
Functional Antagonism of Junctional Adhesion Molecule-A (JAM-A), Overexpressed in Breast Ductal Carcinoma In Situ (DCIS), Reduces HER2-Positive Tumor Progression

 ,
,  , , , , , , , , , and
, , , , , , , , , and  add
Show full author list
add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. JAM-A Immunohistochemistry
2.2. Design, Molecular Modelling, and Synthesis of JAM-A-Interacting Peptide JBS2
2.3. JBS2 Binding Assay
2.4. Cell Culture
2.5. Mammary Fat Pad (mfp) Murine Model of Breast Cancer
2.6. Murine Intraductal Model of DCIS
2.7. Reverse-Phase Protein Array (RPPA) Analysis
2.8. Multiplex Protein Array Profiling in Tissues
2.9. Chorioallantoic Membrane (CAM) Chick Embryo Xenograft Model
2.10. Statistical Analysis
3. Results
3.1. JAM-A Expression Is Elevated in DCIS
3.2. Designing a Peptide Antagonist to JAM-A
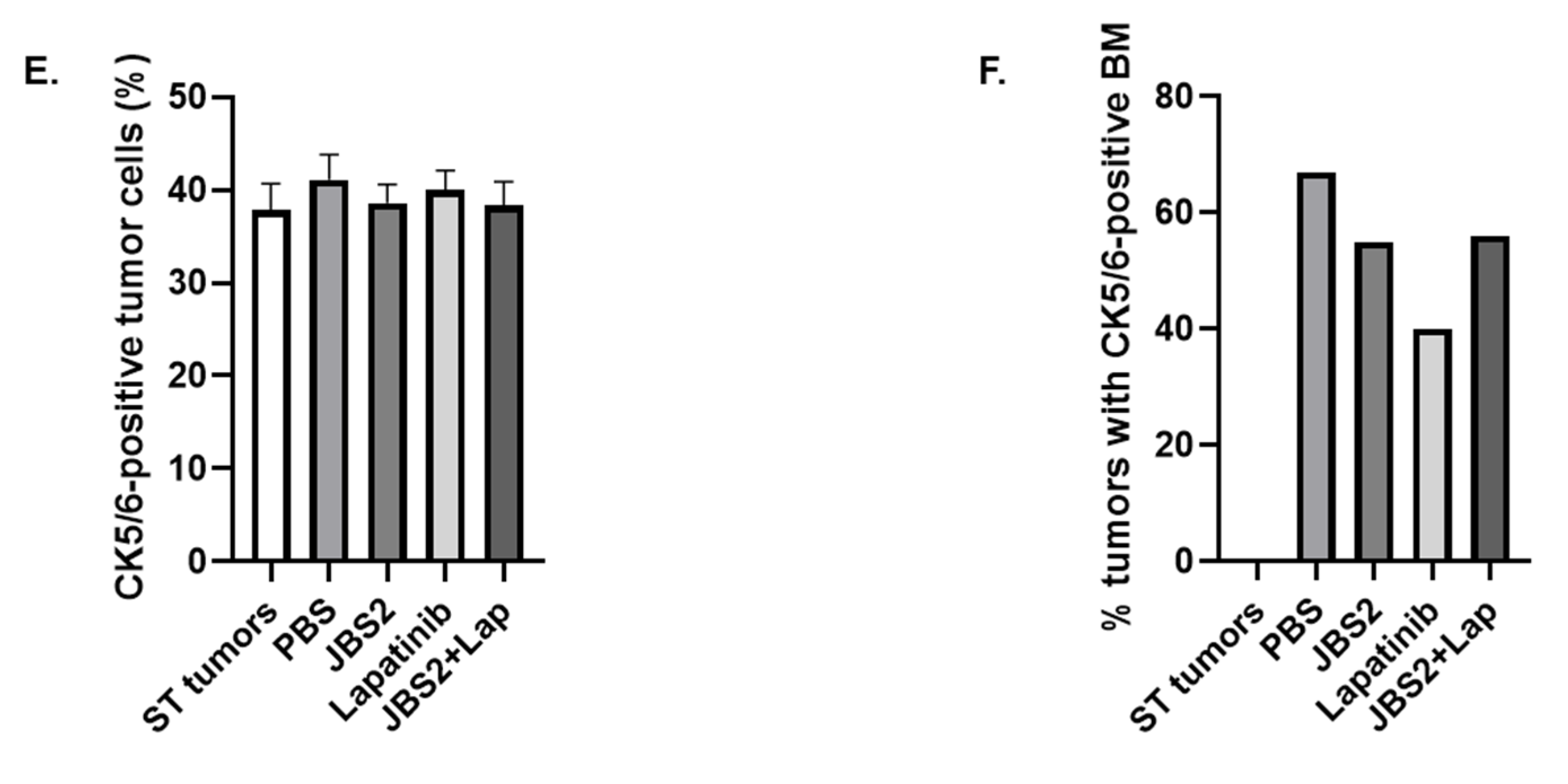
3.3. JAM-A Antagonism Reduces Tumor Growth In Vivo
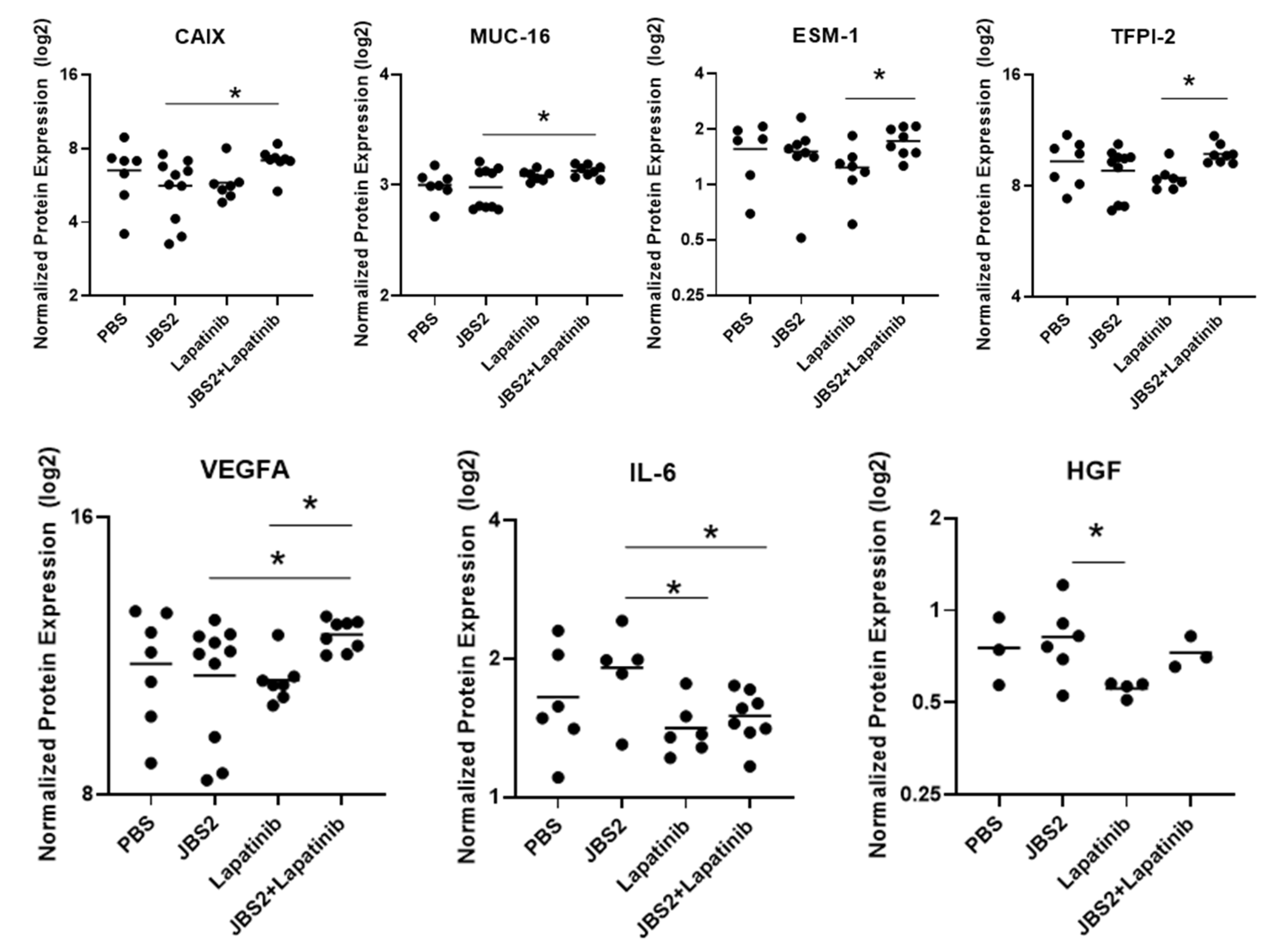
3.4. Proteomic Changes in JAM-A-Antagonized Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Doke, K.; Butler, S.; Mitchell, M.P. Current Therapeutic Approaches to DCIS. J. Mammary Gland. Biol. Neoplasia 2018, 23, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Burstein, H.J.; Polyak, K.; Wong, J.S.; Lester, S.C.; Kaelin, C.M. Ductal carcinoma in situ of the breast. N. Engl. J. Med. 2004, 350, 1430–1441. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.J.; Salunga, R.; Tuggle, J.T.; Gaudet, J.; Enright, E.; McQuary, P.; Payette, T.; Pistone, M.; Stecker, K.; Zhang, B.M.; et al. Gene expression profiles of human breast cancer progression. Proc. Natl. Acad. Sci. USA 2003, 100, 5974–5979. [Google Scholar] [CrossRef]
- Sanders, M.E.; Schuyler, P.A.; Simpson, J.F.; Page, D.L.; Dupont, W.D. Continued observation of the natural history of low-grade ductal carcinoma in situ reaffirms proclivity for local recurrence even after more than 30 years of follow-up. Mod. Pathol. 2015, 28, 662–669. [Google Scholar] [CrossRef]
- Solin, L.J.; Gray, R.; Baehner, F.L.; Butler, S.M.; Hughes, L.L.; Yoshizawa, C.; Cherbavaz, D.B.; Shak, S.; Page, D.L.; Sledge, G.W., Jr.; et al. A multigene expression assay to predict local recurrence risk for ductal carcinoma in situ of the breast. J. Natl. Cancer Inst. 2013, 105, 701–710. [Google Scholar] [CrossRef]
