
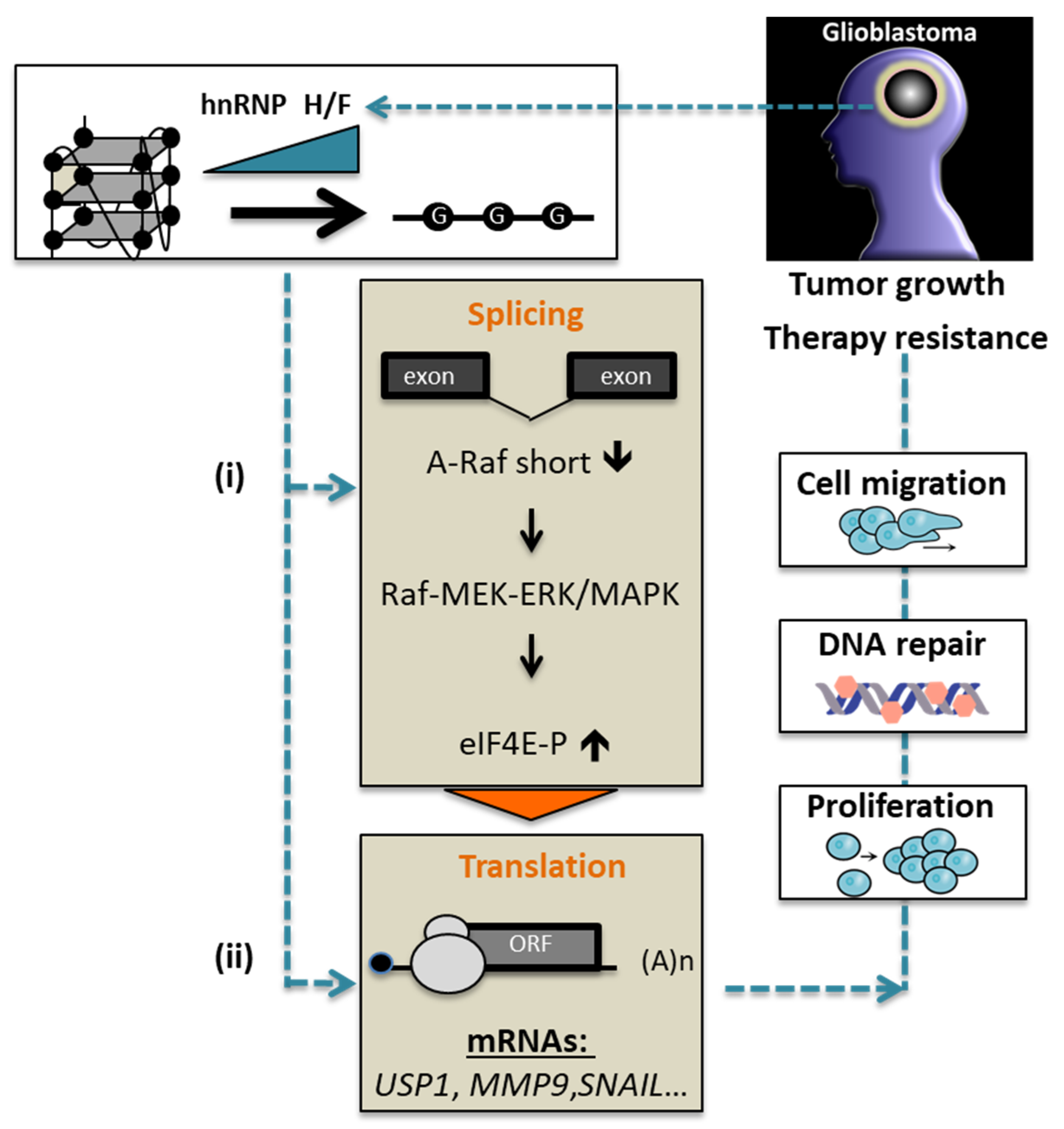
Translational Regulation by hnRNP H/F Is Essential for the Proliferation and Survival of Glioblastoma

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Cell Transfection
2.3. Western Blotting
2.4. RT-qPCR and RT-PCR
2.5. Immunoprecipitation
2.6. Preparation of RNA–Protein Complexes and Analysis by RT-qPCR
2.7. SUnSET
2.8. Polysome Profiling
2.9. Proliferation Assay
2.10. Colony Formation Assay
2.11. Xenograft Tumors in Nude Mice
2.12. Flow Cytometry
2.13. Migration/In Vitro Wound Closure Assay
3. Results
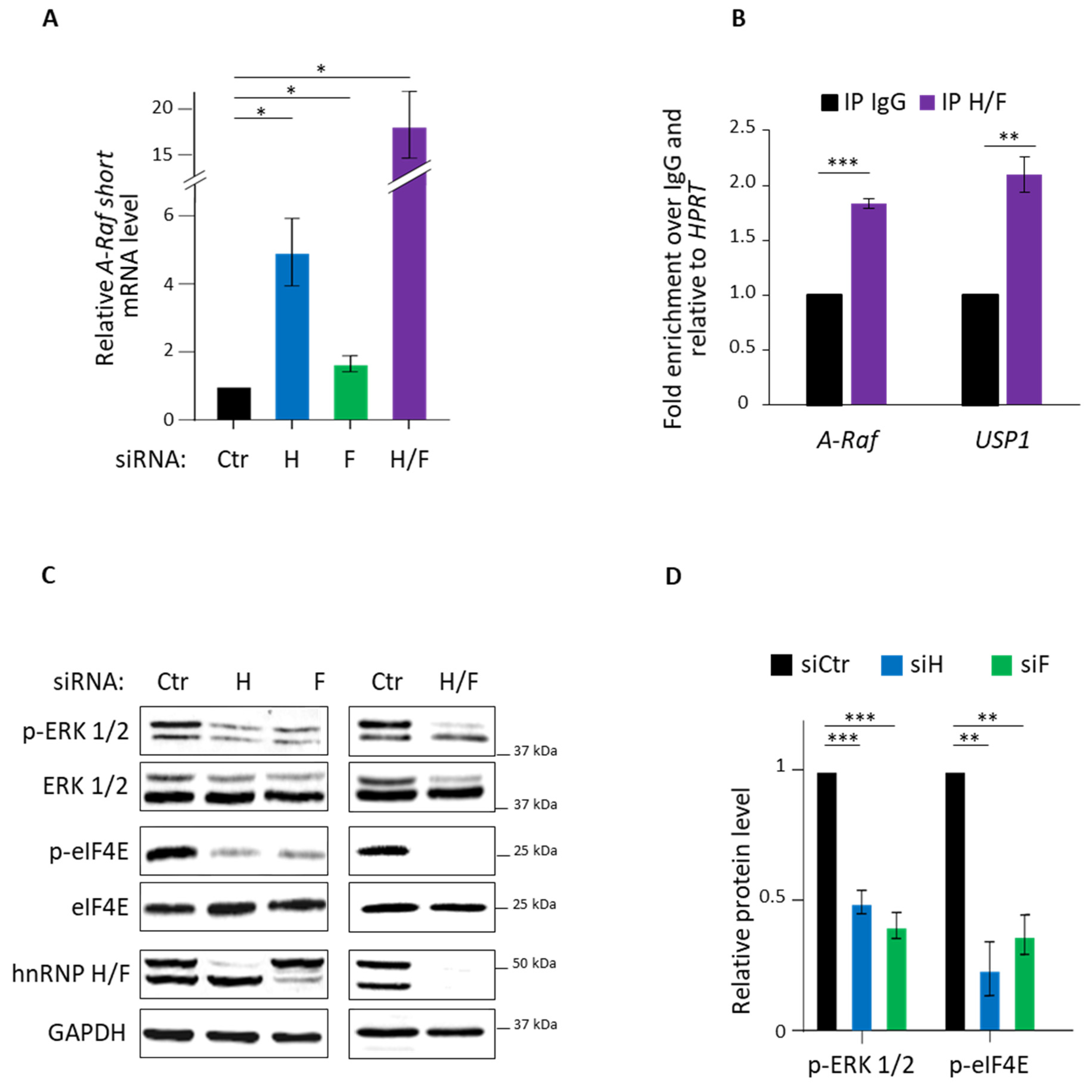
3.1. hnRNP H/F Impact on A-Raf Splicing and eIF4E Phosporylation
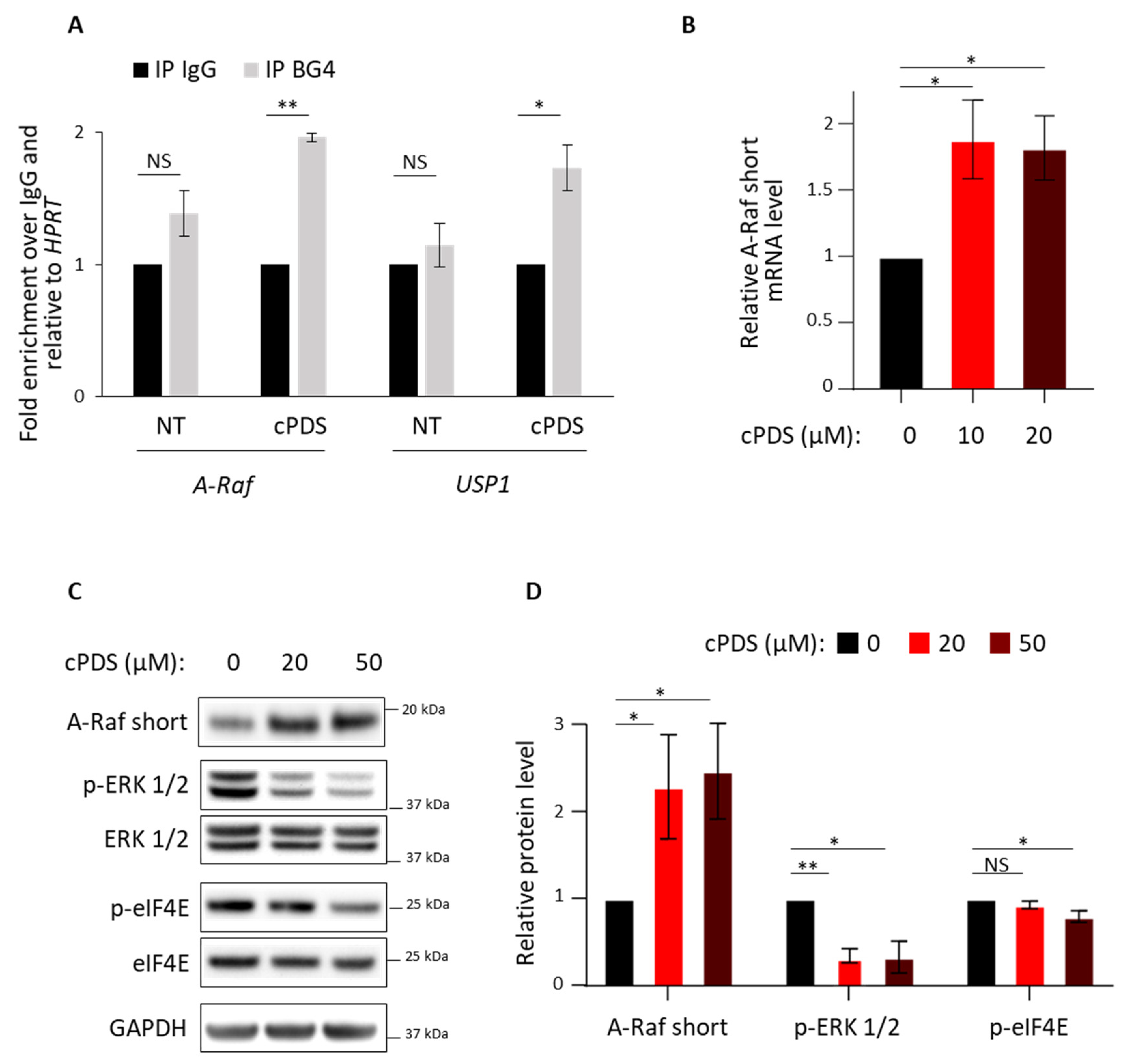
3.2. Role of RG4 Stabilization on A-Raf Splicing and eIF4E Phosphorylation
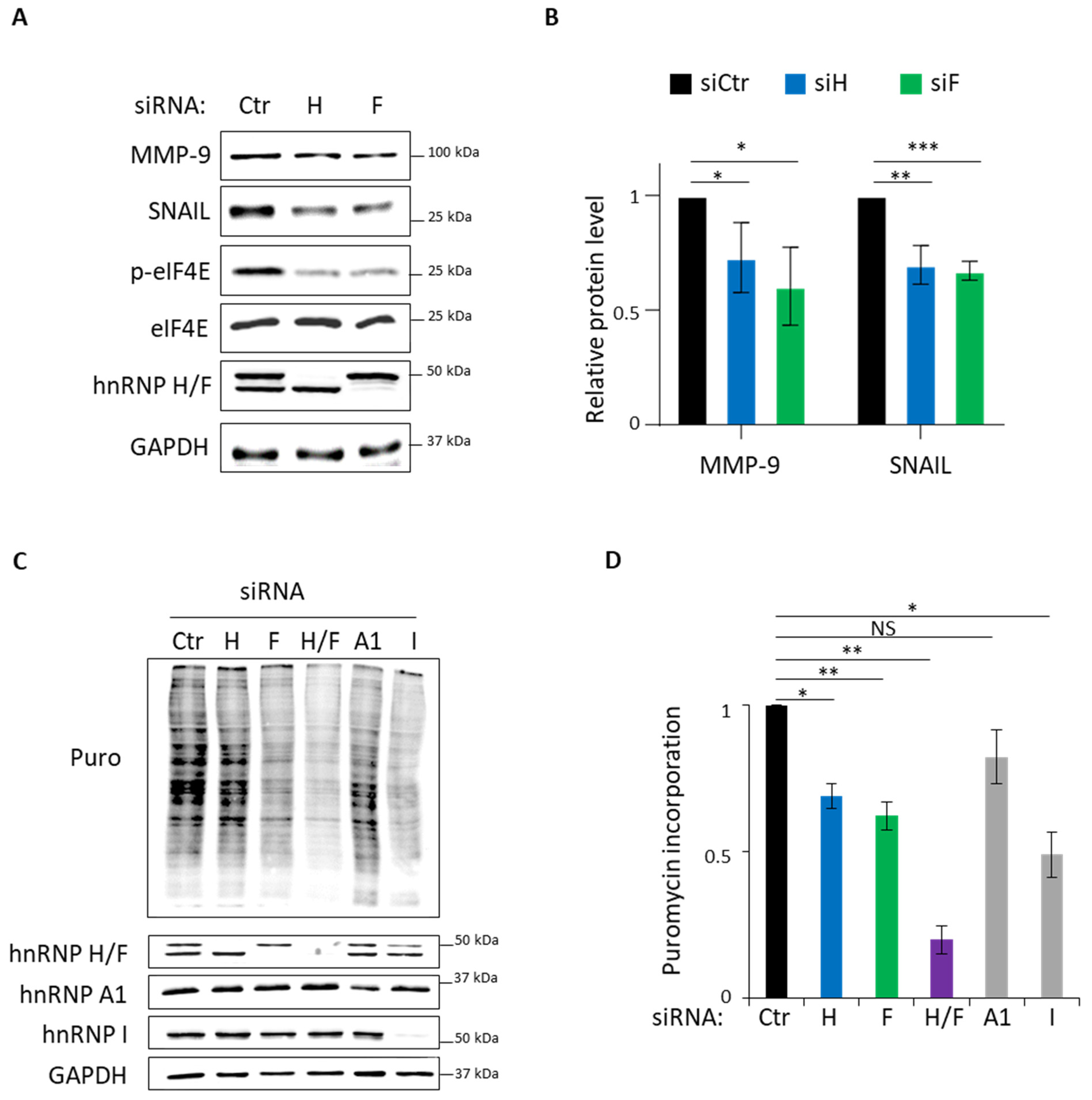
3.3. Role of hnRNP H/F in Translational Control in GBM
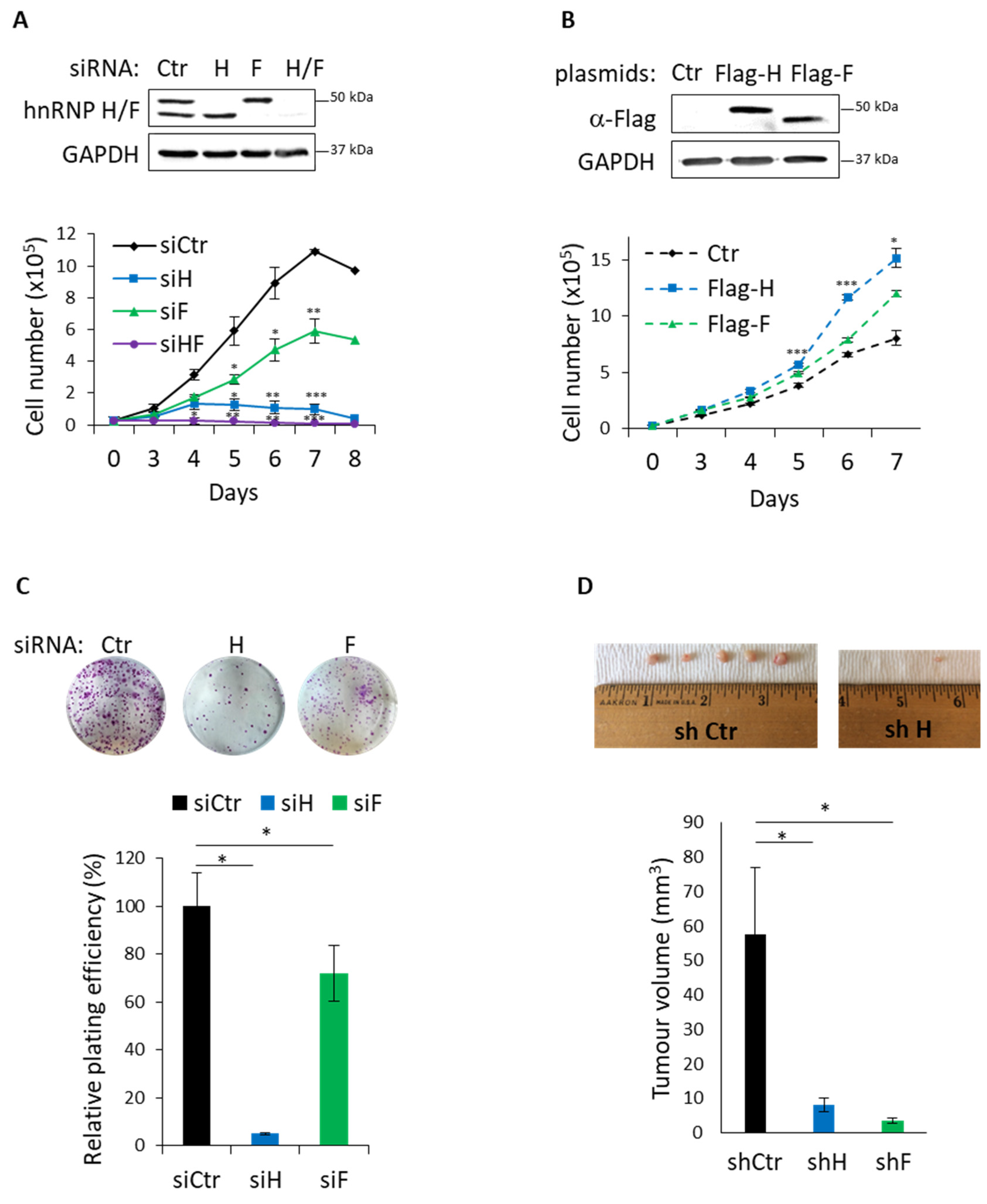
3.4. Functional Impact of hnRNP H/F on GBM Cell Migration and Proliferation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Rajaratnam, V.; Islam, M.M.; Yang, M.; Slaby, R.; Ramirez, H.M.; Mirza, S.P. Glioblastoma: Pathogenesis and Current Status of Chemotherapy and Other Novel Treatments. Cancers 2020, 12, 937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvera, D.; Formenti, S.C.; Schneider, R.J. Translational control in cancer. Nat. Rev. Cancer 2010, 10, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Truitt, M.L.; Ruggero, D. New frontiers in translational control of the cancer genome. Nat. Rev. Cancer 2016, 16, 288–304. [Google Scholar] [CrossRef] [Green Version]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 2015, 14, 261–278. [Google Scholar] [CrossRef]
- Blagden, S.P.; Willis, A.E. The biological and therapeutic relevance of mRNA translation in cancer. Nat. Rev. Clin. Oncol. 2011, 8, 280–291. [Google Scholar] [CrossRef]
