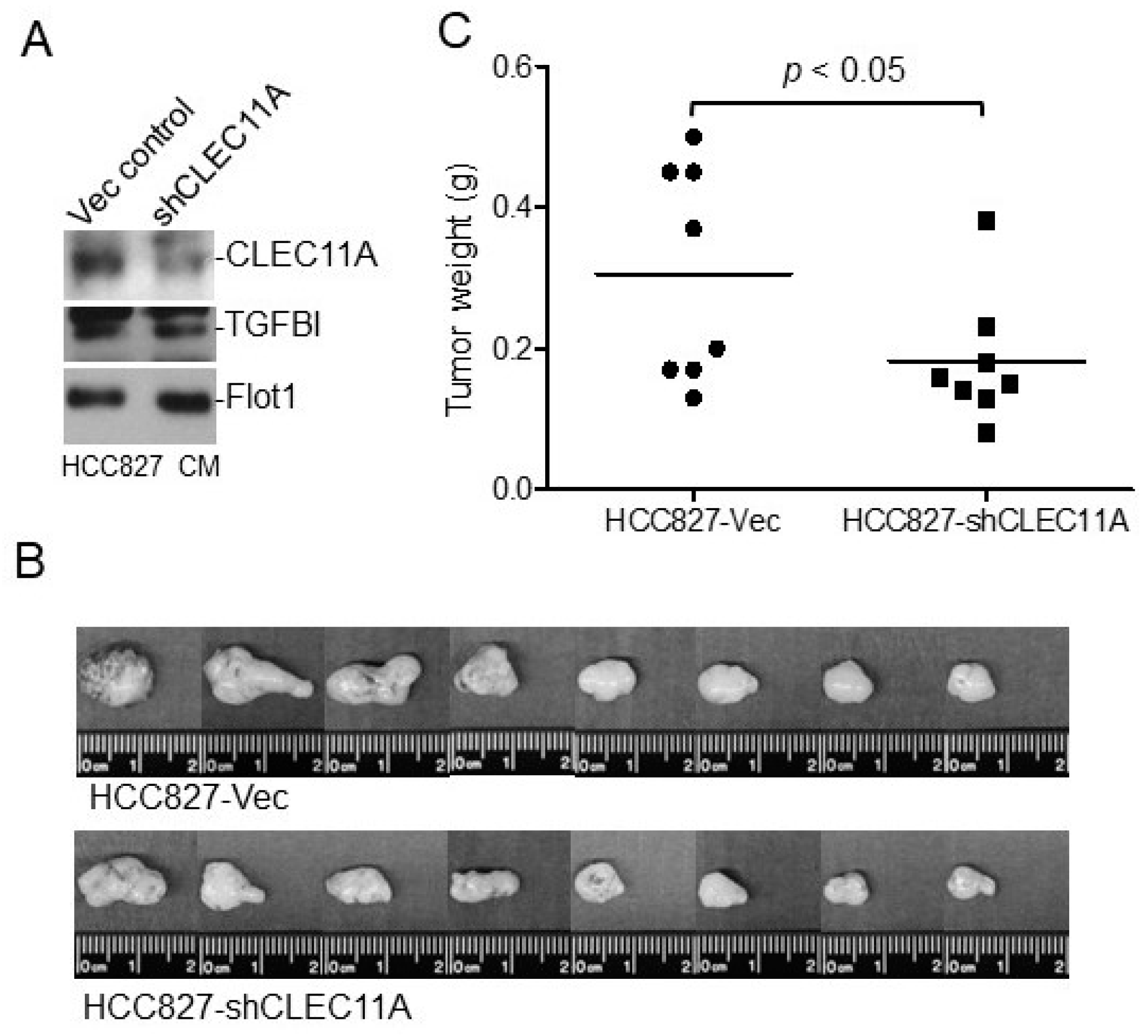
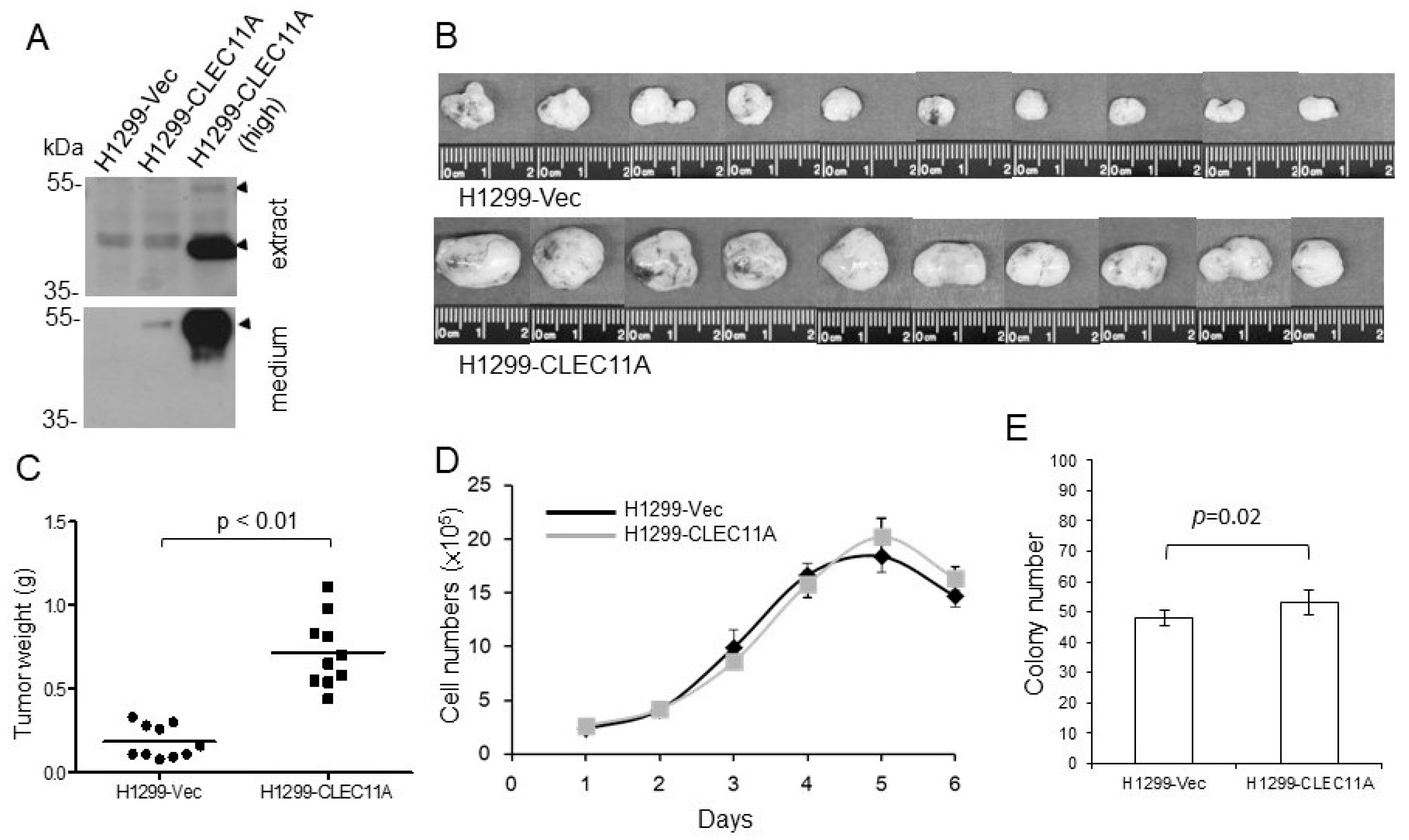
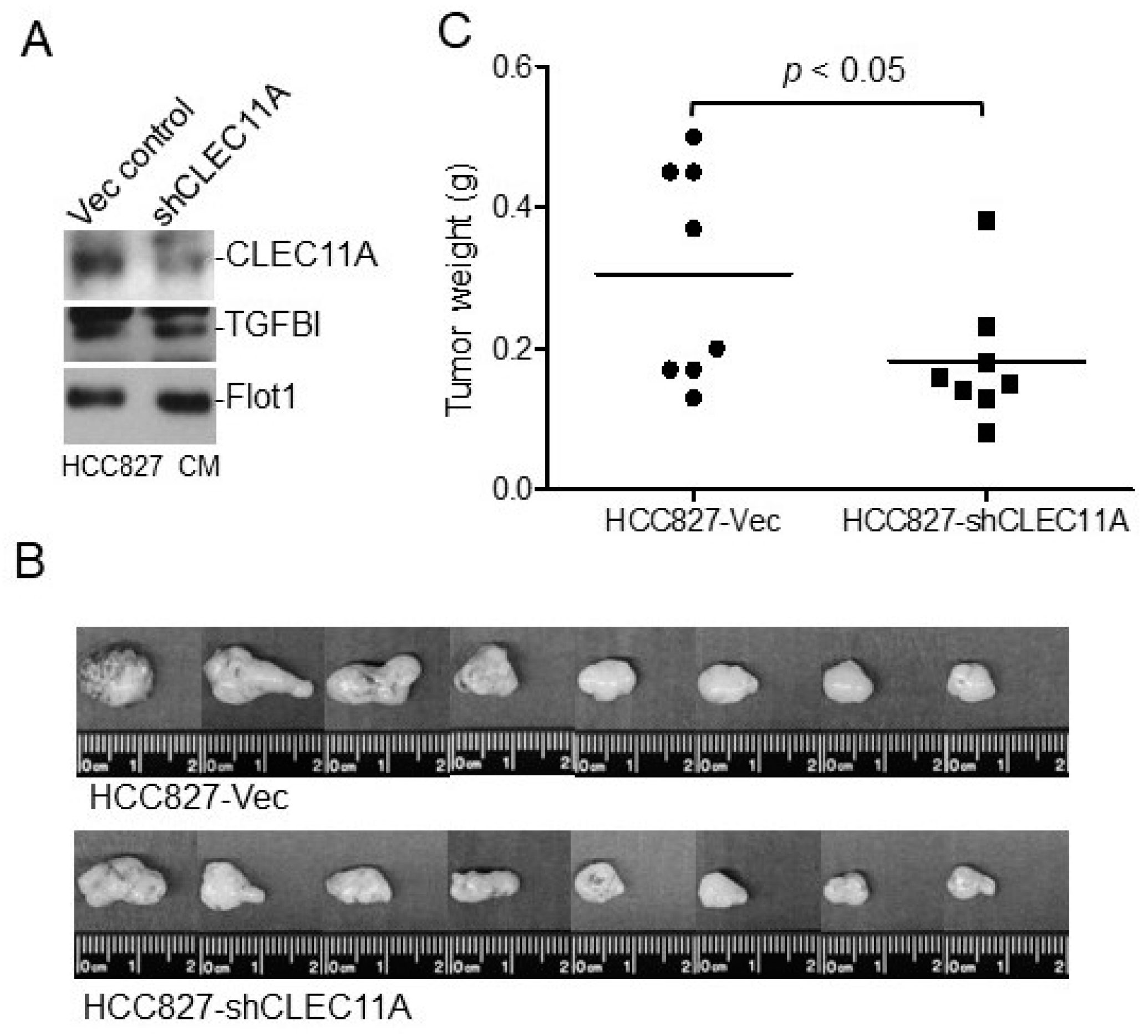
Eight-week-old female BALB/c nude mice or C57BL/6 mice were purchased from the National Laboratory Animal Center, Taiwan. The animals had access to food and water ad libitum. Experimental procedures using animals were approved by the Institutional Animal Care and Use Committees of the National Health Research Institutes. The nude mice were injected subcutaneously with H1299-Vec, H1299-CLEC11A, HCC827-Vec, or HCC827-shCLEC11A cells. Each injection contained 2 × 106 cells suspended in 150 μL of RPMI 1640 medium with 30% of Matrigel. At four to five weeks post-injection, the tumors were harvested, weighed, and processed for immunohistochemistry or Western blot analyses. C57BL/6 mice were injected subcutaneously with Lewis lung carcinoma (LL/2) cells. Each injection contained 1 × 106 cells suspended in 150 μL of RPMI 1640 medium with 30% of Matrigel with or without CLEC11A (1 μg/mL). At two weeks post-injection, the tumors were harvested, weighed, and processed for further analyses.
2.10. LAC Tissue Sections and Immunohistochemical (IHC) Staining
Parafilm-embedded LAC tissues sections were obtained from Chang Gung Memorial Hospital (CGMH) tissue bank. The study protocol was reviewed and approved by the Institutional Review Boards of CGMH and NHRI. The
EGFR mutation status of the LAC tissues was determined as previously described [
18]. IHC staining was performed at the Pathology Core Laboratory, National Health Research Institutes. The assays were performed on 4 μm thick formalin-fixed paraffin-embedded tissue sections. The sections were deparaffinized twice in xylene for 10 min and twice in ethanol for 2 min, and then placed in 10 mM citric buffer solution (pH 6.0) and heated at 95 °C for 15 min. The samples were washed once with PBS and the endogenous peroxidase activity was blocked by 3% H
2O
2 for 5 min. The slides were rinsed twice with PBS and then incubated with an anti-CLEC11A monoclonal antibody (MAB1904; R&D System) at 1:50 dilution for 2 h at room temperature. The slides were washed three times with PBS, and then incubated with an anti-mouse antibody conjugated with peroxidase (1:500; Santa Cruz) for 1 h. The slides were then washed with PBS three times and incubated with 3,3′ Diaminobenzidine (DAB) chromogen for 10 min. After the reaction, the slides were washed with water three times and counterstained with hematoxylin.
