
Metformin and ICG-001 Act Synergistically to Abrogate Cancer Stem Cells-Mediated Chemoresistance in Colorectal Cancer by Promoting Apoptosis and Autophagy



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Drugs, Antibodies, and Primers
2.2. Cell Culture
2.3. Cell Viability Assay
2.4. Isobologram Analysis
2.5. Clonogenic Cell Survival Assay
2.6. Apoptosis and Cell Cycle Analysis
2.7. Spheroid Formation Assay
2.8. Flow Cytometric Analysis of CD44
2.9. Wound Healing Assay
2.10. Invasion Assay
2.11. CRC Patient-Derived Tumor Organoids
2.12. RNA Isolation, cDNA Synthesis, and Gene Expression Profiling by qPCR
2.13. Western Blot Analysis
2.14. Genome-Wide Transcriptomic Profiling and Pathway Enrichment Analysis
2.15. Statistical Analysis
3. Results
3.1. Metformin Treatment Inhibits Cell Proliferation and Colony Formation in CRC Cells
3.2. Metformin Treatment Induces Cell Cycle Arrest and Apoptosis and Inhibits Spheroid Formation in CRC Cells
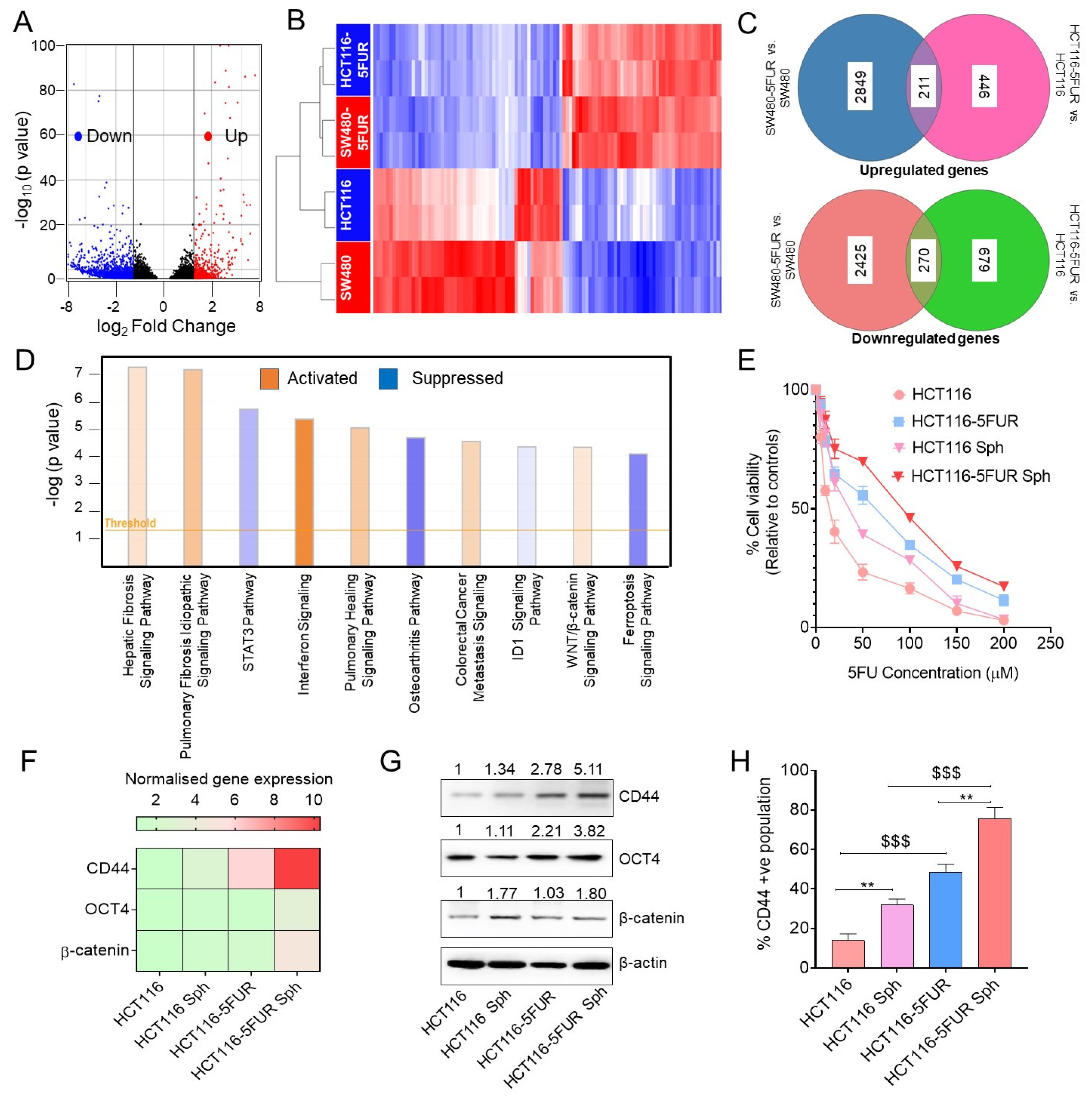
3.3. Transcriptomic Analysis Revealed Enrichment of Wnt/β-Catenin Signaling in 5FU-Resistant CRC Cells
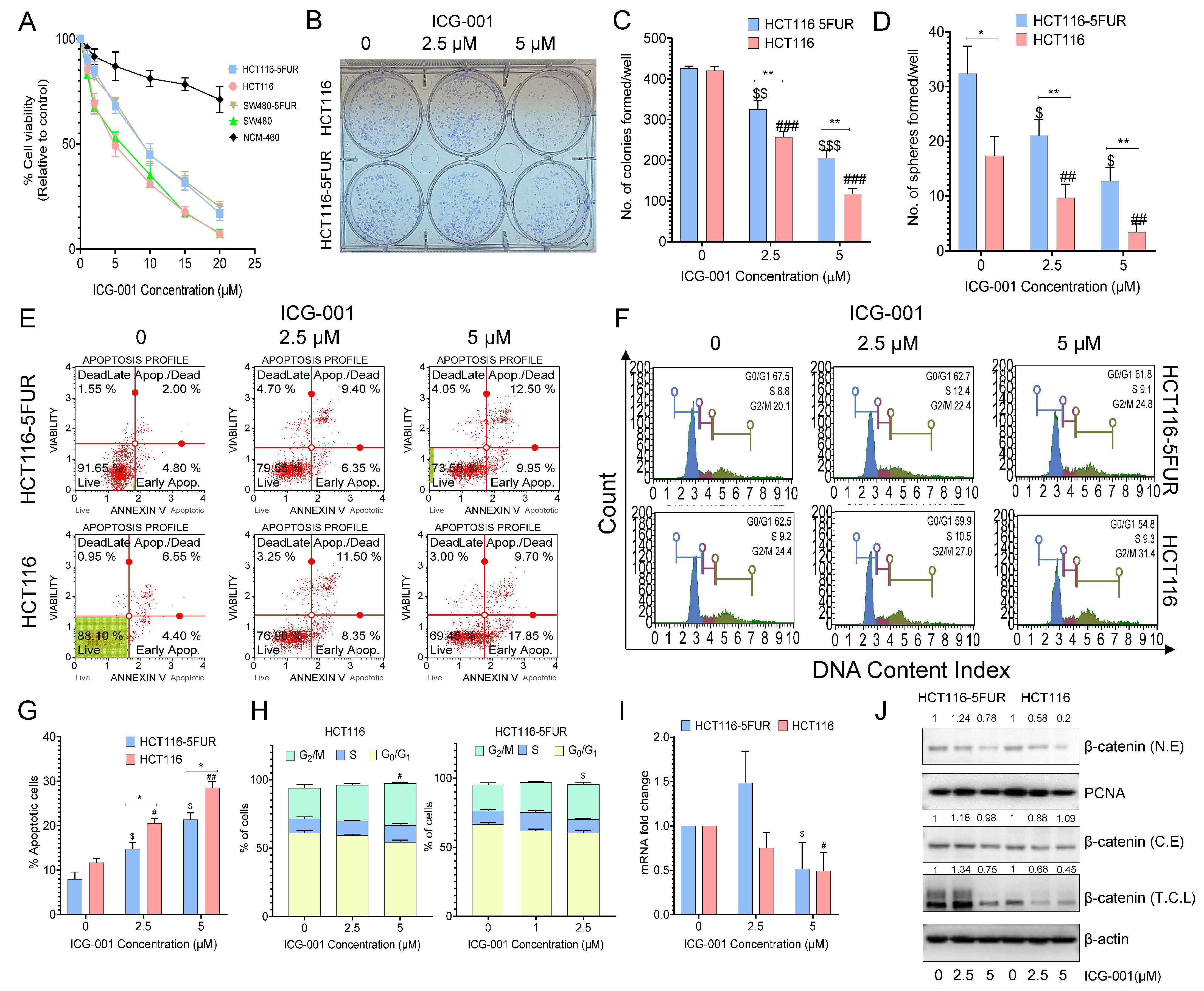
3.4. Selective Targeting of the Wnt Signaling Pathway by ICG-001 Promotes Chemosensitivity in CRC Cells
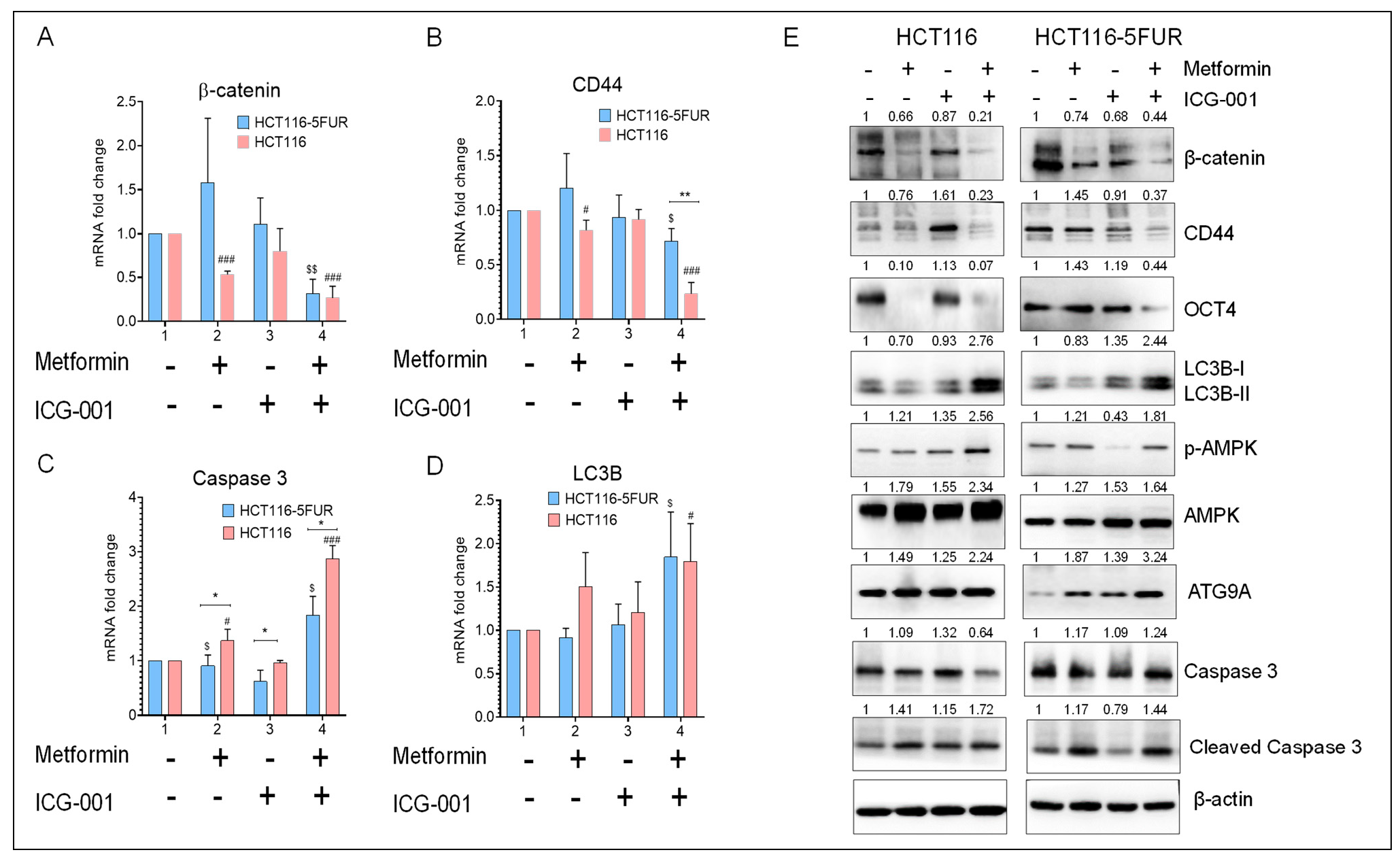
3.5. Combined Treatment with Metformin and ICG-001 Abrogates Chemoresistance in a Synergistic Manner
3.6. Combined Treatment with Metformin and ICG-001 Decreases the Expression of CSC Markers and Promotes Autophagy and Apoptosis in CRC Cells
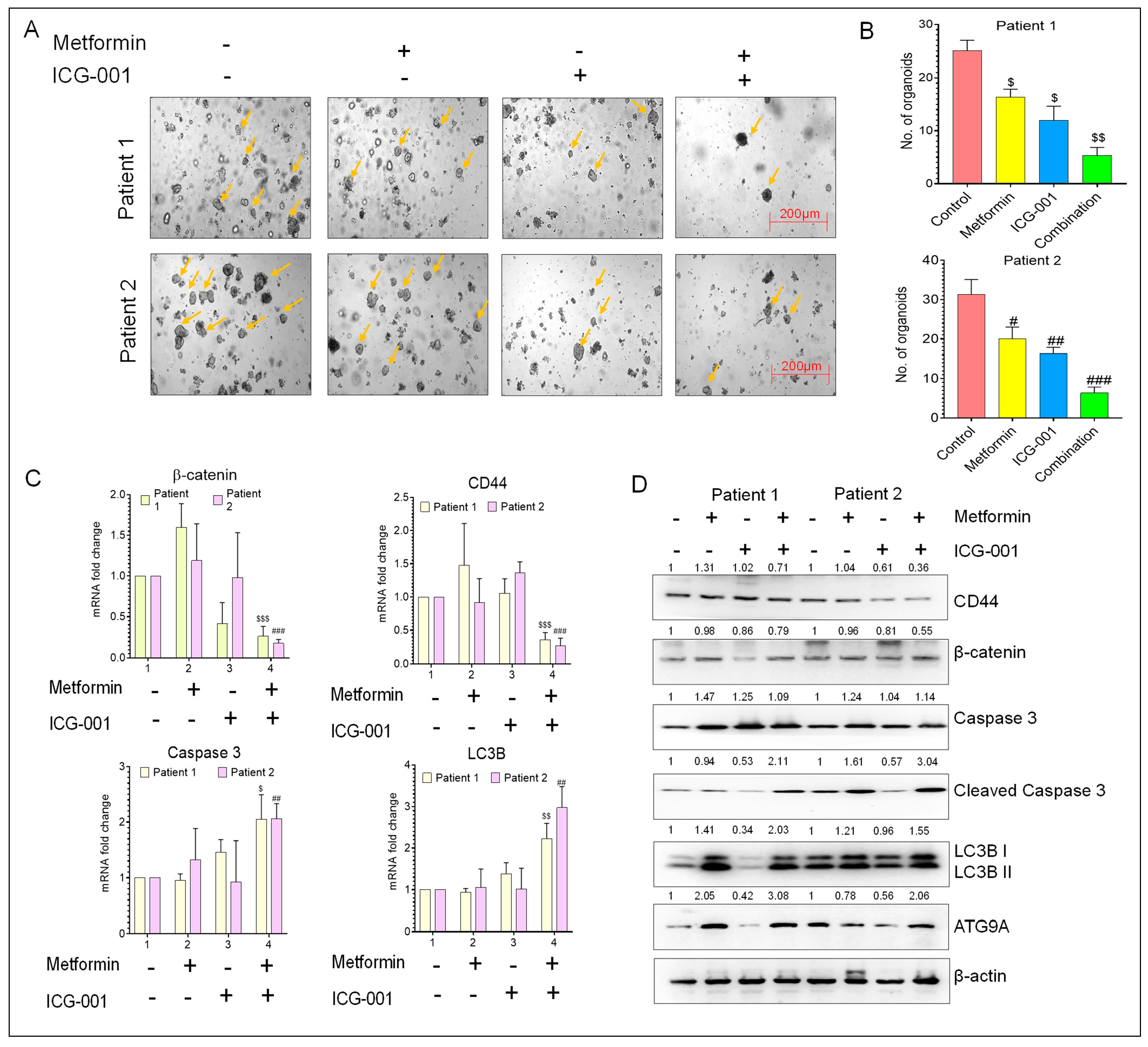