- Solassol, J.; Rouanet, P.; Lamy, P.J.; Allal, C.; Favre, G.; Maudelonde, T.; Mange, A. Serum protein signature may improve detection of ductal carcinoma in situ of the breast. Oncogene 2010, 29, 550–560. [Google Scholar] [CrossRef]
- Ebnet, K. Junctional Adhesion Molecules (JAMs): Cell Adhesion Receptors With Pleiotropic Functions in Cell Physiology and Development. Physiol. Rev. 2017, 97, 1529–1554. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.; McSherry, E.A.; Hudson, L.; Kay, E.W.; Hill, A.D.; Young, L.S.; Hopkins, A.M. Junctional adhesion molecule-A is co-expressed with HER2 in breast tumors and acts as a novel regulator of HER2 protein degradation and signaling. Oncogene 2013, 32, 2799–2804. [Google Scholar] [CrossRef]
- Goetsch, L.; Haeuw, J.F.; Beau-Larvor, C.; Gonzalez, A.; Zanna, L.; Malissard, M.; Lepecquet, A.M.; Robert, A.; Bailly, C.; Broussas, M.; et al. A novel role for junctional adhesion molecule-A in tumor proliferation: Modulation by an anti-JAM-A monoclonal antibody. Int. J. Cancer 2013, 132, 1463–1474. [Google Scholar] [CrossRef]
- Leech, A.O.; Vellanki, S.H.; Rutherford, E.J.; Keogh, A.; Jahns, H.; Hudson, L.; O’Donovan, N.; Sabri, S.; Abdulkarim, B.; Sheehan, K.M.; et al. Cleavage of the extracellular domain of junctional adhesion molecule-A is associated with resistance to anti-HER2 therapies in breast cancer settings. Breast Cancer Res. 2018, 20, 140. [Google Scholar] [CrossRef]
- McSherry, E.A.; McGee, S.F.; Jirstrom, K.; Doyle, E.M.; Brennan, D.J.; Landberg, G.; Dervan, P.A.; Hopkins, A.M.; Gallagher, W.M. JAM-A expression positively correlates with poor prognosis in breast cancer patients. Int. J. Cancer 2009, 125, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Giampietro, C.; Giannotta, M.; Corada, M.; Torselli, I.; Orsenigo, F.; Cocito, A.; d’Ario, G.; Mazzarol, G.; Confalonieri, S.; et al. Abrogation of junctional adhesion molecule-A expression induces cell apoptosis and reduces breast cancer progression. PLoS ONE 2011, 6, e21242. [Google Scholar] [CrossRef] [PubMed]
- Czubak-Prowizor, K.; Babinska, A.; Swiatkowska, M. The F11 Receptor (F11R)/Junctional Adhesion Molecule-A (JAM-A) (F11R/JAM-A) in cancer progression. Mol. Cell Biochem. 2021, 477, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.T.; Ding, J.N.; Wang, J.L.; Li, Z.S.; Ding, Y.L.; Ma, R. Differences in pathologic characteristics between ductal carcinoma in situ (DCIS), DCIS with microinvasion and DCIS with invasive ductal carcinoma. Int. J. Clin. Exp. Pathol. 2020, 13, 1066–1072. [Google Scholar] [PubMed]