2.11. Clinical Data Analyses
The data of the LAC, breast cancer (BRCA), as well as head and neck cancer (HNSC) and their respective normal tissues in The Cancer Genome Atlas (TCGA) database were analyzed for CLEC11A mRNA levels through the Boxplot program in Gene Expression Profiling Interactive Analysis (GEPIA). To analyze the CLEC11A expression in the LAC tissues with or without a mutation in EGFR and K-Ras genes, the CLEC11A expression levels of LAC patients were retrieved through the OncoLnc server and were sub-grouped and analyzed according to their EGFR and K-Ras mutation records retrieved from the Genomic Data Commons (GDC) Data Portal, NCI. The correlation of the CLEC11A expression levels (RNA sequencing data) and the survival of LAC patients was examined through the Kaplan–Meier plotter.
4. Discussion
The formation of tumor vasculature for blood supply is achieved through multiple mechanisms, including (but not limited to) (1) the development of blood vessels from endothelial cells (ECs) in pre-existing vessels (angiogenesis), (2) the formation of vessels by EC precursors (vasculogenesis), (3) tumor cells growing along the existing vasculature (vessel co-option), and (4) the formation of vessel-like structures by tumor cells (vasculogenic mimicry) [
1,
2,
19]. Angiogenic factors may participate in most of these processes [
1,