3.7. Metformin and ICG-001 Promote Anti-Tumor Activity in CRC Patient-Derived Organoid Models
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA A Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Kamarudin, M.N.A.; Sarker, M.M.R.; Zhou, J.-R.; Parhar, I. Metformin in colorectal cancer: Molecular mechanism, preclinical and clinical aspects. J. Exp. Clin. Cancer Res. 2019, 38, 491. [Google Scholar] [CrossRef] [PubMed]
- Pisano, M.; Zorcolo, L.; Merli, C.; Cimbanassi, S.; Poiasina, E.; Ceresoli, M.; Agresta, F.; Allievi, N.; Bellanova, G.; Coccolini, F.; et al. 2017 WSES guidelines on colon and rectal cancer emergencies: Obstruction and perforation. World J. Emerg. Surg. 2018, 13, 36. [Google Scholar] [CrossRef]
- Carethers, J.M. Systemic treatment of advanced colorectal cancer: Tailoring therapy to the tumor. Therap. Adv. Gastroenterol. 2008, 1, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Stoehlmacher, J. Prediction of efficacy and side effects of chemotherapy in colorectal cancer. Recent Results Cancer Res. Fortschr. Krebsforschung. Prog. Dans. Rech. Cancer 2007, 176, 81–88. [Google Scholar] [CrossRef]
- Takahashi, M.; Cuatrecasas, M.; Balaguer, F.; Hur, K.; Toiyama, Y.; Castells, A.; Boland, C.R.; Goel, A. The clinical significance of MiR-148a as a predictive biomarker in patients with advanced colorectal cancer. PLoS ONE 2012, 7, e46684. [Google Scholar] [CrossRef]
- Blondy, S.; David, V.; Verdier, M.; Mathonnet, M.; Perraud, A.; Christou, N. 5-Fluorouracil resistance mechanisms in colorectal cancer: From classical pathways to promising processes. Cancer Sci. 2020, 111, 3142–3154. [Google Scholar] [CrossRef]