- Wang, G.; Chen, C.; Pai, P.; Korangath, P.; Sun, S.; Merino, V.F.; Yuan, J.; Li, S.; Nie, G.; Stearns, V.; et al. Intraductal fulvestrant for therapy of ERalpha-positive ductal carcinoma in situ of the breast: A preclinical study. Carcinogenesis 2019, 40, 903–913. [Google Scholar] [CrossRef]
- Kostrewa, D.; Brockhaus, M.; D’Arcy, A.; Dale, G.E.; Nelboeck, P.; Schmid, G.; Mueller, F.; Bazzoni, G.; Dejana, E.; Bartfai, T.; et al. X-ray structure of junctional adhesion molecule: Structural basis for homophilic adhesion via a novel dimerization motif. EMBO J. 2001, 20, 4391–4398. [Google Scholar] [CrossRef]
- Prota, A.E.; Campbell, J.A.; Schelling, P.; Forrest, J.C.; Watson, M.J.; Peters, T.R.; Aurrand-Lions, M.; Imhof, B.A.; Dermody, T.S.; Stehle, T. Crystal structure of human junctional adhesion molecule 1: Implications for reovirus binding. Proc. Natl. Acad. Sci. USA 2003, 100, 5366–5371. [Google Scholar] [CrossRef]
- Monteiro, A.C.; Luissint, A.-C.; Sumagin, R.; Lai, C.; Vielmuth, F.; Wolf, M.F.; Laur, O.; Reiss, K.; Spindler, V.; Stehle, T.; et al. Trans-dimerization of JAM-A regulates Rap2 and is mediated by a domain that is distinct from the cis-dimerization interface. Mol. Biol. Cell 2014, 25, 1574–1585. [Google Scholar] [CrossRef]
- Yoshida, T.; Jin, K.; Song, H.; Park, S.; Huso, D.L.; Zhang, Z.; Liangfeng, H.; Zhu, C.; Bruchertseifer, F.; Morgenstern, A.; et al. Effective treatment of ductal carcinoma in situ with a HER-2- targeted alpha-particle emitting radionuclide in a preclinical model of human breast cancer. Oncotarget 2016, 7, 33306–33315. [Google Scholar] [CrossRef]
- Donatello, S.; Hudson, L.; Cottell, D.C.; Blanco, A.; Aurrekoetxea, I.; Shelly, M.J.; Dervan, P.A.; Kell, M.R.; Stokes, M.; Hill, A.D.; et al. An imbalance in progenitor cell populations reflects tumour progression in breast cancer primary culture models. J. Exp. Clin. Cancer Res. 2011, 30, 45. [Google Scholar] [CrossRef]
- Aslan, O.; Cremona, M.; Morgan, C.; Cheung, L.W.; Mills, G.B.; Hennessy, B.T. Preclinical evaluation and reverse phase protein Array-based profiling of PI3K and MEK inhibitors in endometrial carcinoma in vitro. BMC Cancer 2018, 18, 168. [Google Scholar] [CrossRef]