- Malka-Mahieu, H.; Newman, M.; Desaubry, L.; Robert, C.; Vagner, S. Molecular Pathways: The eIF4F Translation Initiation Complex-New Opportunities for Cancer Treatment. Clin. Cancer Res. 2017, 23, 21–25. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, J.; Graff, J.; Ruggero, D.; Sonenberg, N. Targeting the eIF4F translation initiation complex: A critical nexus for cancer development. Cancer Res. 2015, 75, 250–263. [Google Scholar] [CrossRef] [Green Version]
- Chu, J.; Cargnello, M.; Topisirovic, I.; Pelletier, J. Translation Initiation Factors: Reprogramming Protein Synthesis in Cancer. Trends Cell Biol. 2016, 26, 918–933. [Google Scholar] [CrossRef] [PubMed]
- Kechavarzi, B.; Janga, S.C. Dissecting the expression landscape of RNA-binding proteins in human cancers. Genome Biol. 2014, 15, R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukong, K.E.; Chang, K.W.; Khandjian, E.W.; Richard, S. RNA-binding proteins in human genetic disease. Trends Genet. 2008, 24, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.; Billaud, M.; Almeida, R. RNA-Binding Proteins in Cancer: Old Players and New Actors. Trends Cancer 2017, 3, 506–528. [Google Scholar] [CrossRef]
- Furic, L.; Rong, L.; Larsson, O.; Koumakpayi, I.H.; Yoshida, K.; Brueschke, A.; Petroulakis, E.; Robichaud, N.; Pollak, M.; Gaboury, L.A.; et al. eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc. Natl. Acad. Sci. USA 2010, 107, 14134–14139. [Google Scholar] [CrossRef] [Green Version]
- Robichaud, N.; del Rincon, S.V.; Huor, B.; Alain, T.; Petruccelli, L.A.; Hearnden, J.; Goncalves, C.; Grotegut, S.; Spruck, C.H.; Furic, L.; et al. Phosphorylation of eIF4E promotes EMT and metastasis via translational control of SNAIL and MMP-3. Oncogene 2015, 34, 2032–2042. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, A.C.; Costa, M.; Zollo, O.; Davis, C.; Feldman, M.E.; Testa, J.R.; Meyuhas, O.; Shokat, K.M.; Ruggero, D. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell 2010, 17, 249–261. [Google Scholar] [CrossRef] [Green Version]