- Sakatani, A.; Sonohara, F.; Goel, A. Melatonin-mediated downregulation of thymidylate synthase as a novel mechanism for overcoming 5-fluorouracil associated chemoresistance in colorectal cancer cells. Carcinogenesis 2019, 40, 422–431. [Google Scholar] [CrossRef]
- González, N.; Prieto, I.; Del Puerto-Nevado, L.; Portal-Nuñez, S.; Ardura, J.A.; Corton, M.; Fernández-Fernández, B.; Aguilera, O.; Gomez-Guerrero, C.; Mas, S.; et al. 2017 update on the relationship between diabetes and colorectal cancer: Epidemiology, potential molecular mechanisms and therapeutic implications. Oncotarget 2017, 8, 18456–18485. [Google Scholar] [CrossRef]
- Joung, K.H.; Jeong, J.-W.; Ku, B.J. The association between type 2 diabetes mellitus and women cancer: The epidemiological evidences and putative mechanisms. Biomed. Res. Int. 2015, 2015, 920618. [Google Scholar] [CrossRef]
- Kheirandish, M.; Mahboobi, H.; Yazdanparast, M.; Kamal, W.; Kamal, M.A. Anti-cancer Effects of Metformin: Recent Evidences for its Role in Prevention and Treatment of Cancer. Curr. Drug Metab. 2018, 19, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Ross, F.A.; Chevtzoff, C.; Green, K.A.; Evans, A.; Fogarty, S.; Towler, M.C.; Brown, L.J.; Ogunbayo, O.A.; Evans, A.M.; et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010, 11, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.H.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005, 310, 1642–1646. [Google Scholar] [CrossRef]