19]. Anti-angiogenic therapeutics, either monoclonal antibody- or chemical-based, against endothelial trophic factors/receptors have been developed for years. However, anti-angiogenic therapy alone is usually not sufficient to achieve a satisfactory effect, and combination with conventional chemotherapy is required for most of the cancer-treating regiments [
1]. The complex mechanisms that solid tumors utilize to evade blockades and achieve blood supply may provide explanations for the current problems in anti-angiogenic therapy. Another possible explanation is that multiple factors (and receptors) are involved in the process of tumor angiogenesis. Although the concept of stopping blood flow to tumors is straightforward, the blocking of a single pathway with a specific antibody or inhibitor proved insufficient in achieving the desired effect. Therefore, a better understanding of the angiogenic mechanisms involved in tumor progression is very important.
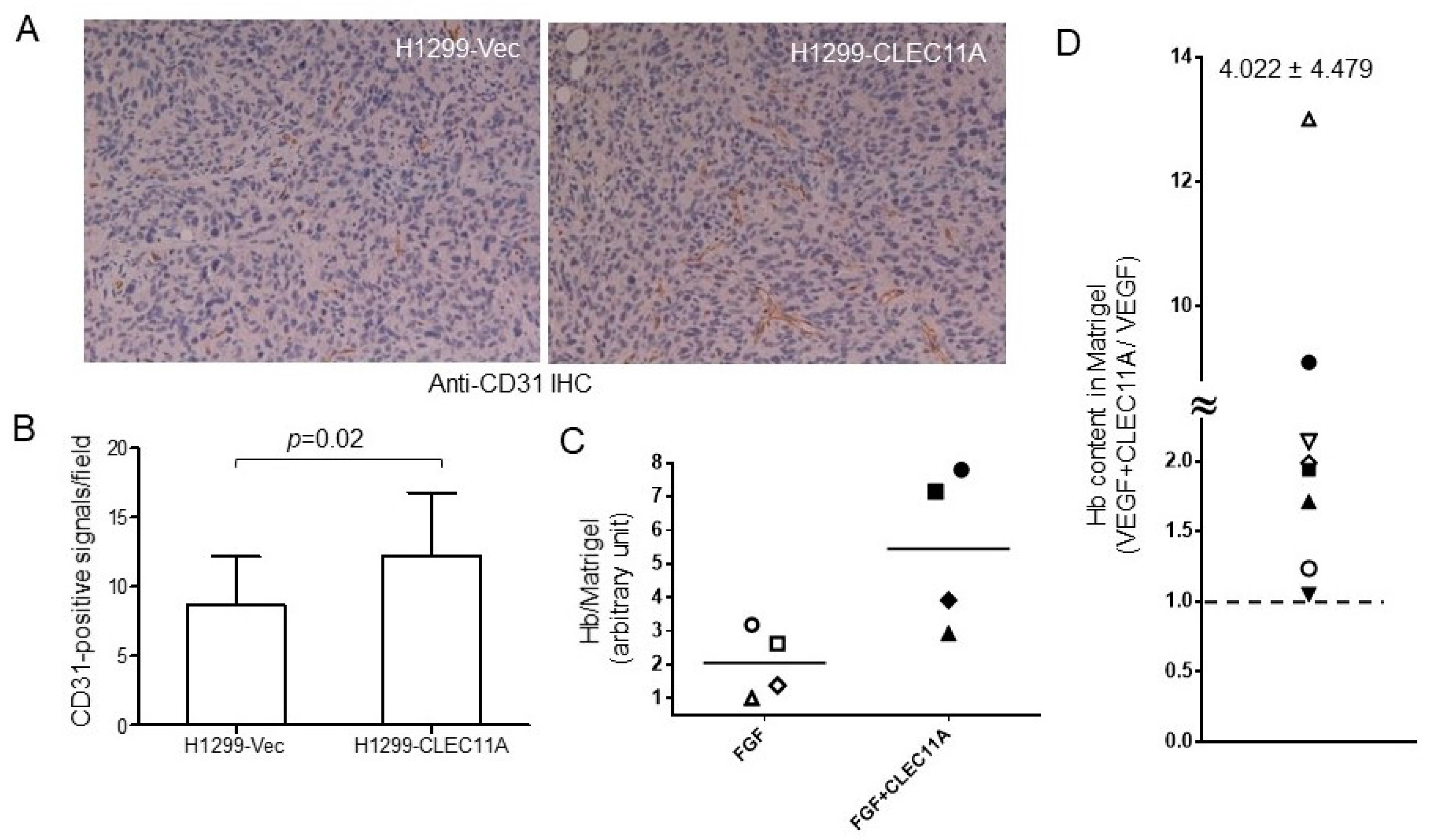
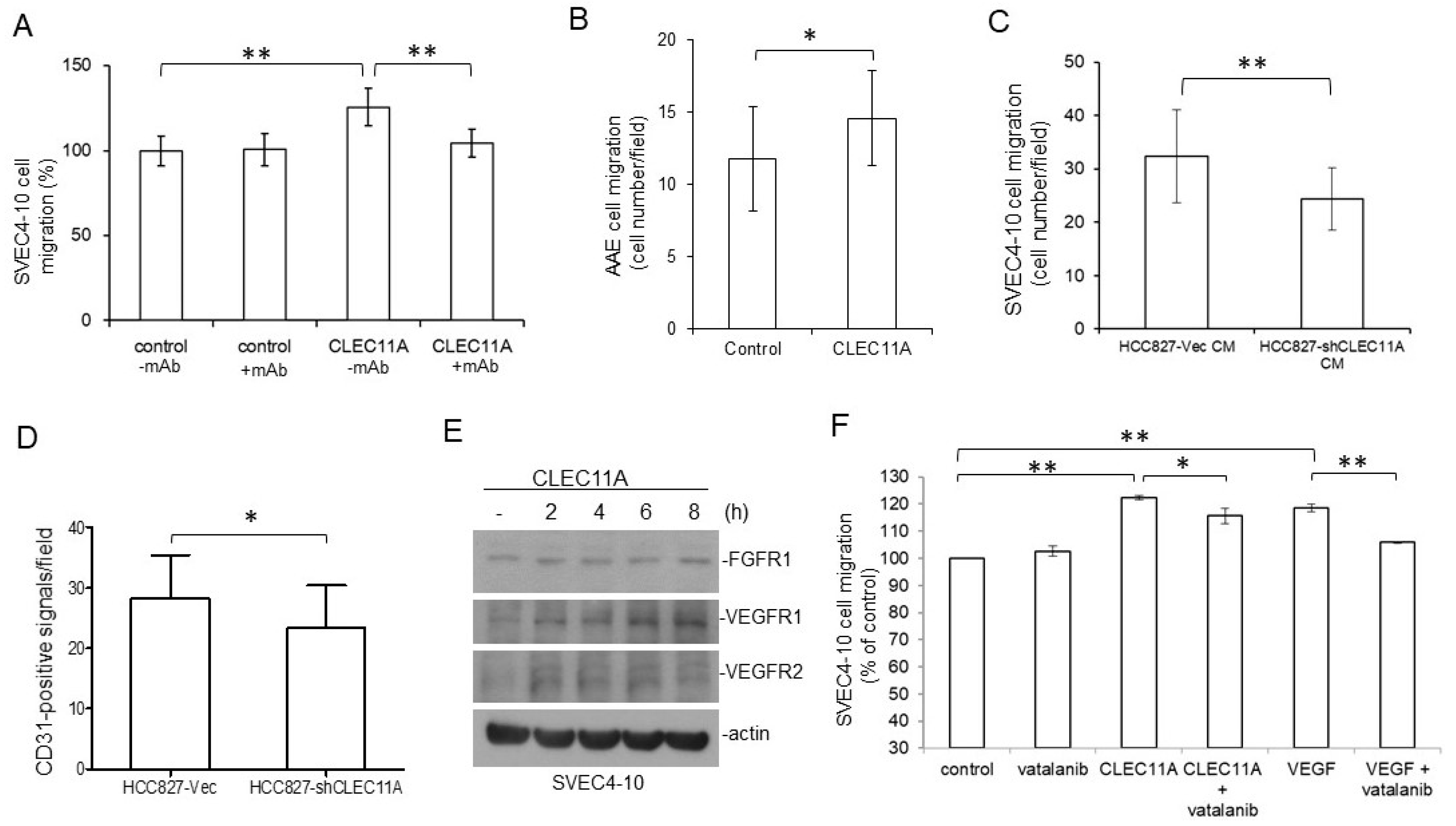
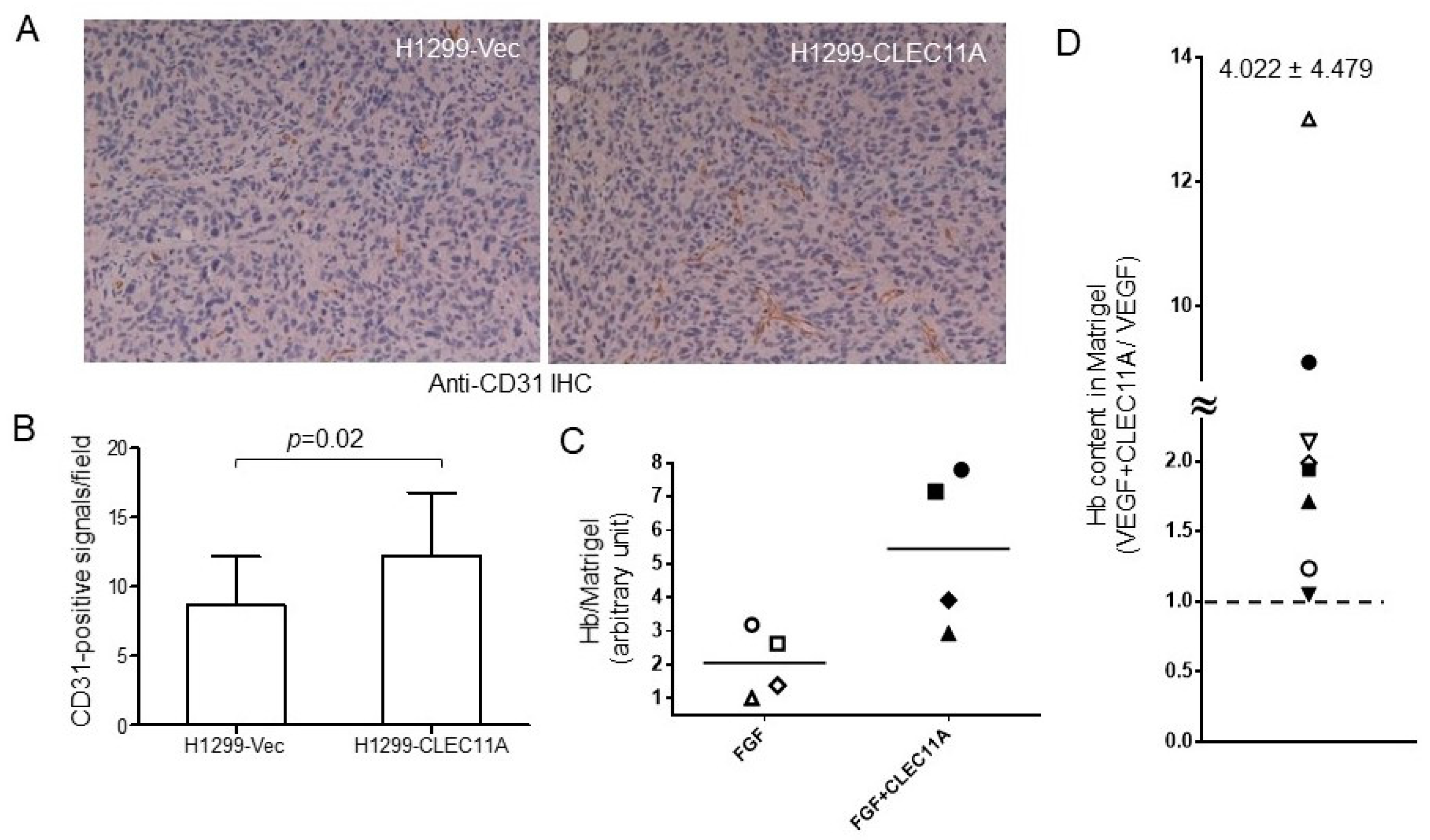
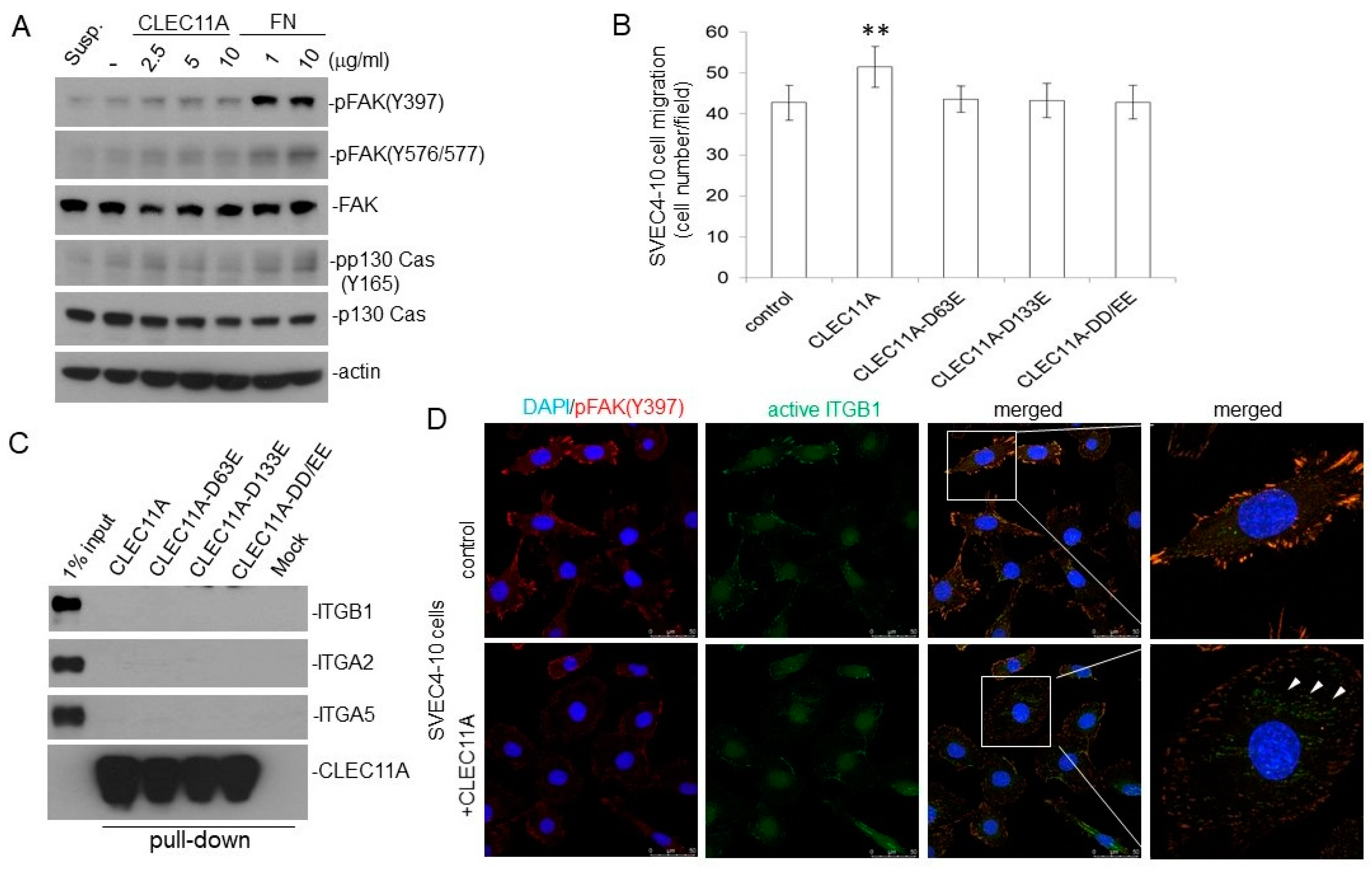
Our findings suggested that CLEC11A produced from LAC may promote tumor vascularization and cancer progression. CLEC11A per se did not show angiogenic activity, since the factor alone could not support significant blood vessel formation in the in vivo angiogenesis assay (data not shown). However, the combination of CLEC11A and VEGF-A (or FGF1) achieved better angiogenic effects than did VEGF-A (or basic FGF) alone (