- Bertorello, J.; Sesen, J.; Gilhodes, J.; Evrard, S.; Courtade-Saïdi, M.; Augustus, M.; Uro-Coste, E.; Toulas, C.; Cohen-Jonathan Moyal, E.; Seva, C.; et al. Translation reprogramming by eIF3 linked to glioblastoma. Nucleic Acids Res. Cancer 2020, 2, zcaa020. [Google Scholar] [CrossRef]
- Sesen, J.; Cammas, A.; Scotland, S.J.; Elefterion, B.; Lemarie, A.; Millevoi, S.; Mathew, L.K.; Seva, C.; Toulas, C.; Moyal, E.C.; et al. Int6/eIF3e is essential for proliferation and survival of human glioblastoma cells. Int. J. Mol. Sci. 2014, 15, 2172–2190. [Google Scholar] [CrossRef] [Green Version]
- Hao, J.; Liang, C.; Jiao, B. Eukaryotic translation initiation factor 3, subunit C is overexpressed and promotes cell proliferation in human glioma U-87 MG cells. Oncol. Lett. 2015, 9, 2525–2533. [Google Scholar] [CrossRef] [Green Version]
- Preukschas, M.; Hagel, C.; Schulte, A.; Weber, K.; Lamszus, K.; Sievert, H.; Pallmann, N.; Bokemeyer, C.; Hauber, J.; Braig, M.; et al. Expression of eukaryotic initiation factor 5A and hypusine forming enzymes in glioblastoma patient samples: Implications for new targeted therapies. PLoS ONE 2012, 7, e43468. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, S.; Patil, V.; Mahalingam, K.; Somasundaram, K. Elucidation of the genetic and epigenetic landscape alterations in RNA binding proteins in glioblastoma. Oncotarget 2017, 8, 16650–16668. [Google Scholar] [CrossRef] [Green Version]
- Correa, B.R.; de Araujo, P.R.; Qiao, M.; Burns, S.C.; Chen, C.; Schlegel, R.; Agarwal, S.; Galante, P.A.; Penalva, L.O. Functional genomics analyses of RNA-binding proteins reveal the splicing regulator SNRPB as an oncogenic candidate in glioblastoma. Genome Biol. 2016, 17, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Jones, L.; Lowery-Norberg, M.; Fowler, M. Expression of eukaryotic initiation factor 4E in astrocytic tumors. Appl. Immunohistochem. Mol. Morphol. 2005, 13, 178–183. [Google Scholar] [CrossRef]