- Carretero, J.; Medina, P.P.; Blanco, R.; Smit, L.; Tang, M.; Roncador, G.; Maestre, L.; Conde, E.; Lopez-Rios, F.; Clevers, H.C.; et al. Dysfunctional AMPK activity, signalling through mTOR and survival in response to energetic stress in LKB1-deficient lung cancer. Oncogene 2007, 26, 1616–1625. [Google Scholar] [CrossRef]
- Lu, G.; Wu, Z.; Shang, J.; Xie, Z.; Chen, C.; Zhang, C. The effects of metformin on autophagy. Biomed. Pharmacother. 2021, 137, 111286. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Zhang, W.; Yi, J.; Xiao, Z.-X. Role of p53 Family Proteins in Metformin Anti-Cancer Activities. J. Cancer 2019, 10, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Ben Sahra, I.; Regazzetti, C.; Robert, G.; Laurent, K.; Le Marchand-Brustel, Y.; Auberger, P.; Tanti, J.F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011, 71, 4366–4372. [Google Scholar] [CrossRef] [PubMed]
- Alimova, I.N.; Liu, B.; Fan, Z.; Edgerton, S.M.; Dillon, T.; Lind, S.E.; Thor, A.D. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle 2009, 8, 909–915. [Google Scholar] [CrossRef]
- Zhang, Y.; Guan, M.; Zheng, Z.; Zhang, Q.; Gao, F.; Xue, Y. Effects of metformin on CD133+ colorectal cancer cells in diabetic patients. PLoS ONE 2013, 8, e81264. [Google Scholar] [CrossRef]
- Toden, S.; Kunitoshi, S.; Cardenas, J.; Gu, J.; Hutchins, E.; Van Keuren-Jensen, K.; Uetake, H.; Toiyama, Y.; Goel, A. Cancer stem cell-associated miRNAs serve as prognostic biomarkers in colorectal cancer. JCI Insight 2019, 4, e125294. [Google Scholar] [CrossRef]
