
Circulating Cell-Free DNA Reflects the Clonal Evolution of Breast Cancer Tumors

 ,
,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients, Sample Material and Clinical Data
2.2. DNA Isolation
2.3. Library Preparation and Sequencing
2.4. Bioinformatics and Variant Calling
2.5. Tumor Evolution Modeling
2.6. Statistical Analysis
3. Results
3.1. Sequencing Performance
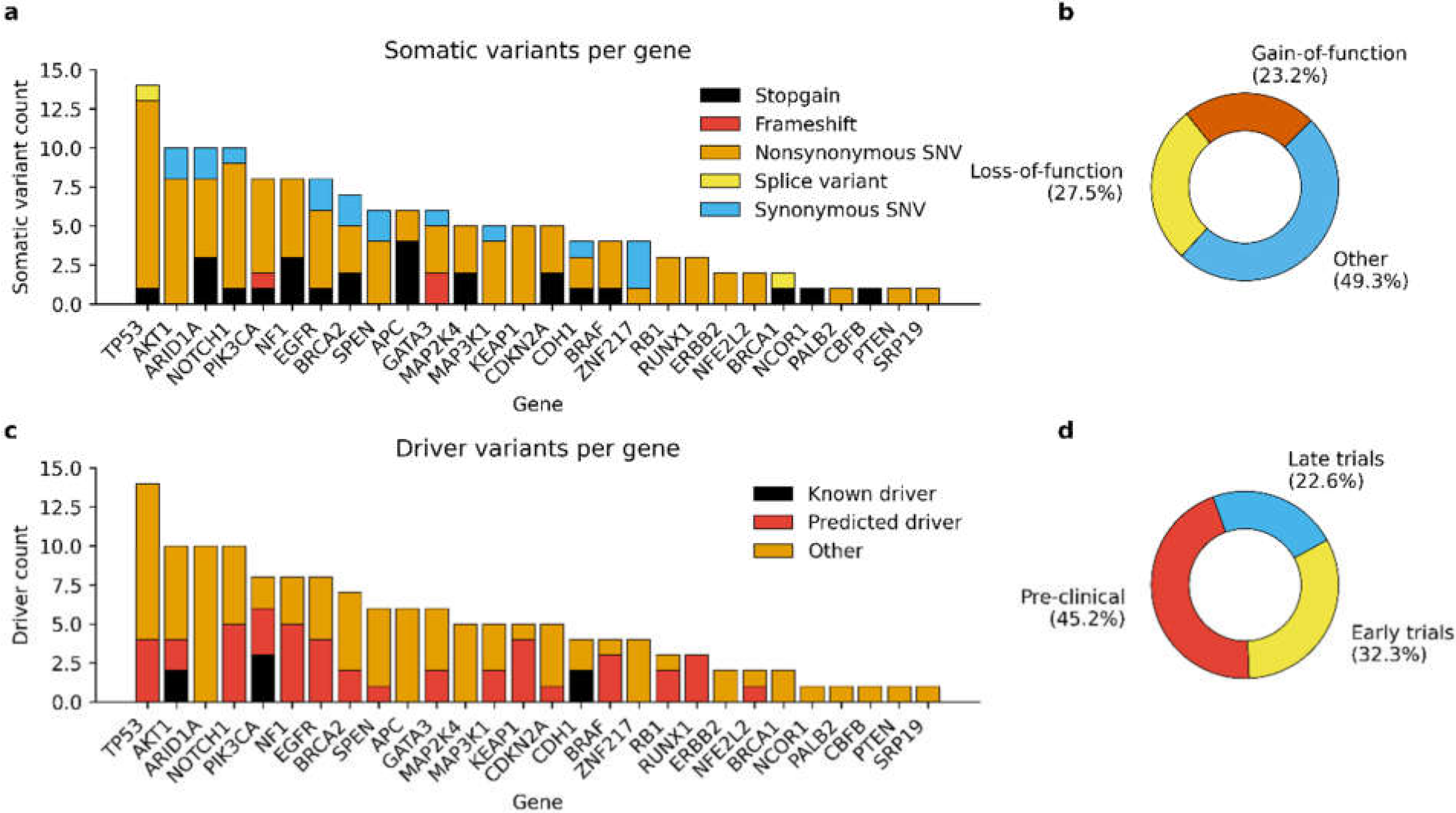
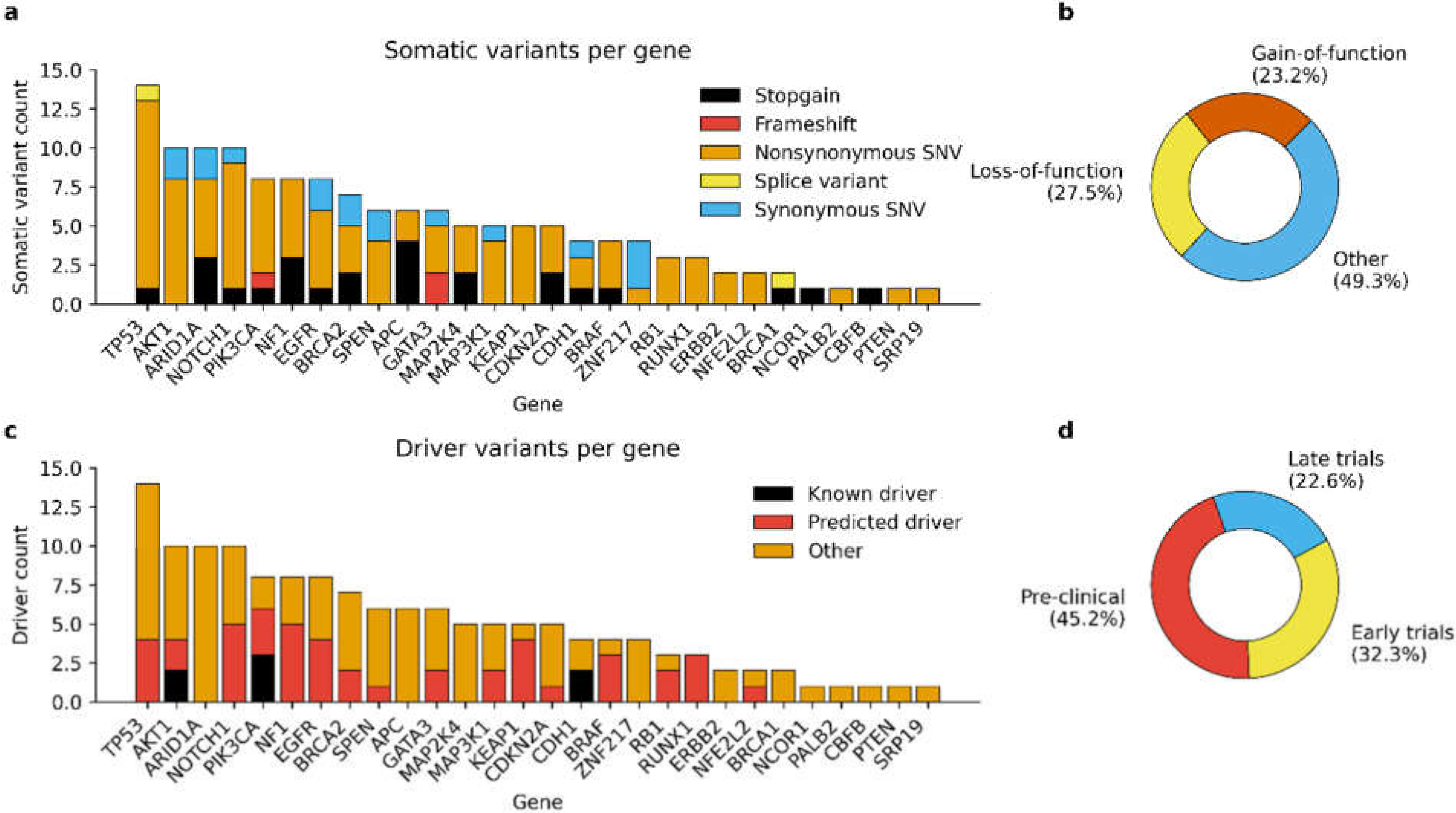
3.2. Detected Somatic Variants
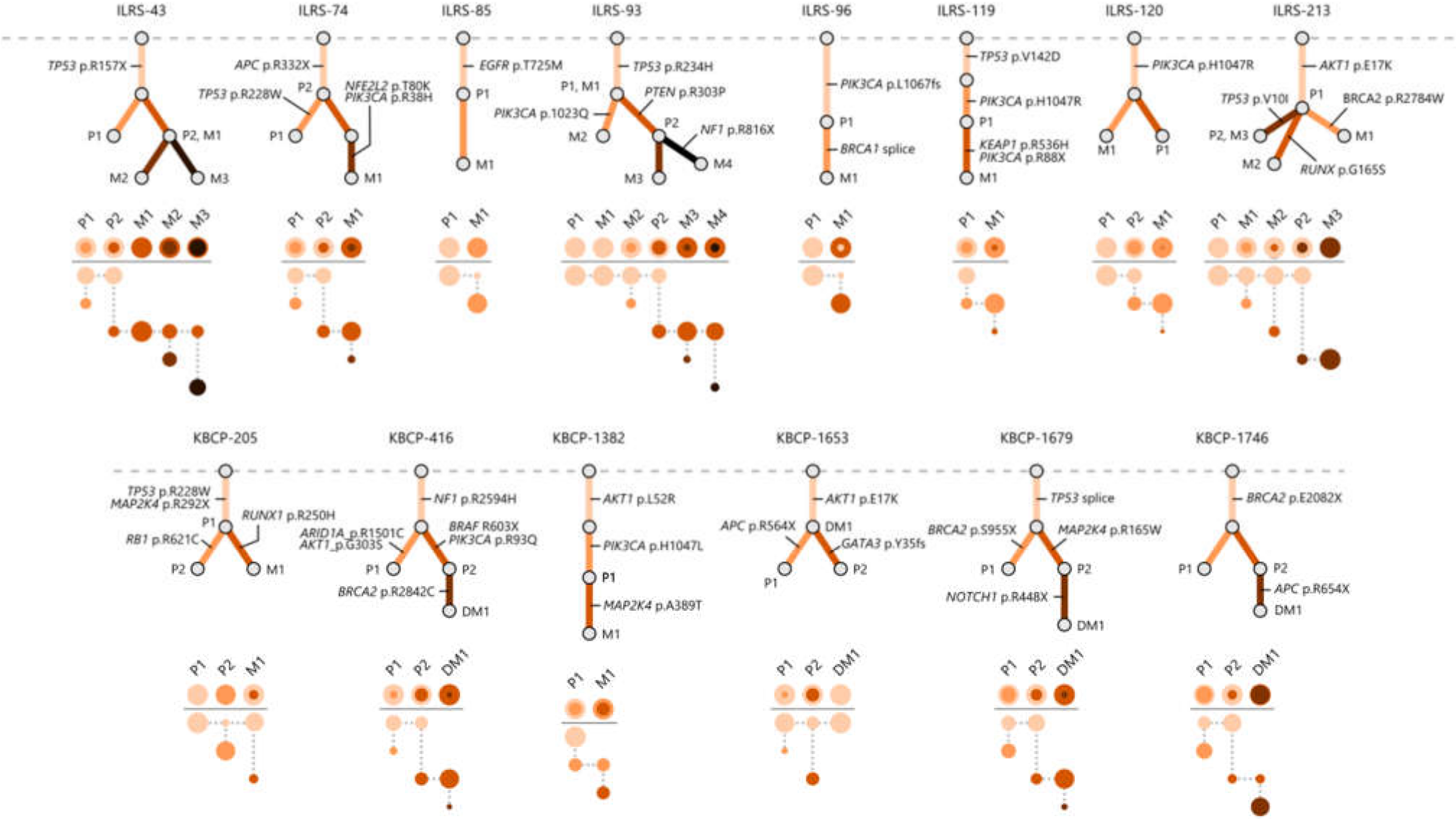
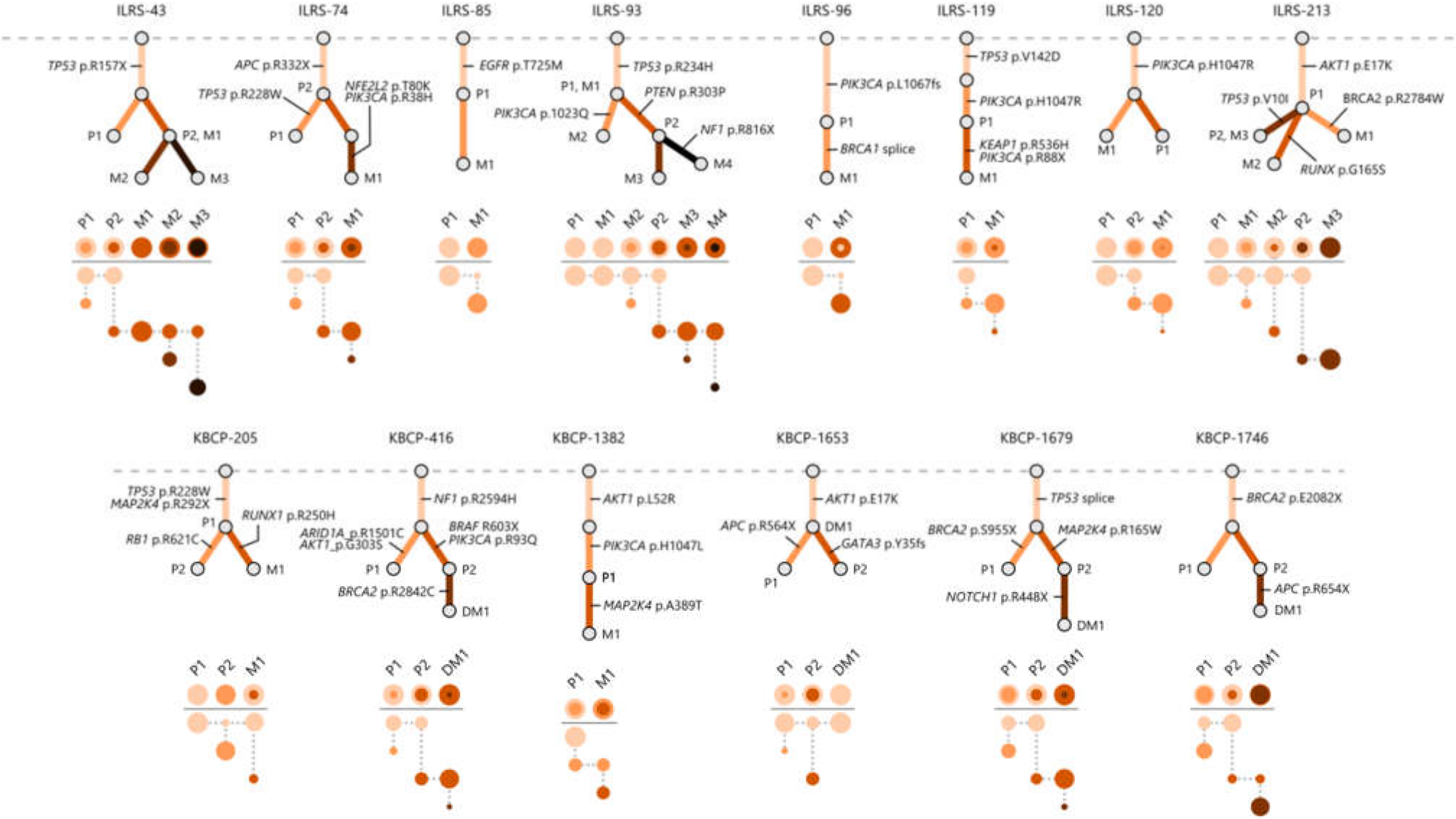
3.3. Multi-Region Sequencing Demonstrates the Clonal Evolution of Metastatic BC
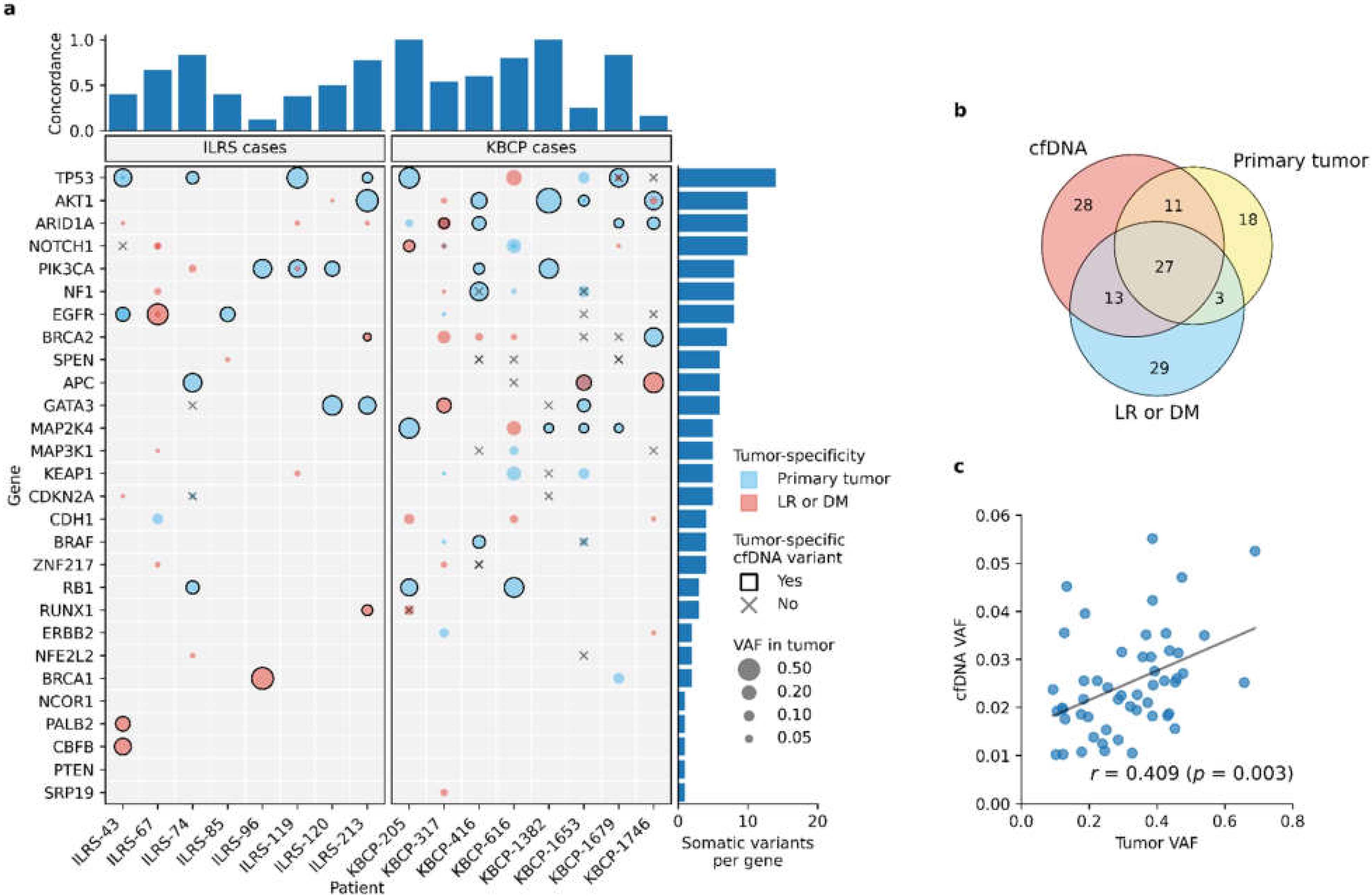
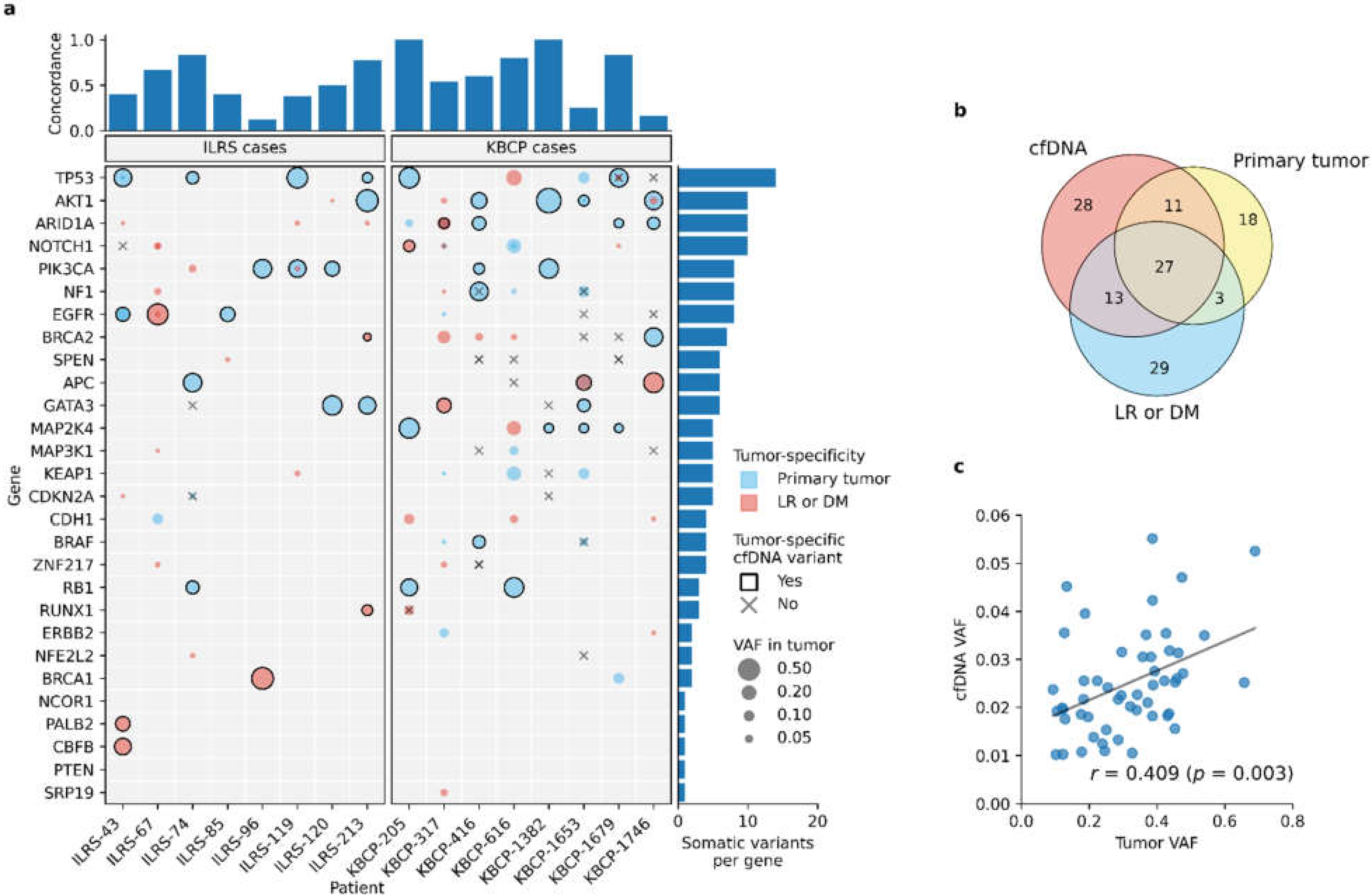
3.4. Liquid Biopsy Detects the Most Prominent Tumor-Specific Somatic Variants
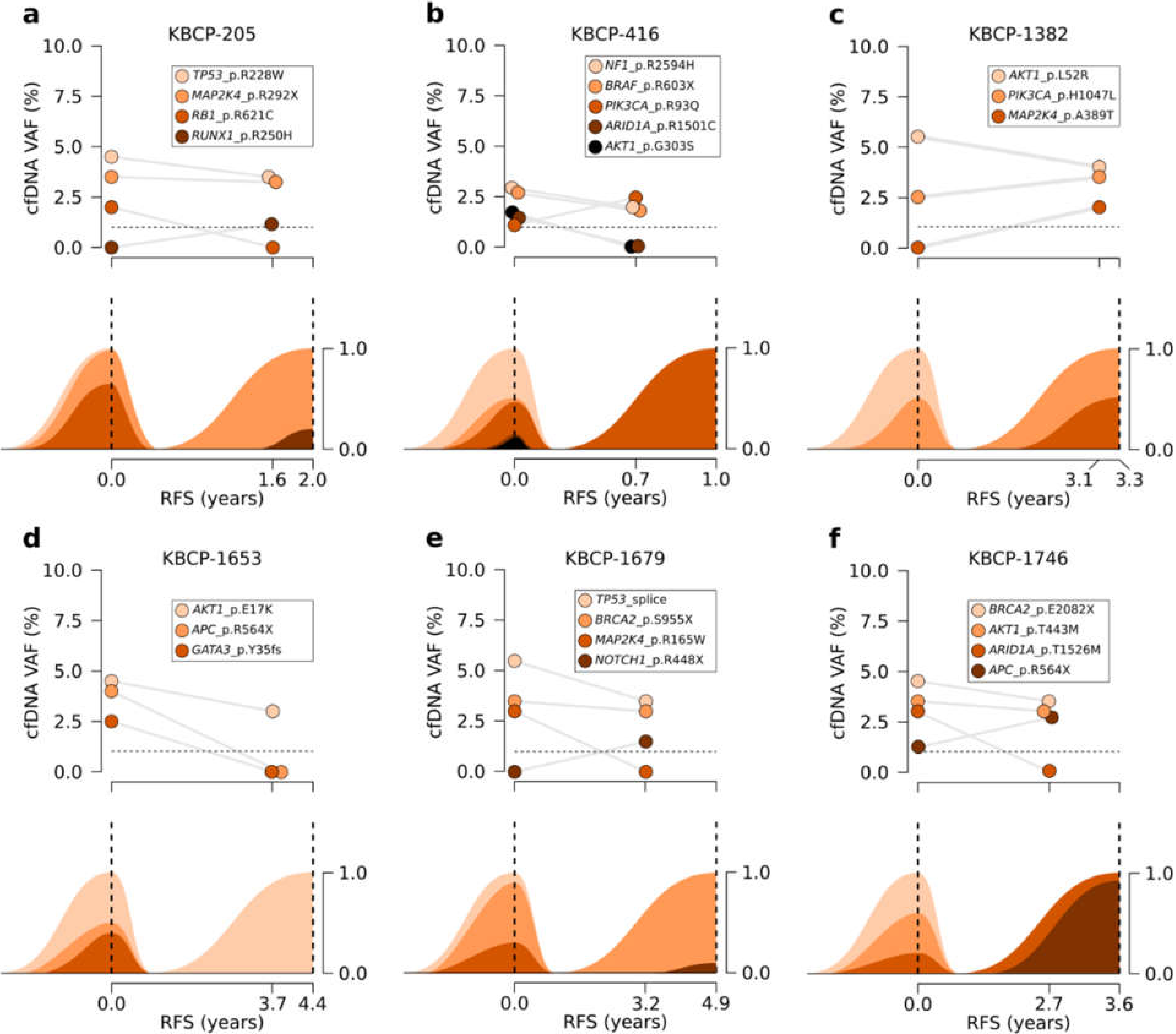
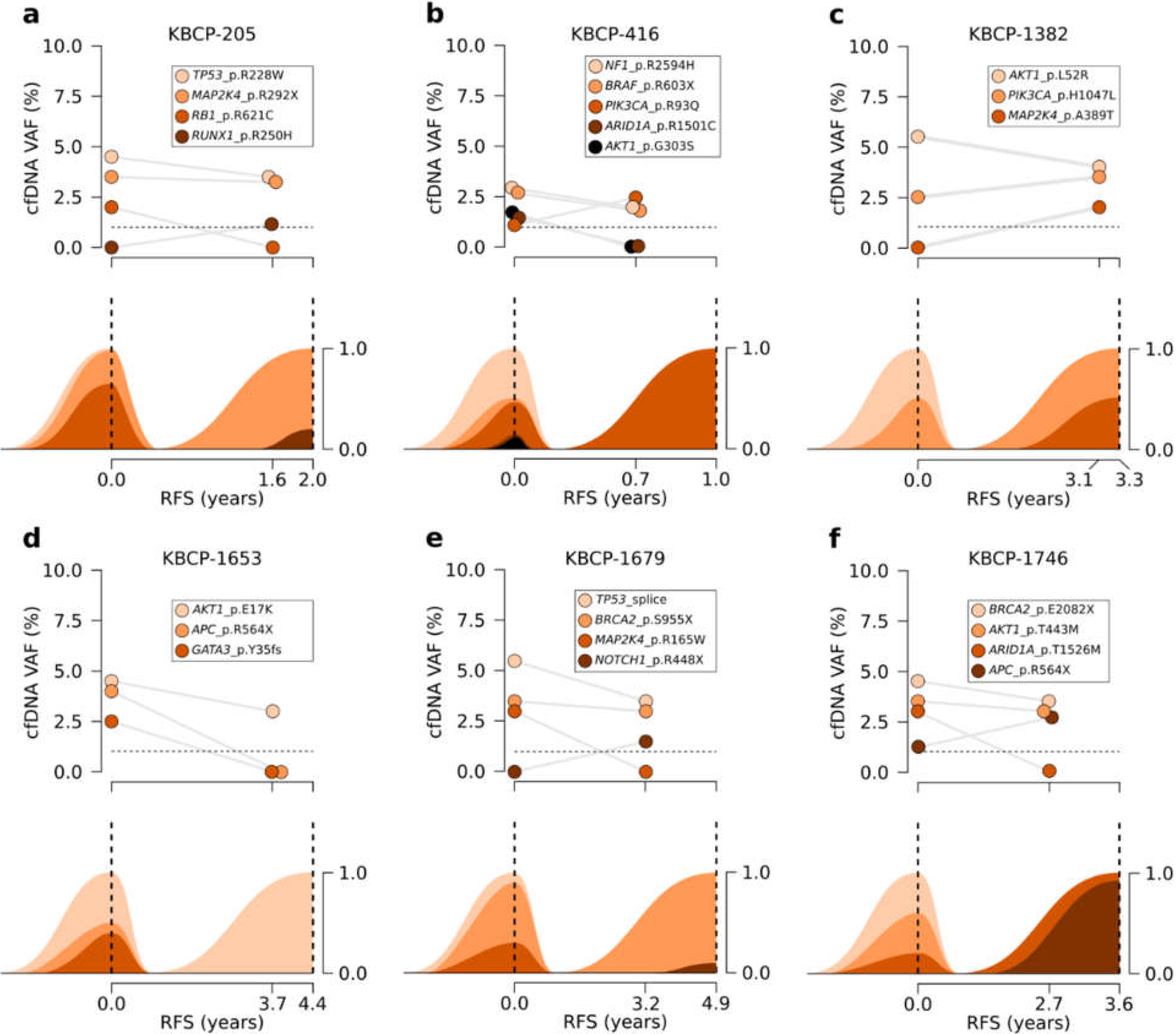
3.5. Serial Sequencing of cfDNA Reflects Changes in Clonal Evolution
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Van Maaren, M.C.; De Munck, L.; Strobbe, L.J.; Sonke, G.; Westenend, P.; Smidt, M.L.; Poortmans, P.M.; Siesling, S. Ten-year recurrence rates for breast cancer subtypes in the Netherlands: A large population-based study. Int. J. Cancer 2019, 144, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Geurts, Y.M.; Witteveen, A.; Bretveld, R.; Poortmans, P.M.; Sonke, G.; Strobbe, L.J.A. Patterns and predictors of first and subsequent recurrence in women with early breast cancer. Breast Cancer Res. Treat. 2017, 165, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Dowsett, M.; Turner, N. Estimating risk of recurrence for early breast cancer: Integrating clinical and genomic risk. J. Clin. Oncol. 2019, 37, 689–692. [Google Scholar] [CrossRef] [Green Version]