- Martinez-Saez, E.; Peg, V.; Ortega-Aznar, A.; Martinez-Ricarte, F.; Camacho, J.; Hernandez-Losa, J.; Ferreres Pinas, J.C.; Ramon, Y.C.S. peIF4E as an independent prognostic factor and a potential therapeutic target in diffuse infiltrating astrocytomas. Cancer Med. 2016, 5, 2501–2512. [Google Scholar] [CrossRef] [Green Version]
- Herviou, P.; Le Bras, M.; Dumas, L.; Hieblot, C.; Gilhodes, J.; Cioci, G.; Hugnot, J.P.; Ameadan, A.; Guillonneau, F.; Dassi, E.; et al. hnRNP H/F drive RNA G-quadruplex-mediated translation linked to genomic instability and therapy resistance in glioblastoma. Nat. Commun. 2020, 11, 2661. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; de la Pena, L.; Barker, C.; Camphausen, K.; Tofilon, P.J. Radiation-induced changes in gene expression involve recruitment of existing messenger RNAs to and away from polysomes. Cancer Res. 2006, 66, 1052–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahba, A.; Rath, B.H.; Bisht, K.; Camphausen, K.; Tofilon, P.J. Polysome Profiling Links Translational Control to the Radioresponse of Glioblastoma Stem-like Cells. Cancer Res. 2016, 76, 3078–3087. [Google Scholar] [CrossRef] [Green Version]
- Bell, J.B.; Eckerdt, F.; Dhruv, H.D.; Finlay, D.; Peng, S.; Kim, S.; Kroczynska, B.; Beauchamp, E.M.; Alley, K.; Clymer, J.; et al. Differential Response of Glioma Stem Cells to Arsenic Trioxide Therapy Is Regulated by MNK1 and mRNA Translation. Mol. Cancer Res. 2018, 16, 32–46. [Google Scholar] [CrossRef] [Green Version]
- Grzmil, M.; Seebacher, J.; Hess, D.; Behe, M.; Schibli, R.; Moncayo, G.; Frank, S.; Hemmings, B.A. Inhibition of MNK pathways enhances cancer cell response to chemotherapy with temozolomide and targeted radionuclide therapy. Cell Signal. 2016, 28, 1412–1421. [Google Scholar] [CrossRef]