- Guo, H.; Liu, W.; Ju, Z.; Tamboli, P.; Jonasch, E.; Mills, G.B.; Lu, Y.; Hennessy, B.T.; Tsavachidou, D. An efficient procedure for protein extraction from formalin-fixed, paraffin-embedded tissues for reverse phase protein arrays. Proteome Sci. 2012, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Assarsson, E.; Lundberg, M.; Holmquist, G.; Bjorkesten, J.; Thorsen, S.B.; Ekman, D.; Eriksson, A.; Rennel Dickens, E.; Ohlsson, S.; Edfeldt, G.; et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS ONE 2014, 9, e95192. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Severson, E.A.; Jiang, L.; Ivanov, A.I.; Mandell, K.J.; Nusrat, A.; Parkos, C.A. Cis-dimerization Mediates Function of Junctional Adhesion Molecule A. Mol. Biol. Cell 2008, 19, 1862–1872. [Google Scholar] [CrossRef] [PubMed]
- Vellanki, S.H.; Cruz, R.G.B.; Jahns, H.; Hudson, L.; Sette, G.; Eramo, A.; Hopkins, A.M. Natural compound Tetrocarcin-A downregulates Junctional Adhesion Molecule-A in conjunction with HER2 and inhibitor of apoptosis proteins and inhibits tumor cell growth. Cancer Lett. 2019, 440–441, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Naik, M.U.; Naik, U.P. Junctional adhesion molecule-A-induced endothelial cell migration on vitronectin is integrin alpha v beta 3 specific. J. Cell Sci. 2006, 119 Pt 3, 490–499. [Google Scholar] [CrossRef]
- Akimoto, T.; Takasawa, A.; Murata, M.; Kojima, Y.; Takasawa, K.; Nojima, M.; Aoyama, T.; Hiratsuka, Y.; Ono, Y.; Tanaka, S.; et al. Analysis of the expression and localization of tight junction transmembrane proteins, claudin-1, -4, -7, occludin and JAM-A, in human cervical adenocarcinoma. Histol. Histopathol. 2016, 31, 921–931. [Google Scholar] [CrossRef]
- Magara, K.; Takasawa, A.; Osanai, M.; Ota, M.; Tagami, Y.; Ono, Y.; Takasawa, K.; Murata, M.; Hirohashi, Y.; Miyajima, M.; et al. Elevated expression of JAM-A promotes neoplastic properties of lung adenocarcinoma. Cancer Sci. 2017, 108, 2306–2314. [Google Scholar] [CrossRef]
- Latta, E.K.; Tjan, S.; Parkes, R.K.; O’Malley, F.P. The role of HER2/neu overexpression/amplification in the progression of ductal carcinoma in situ to invasive carcinoma of the breast. Mod. Pathol. 2002, 15, 1318–1325. [Google Scholar] [CrossRef]
- Gupta, P.B.; Kuperwasser, C. Disease models of breast cancer. Drug Discov. Today Dis. Models 2004, 1, 9–16. [Google Scholar] [CrossRef]