- Pondé, N.F.; Zardavas, D.; Piccart, M. Progress in adjuvant systemic therapy for breast cancer. Nat. Rev. Clin. Oncol. 2019, 16, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Turashvili, G.; Brogi, E. Tumor Heterogeneity in breast cancer. Front. Med. 2017, 4, 227. [Google Scholar] [CrossRef] [Green Version]
- Kalinowski, L.; Saunus, J.M.; McCart Reed, A.E.; Lakhani, S.R. Breast cancer heterogeneity in primary and metastatic disease. Adv. Exp. Med. Biol. 2019, 1152, 75–104. [Google Scholar] [CrossRef]
- Zardavas, D.; Irrthum, A.; Swanton, C.; Piccart, M. Clinical management of breast cancer heterogeneity. Nat. Rev. Clin. Oncol. 2015, 12, 381–394. [Google Scholar] [CrossRef]
- Grabuschnig, S.; Bronkhorst, A.J.; Holdenrieder, S.; Rodriguez, I.R.; Schliep, K.P.; Schwendenwein, D.; Ungerer, V.; Sensen, C.W. Putative origins of cell-free DNA in humans: A review of active and passive nucleic acid release mechanisms. Int. J. Mol. Sci. 2020, 21, 8062. [Google Scholar] [CrossRef]
- Yan, Y.-Y.; Guo, Q.-R.; Wang, F.-H.; Adhikari, R.; Zhu, Z.-Y.; Zhang, H.-Y.; Zhou, W.-M.; Yu, H.; Li, J.-Q.; Zhang, J.-Y. Cell-free DNA: Hope and potential application in cancer. Front. Cell Dev. Biol. 2021, 9, 639233. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Hancock, B.A.; Solzak, J.P.; Brinza, D.; Scafe, C.; Miller, K.D.; Radovich, M. Next-generation sequencing of circulating tumor DNA to predict recurrence in triple-negative breast cancer patients with residual disease after neoadjuvant chemotherapy. npj Breast Cancer 2017, 3, 24. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Murillas, I.; Schiavon, G.; Weigelt, B.; Ng, C.; Hrebien, S.; Cutts, R.J.; Cheang, M.; Osin, P.; Nerurkar, A.; Kozarewa, I.; et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci. Transl. Med. 2015, 7, 302. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, M.; Dawson, S.-J.; Tsui, D.W.Y.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.-F.; Kingsbury, Z.; Wong, A.S.C.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, M.; Baldassarri, M.; Fava, F.; Fabbiani, A.; Campennì, G.M.; Mencarelli, M.A.; Tita, R.; Marsili, S.; Renieri, A.; Frullanti, E. PIK3CA-CDKN2A clonal evolution in metastatic breast cancer and multiple points cell-free DNA analysis. Cancer Cell Int. 2019, 19, 274. [Google Scholar] [CrossRef]
- Weber, Z.T.; Collier, K.A.; Tallman, D.; Forman, J.; Shukla, S.; Asad, S.; Rhoades, J.; Freeman, S.; Parsons, H.A.; Williams, N.O.; et al. Modeling clonal structure over narrow time frames via circulating tumor DNA in metastatic breast cancer. Genome Med. 2021, 13, 89. [Google Scholar] [CrossRef]
- Welter, L.; Xu, L.; McKinley, D.; Dago, A.E.; Prabakar, R.K.; Restrepo-Vassalli, S.; Xu, K.; Rodriguez-Lee, M.; Kolatkar, A.; Nevarez, R.; et al. Treatment response and tumor evolution: Lessons from an extended series of multianalyte liquid biopsies in a metastatic breast cancer patient. Mol. Case Stud. 2020, 6, a005819. [Google Scholar] [CrossRef]