- Honore, B.; Baandrup, U.; Vorum, H. Heterogeneous nuclear ribonucleoproteins F and H/H’ show differential expression in normal and selected cancer tissues. Exp. Cell Res. 2004, 294, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Lefave, C.V.; Squatrito, M.; Vorlova, S.; Rocco, G.L.; Brennan, C.W.; Holland, E.C.; Pan, Y.X.; Cartegni, L. Splicing factor hnRNPH drives an oncogenic splicing switch in gliomas. EMBO J. 2011, 30, 4084–4097. [Google Scholar] [CrossRef] [PubMed]
- Decorsiere, A.; Cayrel, A.; Vagner, S.; Millevoi, S. Essential role for the interaction between hnRNP H/F and a G quadruplex in maintaining p53 pre-mRNA 3’-end processing and function during DNA damage. Genes Dev. 2011, 25, 220–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Zhang, J.; Harvey, S.E.; Hu, X.; Cheng, C. RNA G-quadruplex secondary structure promotes alternative splicing via the RNA-binding protein hnRNPF. Genes Dev. 2017, 31, 2296–2309. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.T.; Stover, P.J. Mechanism of the internal ribosome entry site-mediated translation of serine hydroxymethyltransferase 1. J. Biol. Chem. 2009, 284, 31085–31096. [Google Scholar] [CrossRef] [Green Version]
- Kalifa, Y.; Huang, T.; Rosen, L.N.; Chatterjee, S.; Gavis, E.R. Glorund, a Drosophila hnRNP F/H homolog, is an ovarian repressor of nanos translation. Dev. Cell 2006, 10, 291–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauch, J.; Moran-Jones, K.; Albrecht, V.; Schwarzl, T.; Hunter, K.; Gires, O.; Kolch, W. c-Myc regulates RNA splicing of the A-Raf kinase and its activation of the ERK pathway. Cancer Res. 2011, 71, 4664–4674. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Bakke, J.; Finkelstein, D.; Zeng, H.; Wu, J.; Chen, T. HNRNPH1 is required for rhabdomyosarcoma cell growth and survival. Oncogenesis 2018, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Waskiewicz, A.J.; Johnson, J.C.; Penn, B.; Mahalingam, M.; Kimball, S.R.; Cooper, J.A. Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Mol. Cell Biol 1999, 19, 1871–1880. [Google Scholar] [CrossRef] [Green Version]
- Biffi, G.; Di Antonio, M.; Tannahill, D.; Balasubramanian, S. Visualization and selective chemical targeting of RNA G-quadruplex structures in the cytoplasm of human cells. Nat. Chem. 2014, 6, 75–80. [Google Scholar] [CrossRef]
- Svitkin, Y.V.; Ovchinnikov, L.P.; Dreyfuss, G.; Sonenberg, N. General RNA binding proteins render translation cap dependent. EMBO J. 1996, 15, 7147–7155. [Google Scholar] [CrossRef] [PubMed]
- Uren, P.J.; Bahrami-Samani, E.; de Araujo, P.R.; Vogel, C.; Qiao, M.; Burns, S.C.; Smith, A.D.; Penalva, L.O. High-throughput analyses of hnRNP H1 dissects its multi-functional aspect. RNA Biol. 2016, 13, 400–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goh, E.T.; Pardo, O.E.; Michael, N.; Niewiarowski, A.; Totty, N.; Volkova, D.; Tsaneva, I.R.; Seckl, M.J.; Gout, I. Involvement of heterogeneous ribonucleoprotein F in the regulation of cell proliferation via the mammalian target of rapamycin/S6 kinase 2 pathway. J. Biol. Chem. 2010, 285, 17065–17076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, R.J.; Hellen, C.U.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huelga, S.C.; Vu, A.Q.; Arnold, J.D.; Liang, T.Y.; Liu, P.P.; Yan, B.Y.; Donohue, J.P.; Shiue, L.; Hoon, S.; Brenner, S.; et al. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep. 2012, 1, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Vignon, C.; Debeissat, C.; Georget, M.T.; Bouscary, D.; Gyan, E.; Rosset, P.; Herault, O. Flow cytometric quantification of all phases of the cell cycle and apoptosis in a two-color fluorescence plot. PLoS ONE 2013, 8, e68425. [Google Scholar] [CrossRef] [Green Version]