- Zhang, X.; Cai, J.; Zheng, Z.; Polin, L.; Lin, Z.; Dandekar, A.; Li, L.; Sun, F.; Finley, R.L., Jr.; Fang, D.; et al. A novel ER-microtubule-binding protein, ERLIN2, stabilizes Cyclin B1 and regulates cell cycle progression. Cell Discov. 2015, 1, 15024. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.E.; Barnes, N.L.P.; Cramer, A.; Johnson, R.; Cheema, K.; Morris, J.; Howe, M.; Bundred, N.J. Molecular phenotypes of DCIS predict overall and invasive recurrence. Ann. Oncol. 2015, 26, 1019–1025. [Google Scholar] [CrossRef]
- Tsukabe, M.; Shimazu, K.; Morimoto, K.; Naoi, Y.; Kagara, N.; Shimoda, M.; Shimomura, A.; Maruyama, N.; Kim, S.J.; Noguchi, S. Clinicopathological analysis of breast ductal carcinoma in situ with ALDH1-positive cancer stem cells. Oncology 2013, 85, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Bednarek, R.; Selmi, A.; Wojkowska, D.; Karolczak, K.; Popielarski, M.; Stasiak, M.; Salifu, M.O.; Babinska, A.; Swiatkowska, M. Functional inhibition of F11 receptor (F11R/junctional adhesion molecule-A/JAM-A) activity by a F11R-derived peptide in breast cancer and its microenvironment. Breast Cancer Res. Treat. 2020, 179, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Behbod, F.; Kittrell, F.S.; LaMarca, H.; Edwards, D.; Kerbawy, S.; Heestand, J.C.; Young, E.; Mukhopadhyay, P.; Yeh, H.W.; Allred, D.C.; et al. An intraductal human-in-mouse transplantation model mimics the subtypes of ductal carcinoma in situ. Breast Cancer Res. 2009, 11, R66. [Google Scholar] [CrossRef]
- Sflomos, G.; Dormoy, V.; Metsalu, T.; Jeitziner, R.; Battista, L.; Scabia, V.; Raffoul, W.; Delaloye, J.F.; Treboux, A.; Fiche, M.; et al. A Preclinical Model for ERalpha-Positive Breast Cancer Points to the Epithelial Microenvironment as Determinant of Luminal Phenotype and Hormone Response. Cancer Cell 2016, 29, 407–422. [Google Scholar] [CrossRef]
- Stearns, V.; Mori, T.; Jacobs, L.K.; Khouri, N.F.; Gabrielson, E.; Yoshida, T.; Kominsky, S.L.; Huso, D.L.; Jeter, S.; Powers, P.; et al. Preclinical and clinical evaluation of intraductally administered agents in early breast cancer. Sci. Transl. Med. 2011, 3, 106ra108. [Google Scholar] [CrossRef][Green Version]
- Casamayor, A.; Morrice, N.A.; Alessi, D.R. Phosphorylation of Ser-241 is essential for the activity of 3-phosphoinositide-dependent protein kinase-1: Identification of five sites of phosphorylation in vivo. Biochem. J. 1999, 342 Pt 2, 287–292. [Google Scholar] [CrossRef]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef]