Figure 5D,E). The mechanism by which CLEC11A promoted angiogenesis was intriguing. CLEC11A promotes endothelial cell differentiation, in the presence of VEGF-A, from AC133-positive progenitor cells [
8]. It is possible that CLEC11A secreted from LAC cells facilitated the differentiation of circulating progenitor cells into endothelial cells and the formation of new vessels through vasculogenesis, in the presence of tumor-secreted VEGFs. We found that CLEC11A increased the expression levels of FGFR1 and VEGFRs in SVEC4-10 endothelial cells (
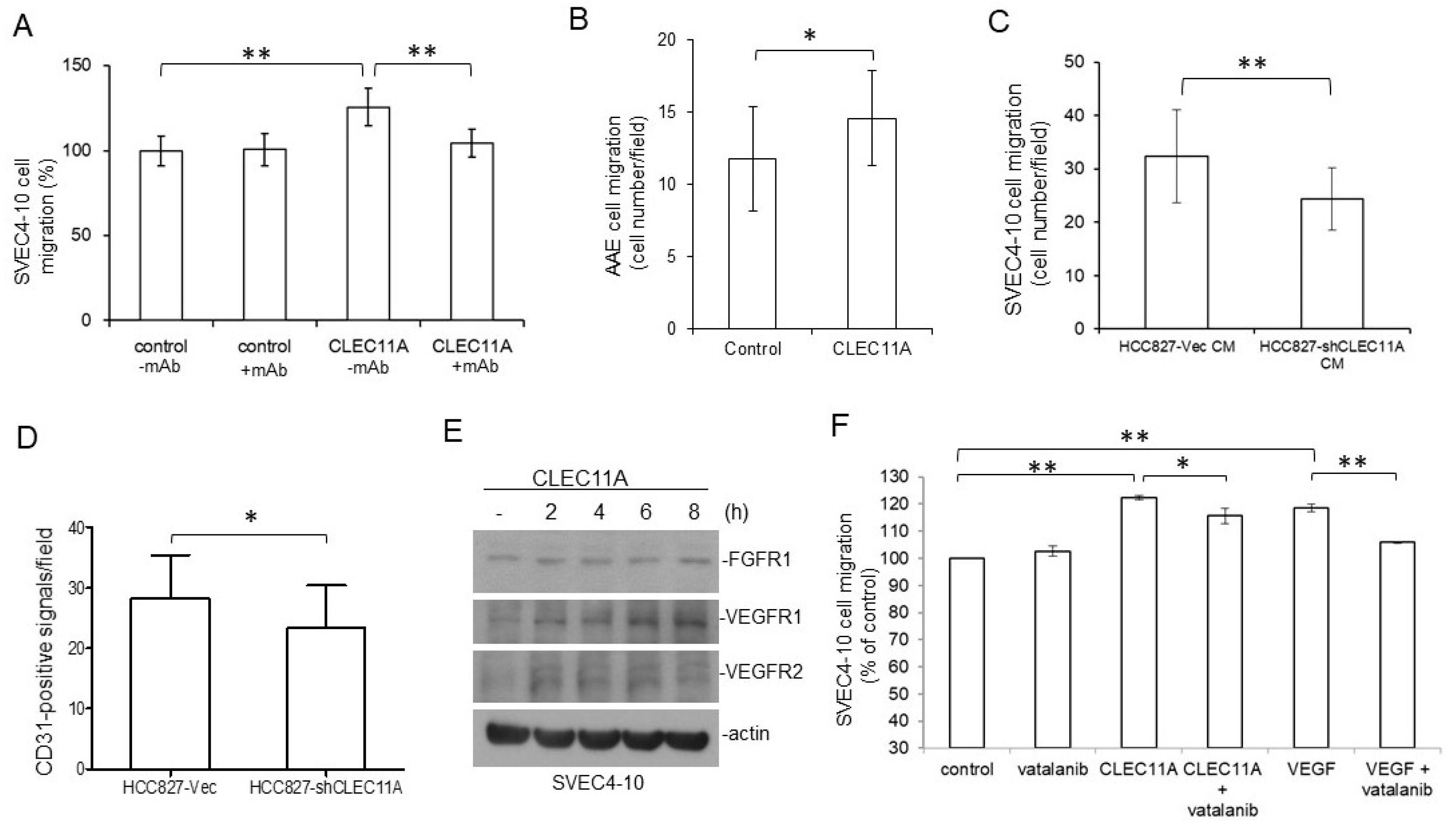
Figure 6E). It was also possible that the presence of CLEC11A triggered the better responses of endothelial cells to angiogenic growth factors. Additionally, our data indicated that CLEC11A is a chemo-attractant for mouse endothelial cells (
Figure 6). This chemo-attractant ability of CLEC11A was not dependent on the VEGF/VEGFR signaling, since the VEGFR inhibitor vatalanib did not abolish this effect (
Figure 6F). It was likely that cancer cells secreted CLEC11A and attracted endothelial cells sprouting from the nearby pre-existing blood vessels and, subsequently, forming new blood vessels in the growing tumor mass. Our data indicated that understanding the biological effect of CLEC11A could be very important in achieving a better understanding of tumor angiogenesis.