- Roy, S.; Kar, M.; Roy, S.; Padhi, S.; Kumar, A.; Thakur, S.; Akhter, Y.; Gatto, G.; Banerjee, B. Inhibition of CD44 sensitizes cisplatin-resistance and affects Wnt/β-catenin signaling in HNSCC cells. Int. J. Biol. Macromol. 2020, 149, 501–512. [Google Scholar] [CrossRef]
- Prieto-Vila, M.; Takahashi, R.U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xia, L.; Wang, H.; Oyang, L.; Su, M.; Liu, Q.; Lin, J.; Tan, S.; Tian, Y.; Liao, Q.; et al. Cancer stem cells in progression of colorectal cancer. Oncotarget 2017, 9, 33403–33415. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, E.; Melo, F.; Vermeulen, L. Wnt Signaling in Cancer Stem Cell Biology. Cancers 2016, 8, 60. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.-H.; Ro, E.J.; Yoon, J.-S.; Mizutani, T.; Kang, D.-W.; Park, J.-C.; Il Kim, T.; Clevers, H.; Choi, K.-Y. 5-FU promotes stemness of colorectal cancer via p53-mediated WNT/β-catenin pathway activation. Nature Commun. 2020, 11, 5321. [Google Scholar] [CrossRef] [PubMed]
- Ravindranathan, P.; Pasham, D.; Goel, A. Oligomeric proanthocyanidins (OPCs) from grape seed extract suppress the activity of ABC transporters in overcoming chemoresistance in colorectal cancer cells. Carcinogenesis 2019, 40, 412–421. [Google Scholar] [CrossRef]
- Toden, S.; Ravindranathan, P.; Gu, J.; Cardenas, J.; Yuchang, M.; Goel, A. Oligomeric proanthocyanidins (OPCs) target cancer stem-like cells and suppress tumor organoid formation in colorectal cancer. Sci. Rep. 2018, 8, 3335. [Google Scholar] [CrossRef]