- Xia, W.; Petricoin, E.F., 3rd; Zhao, S.; Liu, L.; Osada, T.; Cheng, Q.; Wulfkuhle, J.D.; Gwin, W.R.; Yang, X.; Gallagher, R.I.; et al. An heregulin-EGFR-HER3 autocrine signaling axis can mediate acquired lapatinib resistance in HER2+ breast cancer models. Breast Cancer Res. 2013, 15, R85. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Mayer, J.A.; Krisko, T.I.; Speers, C.W.; Wang, T.; Hilsenbeck, S.G.; Brown, P.H. Inhibition of the p38 kinase suppresses the proliferation of human ER-negative breast cancer cells. Cancer Res. 2009, 69, 8853–8861. [Google Scholar] [CrossRef] [PubMed]
- Werden, S.J.; Sphyris, N.; Sarkar, T.R.; Paranjape, A.N.; LaBaff, A.M.; Taube, J.H.; Hollier, B.G.; Ramirez-Pena, E.Q.; Soundararajan, R.; den Hollander, P.; et al. Phosphorylation of serine 367 of FOXC2 by p38 regulates ZEB1 and breast cancer metastasis, without impacting primary tumor growth. Oncogene 2016, 35, 5977–5988. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Gao, S.; Xie, X.; Sun, E.; Zhang, M.; Zhou, Q.; Lu, C. SPARC inhibits breast cancer bone metastasis and may be a clinical therapeutic target. Oncol. Lett. 2017, 14, 5876–5882. [Google Scholar] [CrossRef]
- Martino-Echarri, E.; Fernandez-Rodriguez, R.; Rodriguez-Baena, F.J.; Barrientos-Duran, A.; Torres-Collado, A.X.; Plaza-Calonge Mdel, C.; Amador-Cubero, S.; Cortes, J.; Reynolds, L.E.; Hodivala-Dilke, K.M.; et al. Contribution of ADAMTS1 as a tumor suppressor gene in human breast carcinoma. Linking its tumor inhibitory properties to its proteolytic activity on nidogen-1 and nidogen-2. Int. J. Cancer 2013, 133, 2315–2324. [Google Scholar] [CrossRef]
- Ricciardelli, C.; Frewin, K.M.; Tan Ide, A.; Williams, E.D.; Opeskin, K.; Pritchard, M.A.; Ingman, W.V.; Russell, D.L. The ADAMTS1 protease gene is required for mammary tumor growth and metastasis. Am. J. Pathol. 2011, 179, 3075–3085. [Google Scholar] [CrossRef]
- Ehrenreiter, K.; Piazzolla, D.; Velamoor, V.; Sobczak, I.; Small, J.V.; Takeda, J.; Leung, T.; Baccarini, M. Raf-1 regulates Rho signaling and cell migration. J. Cell. Biol. 2005, 168, 955–964. [Google Scholar] [CrossRef]
- McSherry, E.A.; Brennan, K.; Hudson, L.; Hill, A.D.; Hopkins, A.M. Breast cancer cell migration is regulated through junctional adhesion molecule-A-mediated activation of Rap1 GTPase. Breast Cancer Res. 2011, 13, R31. [Google Scholar] [CrossRef]