- Kauppinen, J.M.; Kosma, V.-M.; Soini, Y.; Sironen, R.; Nissinen, M.; Nykopp, T.K.; Kärjä, V.; Eskelinen, M.; Kataja, V.; Mannermaa, A. ST14 gene variant and decreased matriptase protein expression predict poor breast cancer survival. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2133–2142. [Google Scholar] [CrossRef] [Green Version]
- Pellikainen, M.J.; Pekola, T.T.; Ropponen, K.M.; Kataja, V.V.; Kellokoski, J.K.; Eskelinen, M.J.; Kosma, V.-M. p21WAF1 expression in invasive breast cancer and its association with p53, AP-2, cell proliferation, and prognosis. J. Clin. Pathol. 2003, 56, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Hartikainen, J.M.; Tuhkanen, H.; Kataja, V.; Dunning, A.M.; Antoniou, A.; Smith, P.; Arffman, A.; Pirskanen, M.; Easton, D.F.; Uusitupa, M.; et al. An autosome-wide scan for linkage disequilibrium-based association in sporadic breast cancer cases in eastern Finland: Three candidate regions found. Cancer Epidemiol. Biomark. Prev. 2005, 14, 75–80. [Google Scholar]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–111033. [Google Scholar] [CrossRef]
- Andrews, S. FastQC. 2012. Available online: http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc (accessed on 7 June 2019).
- Broad Institute. Picard Toolkit. 2019. Available online: https://broadinstitute.github.io/picard/ (accessed on 7 June 2019).
- Koboldt, D.C.; Larson, D.E.; Wilson, R.K. Using VarScan 2 for germline variant calling and somatic mutation detection. Curr. Protoc. Bioinform. 2013, 44, 5.4.1–15417. [Google Scholar] [CrossRef] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Tamborero, D.; Rubio-Perez, C.; Deu-Pons, J.; Schroeder, M.P.; Vivancos, A.; Rovira, A.; Tusquets, I.; Albanell, J.; Rodon, J.; Tabernero, J.; et al. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 2018, 10, 25. [Google Scholar] [CrossRef]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef]
- Deshwar, A.G.; Vembu, S.; Yung, C.K.; Jang, G.H.; Stein, L.; Morris, Q. PhyloWGS: Reconstructing subclonal composition and evolution from whole-genome sequencing of tumors. Genome Biol. 2015, 16, 35. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.; White, B.S.; Dees, N.D.; Griffith, M.; Welch, J.S.; Griffith, O.; Vij, R.; Tomasson, M.; Graubert, T.; Walter, M.J.; et al. SciClone: Inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution. PLoS Comput. Biol. 2014, 10, e1003665. [Google Scholar] [CrossRef]
- Swift, S.L.; Duffy, S.; Lang, S.H. Impact of tumor heterogeneity and tissue sampling for genetic mutation testing: A systematic review and post hoc analysis. J. Clin. Epidemiol. 2020, 126, 45–55. [Google Scholar] [CrossRef]
- Cheng, F.T.-F.; Lapke, N.; Wu, C.-C.; Lu, Y.-J.; Chen, S.-J.; Yu, P.-N.; Liu, Y.-T.; Tan, K.T. Liquid biopsy detects relapse five months earlier than regular clinical follow-up and guides targeted treatment in breast cancer. Case Rep. Oncol. Med. 2019, 2019, 6545298. [Google Scholar] [CrossRef] [PubMed]
- Olsson, E.; Winter, C.; George, A.; Chen, Y.; Howlin, J.; Tang, M.-H.E.; Dahlgren, M.; Schulz, R.; Grabau, D.; van Westen, D.; et al. Serial monitoring of circulating tumor DNA in patients with primary breast cancer for detection of occult metastatic disease. EMBO Mol. Med. 2015, 7, 1034–1047. [Google Scholar] [CrossRef]