- Braun, S.; Enculescu, M.; Setty, S.T.; Cortes-Lopez, M.; de Almeida, B.P.; Sutandy, F.X.R.; Schulz, L.; Busch, A.; Seiler, M.; Ebersberger, S.; et al. Decoding a cancer-relevant splicing decision in the RON proto-oncogene using high-throughput mutagenesis. Nat. Commun. 2018, 9, 3315. [Google Scholar] [CrossRef]
- Shilo, A.; Ben Hur, V.; Denichenko, P.; Stein, I.; Pikarsky, E.; Rauch, J.; Kolch, W.; Zender, L.; Karni, R. Splicing factor hnRNP A2 activates the Ras-MAPK-ERK pathway by controlling A-Raf splicing in hepatocellular carcinoma development. RNA 2014, 20, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Dardenne, E.; Polay Espinoza, M.; Fattet, L.; Germann, S.; Lambert, M.P.; Neil, H.; Zonta, E.; Mortada, H.; Gratadou, L.; Deygas, M.; et al. RNA helicases DDX5 and DDX17 dynamically orchestrate transcription, miRNA, and splicing programs in cell differentiation. Cell Rep. 2014, 7, 1900–1913. [Google Scholar] [CrossRef] [Green Version]
- Blaustein, M.; Pelisch, F.; Tanos, T.; Munoz, M.J.; Wengier, D.; Quadrana, L.; Sanford, J.R.; Muschietti, J.P.; Kornblihtt, A.R.; Caceres, J.F.; et al. Concerted regulation of nuclear and cytoplasmic activities of SR proteins by AKT. Nat. Struct. Mol. Biol. 2005, 12, 1037–1044. [Google Scholar] [CrossRef]
- Graveley, B.R. Coordinated control of splicing and translation. Nat. Struct. Mol. Biol. 2005, 12, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Keene, J.D. RNA regulons: Coordination of post-transcriptional events. Nat. Rev. Genet. 2007, 8, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Maslon, M.M.; Heras, S.R.; Bellora, N.; Eyras, E.; Caceres, J.F. The translational landscape of the splicing factor SRSF1 and its role in mitosis. eLife 2014, 3, e02028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterne-Weiler, T.; Martinez-Nunez, R.T.; Howard, J.M.; Cvitovik, I.; Katzman, S.; Tariq, M.A.; Pourmand, N.; Sanford, J.R. Frac-seq reveals isoform-specific recruitment to polyribosomes. Genome Res. 2013, 23, 1615–1623. [Google Scholar] [CrossRef] [Green Version]