- Wulfing, P.; Kersting, C.; Buerger, H.; Mattsson, B.; Mesters, R.; Gustmann, C.; Hinrichs, B.; Tio, J.; Bocker, W.; Kiesel, L. Expression patterns of angiogenic and lymphangiogenic factors in ductal breast carcinoma in situ. Br. J. Cancer 2005, 92, 1720–1728. [Google Scholar] [CrossRef]
- Vermeulen, M.A.; van Deurzen, C.H.; Schroder, C.P.; Martens, J.W.; van Diest, P.J. Expression of hypoxia-induced proteins in ductal carcinoma in situ and invasive cancer of the male breast. J. Clin. Pathol. 2020, 73, 204–208. [Google Scholar] [CrossRef]
- Lakshmanan, I.; Ponnusamy, M.P.; Das, S.; Chakraborty, S.; Haridas, D.; Mukhopadhyay, P.; Lele, S.M.; Batra, S.K. MUC16 induced rapid G2/M transition via interactions with JAK2 for increased proliferation and anti-apoptosis in breast cancer cells. Oncogene 2012, 31, 805–817. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, Y.E.; Wang, G.; Flynn, C.L.; Madden, S.F.; MacEneaney, O.; Cruz, R.G.B.; Richards, C.E.; Jahns, H.; Brennan, M.; Cremona, M.; et al. Functional Antagonism of Junctional Adhesion Molecule-A (JAM-A), Overexpressed in Breast Ductal Carcinoma In Situ (DCIS), Reduces HER2-Positive Tumor Progression. Cancers 2022, 14, 1303. https://doi.org/10.3390/cancers14051303
Smith YE, Wang G, Flynn CL, Madden SF, MacEneaney O, Cruz RGB, Richards CE, Jahns H, Brennan M, Cremona M, et al. Functional Antagonism of Junctional Adhesion Molecule-A (JAM-A), Overexpressed in Breast Ductal Carcinoma In Situ (DCIS), Reduces HER2-Positive Tumor Progression. Cancers. 2022; 14(5):1303. https://doi.org/10.3390/cancers14051303
Chicago/Turabian StyleSmith, Yvonne E., Guannan Wang, Ciara L. Flynn, Stephen F. Madden, Owen MacEneaney, Rodrigo G. B. Cruz, Cathy E. Richards, Hanne Jahns, Marian Brennan, Mattia Cremona, and et al. 2022. "Functional Antagonism of Junctional Adhesion Molecule-A (JAM-A), Overexpressed in Breast Ductal Carcinoma In Situ (DCIS), Reduces HER2-Positive Tumor Progression" Cancers 14, no. 5: 1303. https://doi.org/10.3390/cancers14051303
APA StyleSmith, Y. E., Wang, G., Flynn, C. L., Madden, S. F., MacEneaney, O., Cruz, R. G. B., Richards, C. E., Jahns, H., Brennan, M., Cremona, M., Hennessy, B. T., Sheehan, K., Casucci, A., Sani, F. A., Hudson, L., Fay, J., Vellanki, S. H., O’Flaherty, S., Devocelle, M., ... Hopkins, A. M. (2022). Functional Antagonism of Junctional Adhesion Molecule-A (JAM-A), Overexpressed in Breast Ductal Carcinoma In Situ (DCIS), Reduces HER2-Positive Tumor Progression. Cancers, 14(5), 1303. https://doi.org/10.3390/cancers14051303