- Henriksen, T.V.; Reinert, T.; Christensen, E.; Sethi, H.; Birkenkamp-Demtröder, K.; Gögenur, M.; Gögenur, I.; Zimmermann, B.G.; Dyrskjøt, L.; Andersen, C.L.; et al. The effect of surgical trauma on circulating free DNA levels in cancer patients—implications for studies of circulating tumor DNA. Mol. Oncol. 2020, 14, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Peled, M.; Agassi, R.; Czeiger, D.; Ariad, S.; Riff, R.; Rosenthal, M.; Lazarev, I.; Novack, V.; Yarza, S.; Mizrakli, Y.; et al. Cell-free DNA concentration in patients with clinical or mammographic suspicion of breast cancer. Sci. Rep. 2020, 10, 14601. [Google Scholar] [CrossRef] [PubMed]
- André, F.; Hurvitz, S.; Fasolo, A.; Tseng, L.-M.; Jerusalem, G.; Wilks, S.; O’Regan, R.; Isaacs, C.; Toi, M.; Burris, H.A.; et al. Molecular alterations and everolimus efficacy in human epidermal growth factor receptor 2–Overexpressing metastatic breast cancers: Combined exploratory biomarker analysis from BOLERO-1 and BOLERO-3. J. Clin. Oncol. 2016, 34, 2115–2124. [Google Scholar] [CrossRef]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef] [Green Version]
- Kilgour, E.; Rothwell, D.G.; Brady, G.; Dive, C. Liquid biopsy-based biomarkers of treatment response and resistance. Cancer Cell 2020, 37, 485–495. [Google Scholar] [CrossRef]
- Jahangiri, L.; Hurst, T. Assessing the concordance of genomic alterations between circulating-free DNA and tumour tissue in cancer patients. Cancers 2019, 11, 1938. [Google Scholar] [CrossRef] [Green Version]