Despite the fact that CLEC11A has been identified for more than three decades, we do not understand much about the biological functions of CLEC11A and how those functions link to its structural configuration. A major difference between human and mouse CLEC11A is that the mouse protein does not contain an RGD motif [
6,
10]. This motif, along with the LDT motif, is proposed as the binding motifs on CLEC11A to integrin receptors [
10]. We found that CLEC11A mutated at the RGD and/or LDT motifs lost the chemo-attractant ability toward endothelial cells, suggesting that these motifs, directly or indirectly, were involved in this function (
Figure 7B). Contrary to the previous report, we did not observe the physical interaction between CLEC11A and integrin receptors (
Figure 7C). We also did not detect significant activation of FAK in the endothelial cells by CLEC11A stimulation, unlike fibronectin (
Figure 7A). Nevertheless, we detected the activation of integrin β1 by CLEC11A using a conformation-specific antibody (
Figure 7D). Our collective data suggested that the integrin(s) might participate in the biological effects of CLEC11A in endothelial cells, but conventional integrin signaling may not be important. It should be noted that the promoting effects of CLEC11A on osteoblast differentiation were observed in assays using human, not mouse, CLEC11A [
9,
10]. It would be interesting to know whether the RGD and LDT motifs are essential for all of the biological effects of CLEC11A.