- Dassi, E. Handshakes and Fights: The Regulatory Interplay of RNA-Binding Proteins. Front. Mol. Biosci. 2017, 4, 67. [Google Scholar] [CrossRef] [Green Version]
- Havens, M.A.; Reich, A.A.; Hastings, M.L. Drosha promotes splicing of a pre-microRNA-like alternative exon. PLoS Genet. 2014, 10, e1004312. [Google Scholar] [CrossRef]
- Chen, L.; Wang, W.; Zhu, S.; Jin, X.; Wang, J.; Zhu, J.; Zhou, Y. MicroRNA-590-3p enhances the radioresistance in glioblastoma cells by targeting LRIG1. Exp. Ther. Med. 2017, 14, 1818–1824. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le Bras, M.; Gorelick, N.; Pautet, S.; Tyler, B.; Manenti, S.; Skuli, N.; Millevoi, S.; Cammas, A. Translational Regulation by hnRNP H/F Is Essential for the Proliferation and Survival of Glioblastoma. Cancers 2022, 14, 1283. https://doi.org/10.3390/cancers14051283
Le Bras M, Gorelick N, Pautet S, Tyler B, Manenti S, Skuli N, Millevoi S, Cammas A. Translational Regulation by hnRNP H/F Is Essential for the Proliferation and Survival of Glioblastoma. Cancers. 2022; 14(5):1283. https://doi.org/10.3390/cancers14051283
Chicago/Turabian StyleLe Bras, Morgane, Noah Gorelick, Sylvain Pautet, Betty Tyler, Stéphane Manenti, Nicolas Skuli, Stefania Millevoi, and Anne Cammas. 2022. "Translational Regulation by hnRNP H/F Is Essential for the Proliferation and Survival of Glioblastoma" Cancers 14, no. 5: 1283. https://doi.org/10.3390/cancers14051283
APA StyleLe Bras, M., Gorelick, N., Pautet, S., Tyler, B., Manenti, S., Skuli, N., Millevoi, S., & Cammas, A. (2022). Translational Regulation by hnRNP H/F Is Essential for the Proliferation and Survival of Glioblastoma. Cancers, 14(5), 1283. https://doi.org/10.3390/cancers14051283