- Merker, J.D.; Oxnard, G.R.; Compton, C.; Diehn, M.; Hurley, P.; Lazar, A.; Lindeman, N.; Lockwood, C.; Rai, A.J.; Schilsky, R.L.; et al. Circulating tumor DNA analysis in patients with cancer: American society of clinical oncology and college of American pathologists joint review. J. Clin. Oncol. 2018, 36, 1631–1641. [Google Scholar] [CrossRef]
- Bohers, E.; Viailly, P.-J.; Jardin, F. cfDNA Sequencing: Technological approaches and bioinformatic issues. Pharmaceuticals 2021, 14, 596. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Grouping | KBCP Cases | ILRS Cases | All Cases |
|---|---|---|---|---|
| Age at diagnosis | ≤39 years | 0 (0.0%) | 1 (11.1%) | 1 (5.6%) |
| 40–49 years | 1 (11.1%) | 1 (11.1%) | 2 (11.1%) | |
| 50–59 years | 6 (66.7%) | 0 (0.0%) | 6 (33.3%) | |
| 60–69 years | 2 (22.2%) | 4 (44.5%) | 6 (33.3%) | |
| ≥70 years | 0 (0.0%) | 3 (33.3%) | 3 (16.7%) | |
| ER status | Positive | 7 (77.8%) | 6 (66.7%) | 13 (72.2%) |
| Negative | 2 (22.2%) | 3 (33.3%) | 5 (27.8%) | |
| PR status | Positive | 7 (77.8%) | 7 (77.8%) | 14 (77.8%) |
| Negative | 2 (22.2%) | 2 (22.2%) | 4 (22.2%) | |
| HER2 status | Positive | 1 (11.1%) | 3 (33.3%) | 4 (22.2%) |
| Negative | 8 (88.9%) | 6 (66.7%) | 14 (77.8%) | |
| Tumor grade | I | 3 (33.3%) | 0 (0.0%) | 3 (16.7%) |
| II | 3 (33.3%) | 4 (44.4%) | 7 (38.9%) | |
| III | 3 (33.3%) | 5 (55.6%) | 8 (44.4%) | |
| Tumor size | T1 | 6 (66.7%) | 1 (11.1%) | 7 (38.9%) |
| T2 | 3 (33.3%) | 7 (77.8%) | 10 (55.5%) | |
| T3 | 0 (0.0%) | 1 (11.1%) | 1 (5.6%) | |
| Lymph node status | N0 | 9 (100.0%) | 0 (0.0%) | 9 (50.0%) |
| N1 | 0 (0.0%) | 6 (66.7%) | 6 (33.3%) | |