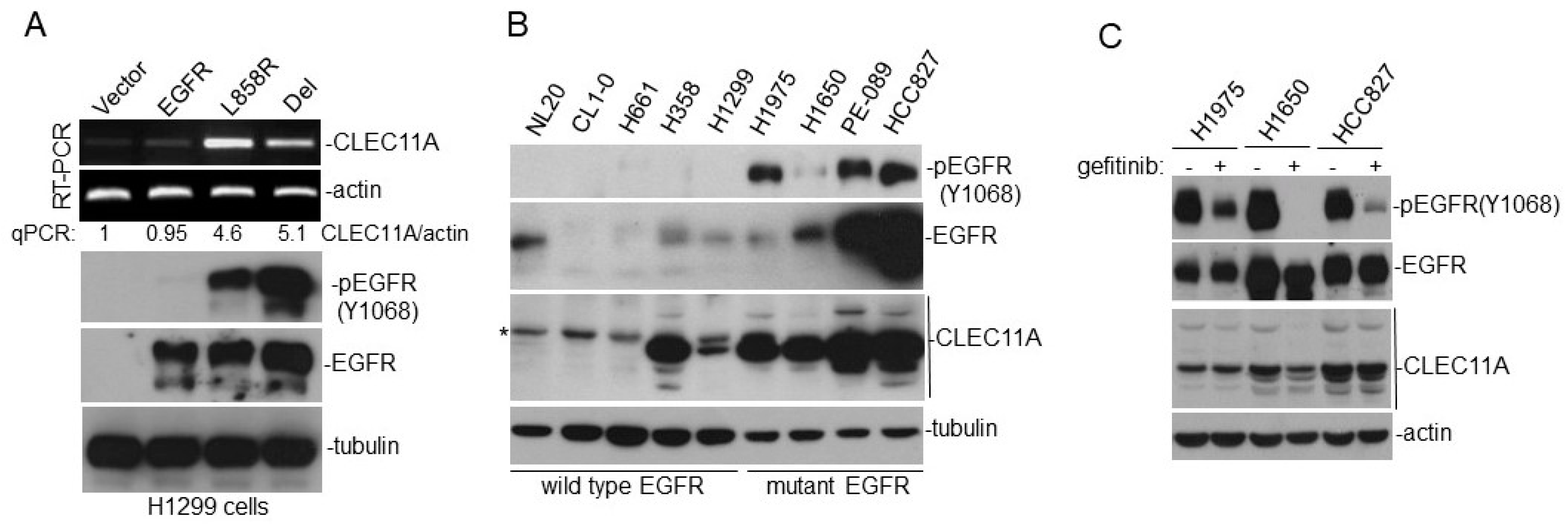
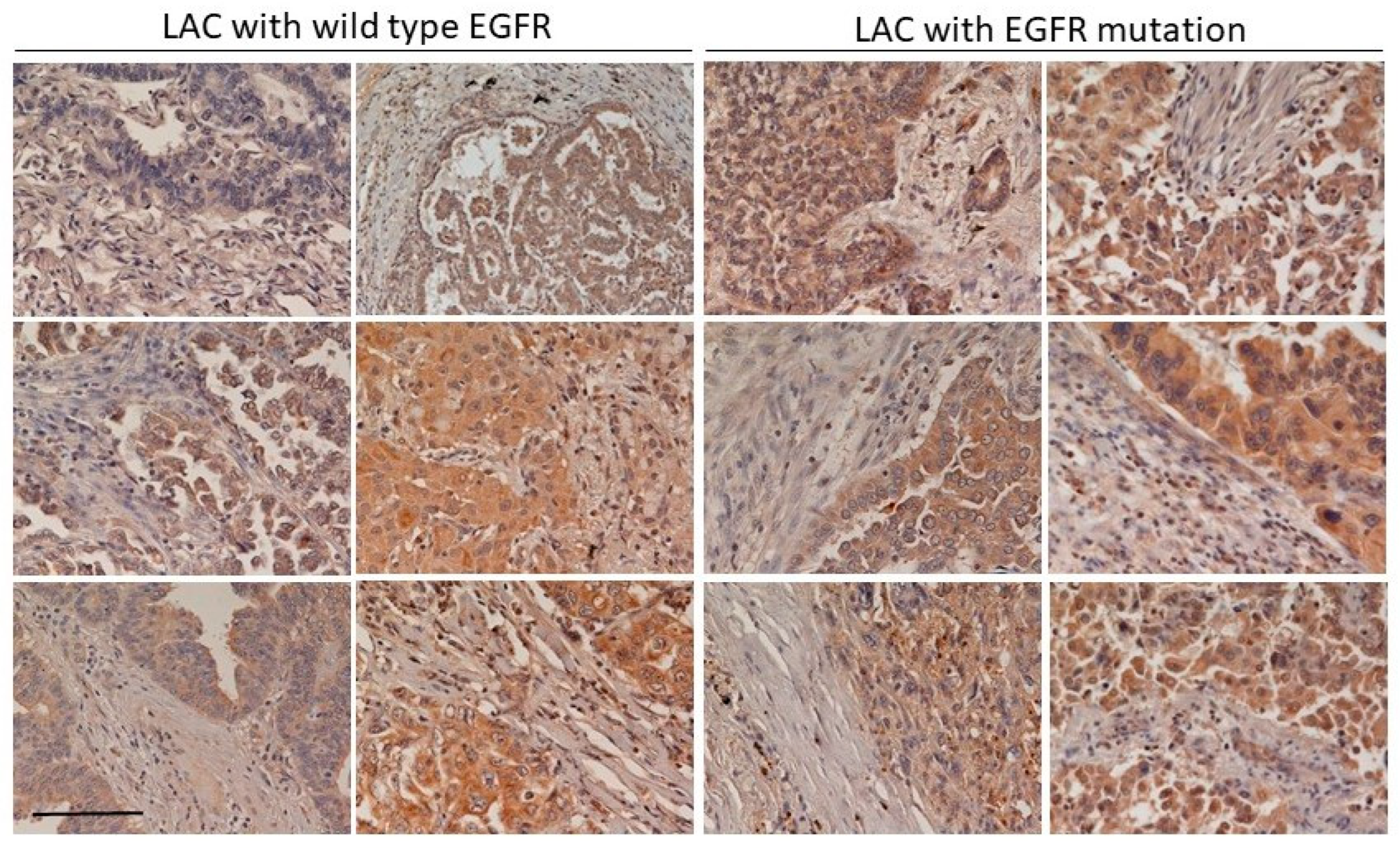
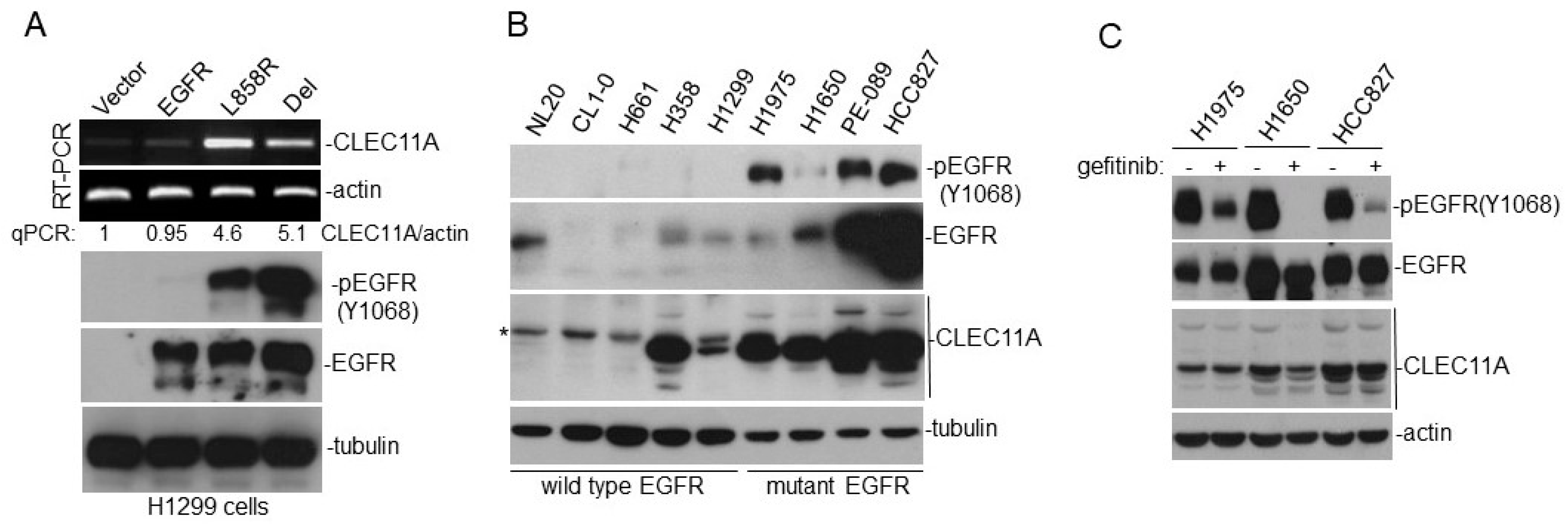
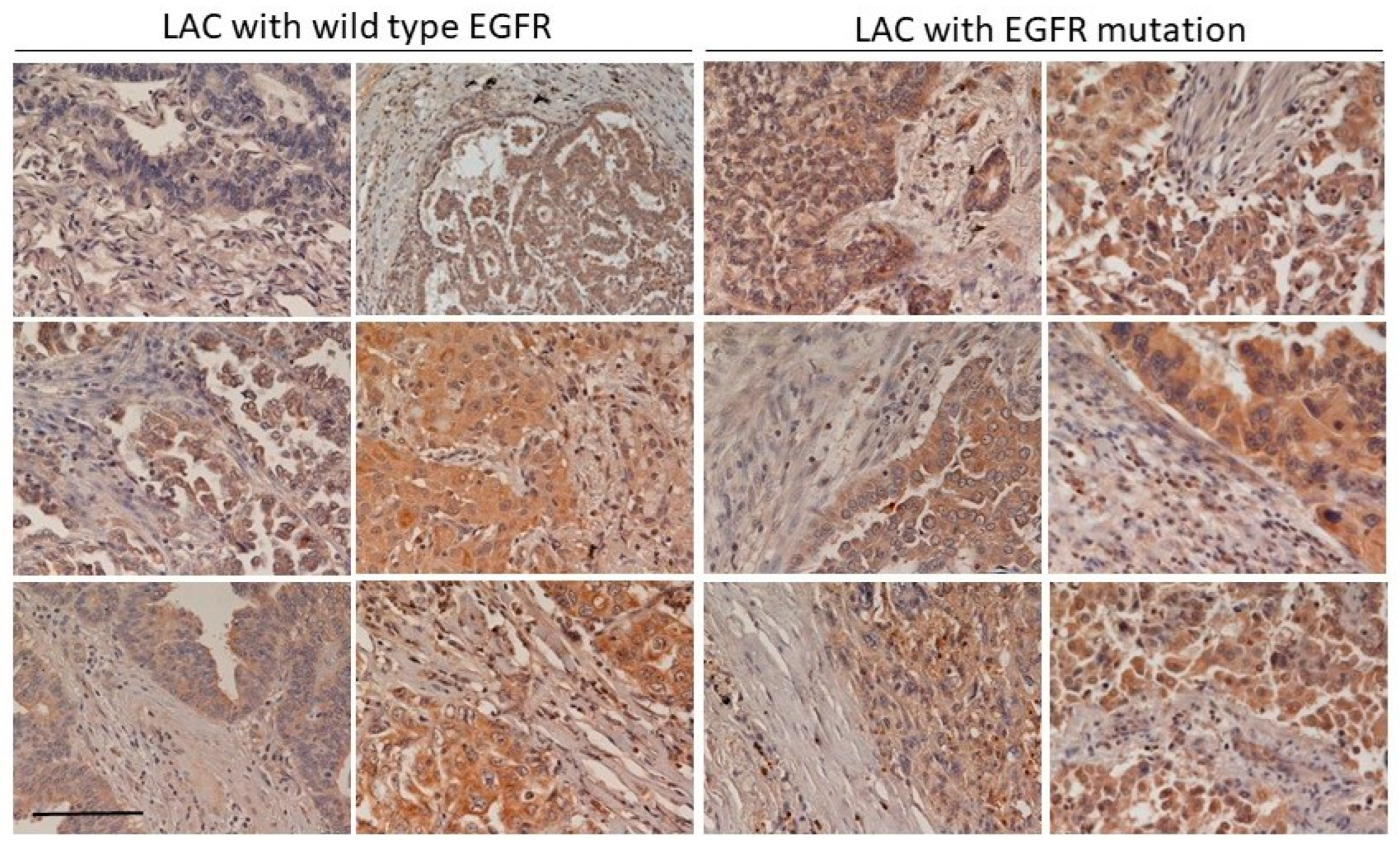
We found that the increase of CLEC11A in LAC was more common in LAC with
EGFR mutation (
Figure 1 and
Figure 2). Mutations of EGFR at its kinase domain, which frequently occur in LAC patients in east Asian populations, are associated with the clinical responsiveness to EGFR tyrosine kinase inhibitors (TKIs) [
20,
21]. Despite the initial effectiveness of TKIs in treating LAC with
EGFR mutation, drug resistance frequently develops within one year [
22]. New therapeutic approaches remain desirable for the better treatment of LAC patients [
23]. Interestingly, gefitinib failed to effectively abolish CLEC11A expression in LAC cells with
EGFR mutation, suggesting that CLEC11A was not induced by signaling immediately downstream of the mutated EGFR (
Figure 1C). The EGFR protein is capable of achieving some biological functions independent of its kinase activity [
24]. EGFR itself can also localize to nuclei and serve as a transcriptional factor [
25]. It will be interesting to know whether wild-type and mutated EGFR proteins are different in these aspects. Would it be possible for CLEC11A to be targeted for cancer therapeutic purposes? The initial studies on CLEC11A (previously named SCGF) implicated its role in the survival of leukemia cells [
5]. The neutralization antibody against CLEC11A induces cell death in multiple leukemia cell lines [
26]. However, later studies did not support the biological function of CLEC11A in regulating hematopoiesis [
9]. Several studies have reported the increase of CLEC11A in various malignant diseases, but the pathological effect of this increase remains unclear [
27,
28]. From the clinical information in the TCGA database, we found that a significant increase of CLEC11A also occurred in other types of carcinomas, such as head and neck cancer and breast cancer (
Supplementary Figure S1). It is possible that the increase of CLEC11A is a common feature in the progression of various cancers. Since CLEC11A-knockout mice did not show significant defects in the hematopoietic system, and the most significant phenotype observed in CLEC11A-null mice is the defect in bone formation in vertebrates and limb bones, the mice could survive without CLEC11A [
9]. It is likely that the loss of CLEC11A is bearable for a living organism, and that targeting the CLEC11A over-produced by cancers is a potential treatment strategy. The evidence provided in our study warrants further studies of the role of CLEC11A in tumor angiogenesis and the potential of targeting CLEC11A for cancer therapy.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}