| N2 | 0 (0.0%) | 1 (11.1%) | 1 (5.6%) | |
| N3 | 0 (0.0%) | 2 (22.2%) | 2 (11.1%) | |
| Distant metastases | M0 | 9 (100.0%) | 0 (0.0%) | 9 (50.0%) |
| M1 | 0 (0.0%) | 9 (100.0%) | 9 (50.0%) | |
| Histological subtype | Ductal carcinoma | 8 (88.8%) | 9 (100.0%) | 17 (94.4%) |
| Tubular carcinoma | 1 (11.2%) | 0 (0.0%) | 1 (5.6%) | |
| Outcome | Locoregional recurrence | 2 (22.2%) | 1 (11.1%) | 3 (16.7%) |
| Distant metastasis | 7 (77.8%) | 3 (33.3%) | 10 (55.5%) | |
| Disease-free | 0 (0.0%) | 5 (55.6%) | 5 (27.8%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kujala, J.; Hartikainen, J.M.; Tengström, M.; Sironen, R.; Auvinen, P.; Kosma, V.-M.; Mannermaa, A. Circulating Cell-Free DNA Reflects the Clonal Evolution of Breast Cancer Tumors. Cancers 2022, 14, 1332. https://doi.org/10.3390/cancers14051332
Kujala J, Hartikainen JM, Tengström M, Sironen R, Auvinen P, Kosma V-M, Mannermaa A. Circulating Cell-Free DNA Reflects the Clonal Evolution of Breast Cancer Tumors. Cancers. 2022; 14(5):1332. https://doi.org/10.3390/cancers14051332
Chicago/Turabian StyleKujala, Jouni, Jaana M. Hartikainen, Maria Tengström, Reijo Sironen, Päivi Auvinen, Veli-Matti Kosma, and Arto Mannermaa. 2022. "Circulating Cell-Free DNA Reflects the Clonal Evolution of Breast Cancer Tumors" Cancers 14, no. 5: 1332. https://doi.org/10.3390/cancers14051332
APA StyleKujala, J., Hartikainen, J. M., Tengström, M., Sironen, R., Auvinen, P., Kosma, V.-M., & Mannermaa, A. (2022). Circulating Cell-Free DNA Reflects the Clonal Evolution of Breast Cancer Tumors. Cancers, 14(5), 1332. https://doi.org/10.3390/cancers14051332