- Sharma, P.; Shimura, T.; Banwait, J.K.; Goel, A. Andrographis-mediated chemosensitization through activation of ferroptosis and suppression of β-catenin/Wnt-signaling pathways in colorectal cancer. Carcinogenesis 2020, 41, 1385–1394. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, C.; Goel, A. Andrographis overcomes 5-fluorouracil-associated chemoresistance through inhibition of DKK1 in colorectal cancer. Carcinogenesis 2021, 42, 814–825. [Google Scholar] [CrossRef]
- Shimura, T.; Sharma, P.; Sharma, G.G.; Banwait, J.K.; Goel, A. Enhanced anti-cancer activity of andrographis with oligomeric proanthocyanidins through activation of metabolic and ferroptosis pathways in colorectal cancer. Sci. Rep. 2021, 11, 7548. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Kar, M.; Roy, S.; Saha, A.; Padhi, S.; Banerjee, B. Role of β-catenin in cisplatin resistance, relapse and prognosis of head and neck squamous cell carcinoma. Cell. Oncol. 2018, 41, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Xing, Y.; Yu, T.; Wu, Y.N.; Roy, M.; Kim, J.; Lee, C. An expectation-maximization algorithm for probabilistic reconstructions of full-length isoforms from splice graphs. Nucleic Acids Res. 2006, 34, 3150–3160. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 2014, 15, 550. [Google Scholar] [CrossRef]
- Mogavero, A.; Maiorana, M.V.; Zanutto, S.; Varinelli, L.; Bozzi, F.; Belfiore, A.; Volpi, C.C.; Gloghini, A.; Pierotti, M.A.; Gariboldi, M. Metformin transiently inhibits colorectal cancer cell proliferation as a result of either AMPK activation or increased ROS production. Sci. Rep. 2017, 7, 15992. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, M.; Zang, X.; Mao, Z.; Chen, Y.; Mao, F.; Qian, H.; Xu, W.; Zhang, X. CircHN1 affects cell proliferation and migration in gastric cancer. J. Clin. Lab. Anal. 2020, 34, e23433. [Google Scholar] [CrossRef]
- Kang, H.E.; Seo, Y.; Yun, J.S.; Song, S.H.; Han, D.; Cho, E.S.; Cho, S.B.; Jeon, Y.; Lee, H.; Kim, H.S.; et al. Metformin and Niclosamide Synergistically Suppress Wnt and YAP in APC-Mutated Colorectal Cancer. Cancers 2021, 13, 3437. [Google Scholar] [CrossRef]
- Herheliuk, T.; Perepelytsina, O.; Ugnivenko, A.; Ostapchenko, L.; Sydorenko, M. Investigation of multicellular tumor spheroids enriched for a cancer stem cell phenotype. Stem. Cell. Investig. 2019, 6, 21. [Google Scholar] [CrossRef]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Moertel, C.G.; Fleming, T.R.; Macdonald, J.S.; Haller, D.G.; Laurie, J.A.; Goodman, P.J.; Ungerleider, J.S.; Emerson, W.A.; Tormey, D.C.; Glick, J.H.; et al. Levamisole and fluorouracil for adjuvant therapy of resected colon carcinoma. N. Engl. J. Med. 1990, 322, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Pettersen, H.S.; Visnes, T.; Vågbø, C.B.; Svaasand, E.K.; Doseth, B.; Slupphaug, G.; Kavli, B.; Krokan, H.E. UNG-initiated base excision repair is the major repair route for 5-fluorouracil in DNA, but 5-fluorouracil cytotoxicity depends mainly on RNA incorporation. Nucleic Acids Res. 2011, 39, 8430–8444. [Google Scholar] [CrossRef]
- Boige, V.; Mendiboure, J.; Pignon, J.P.; Loriot, M.A.; Castaing, M.; Barrois, M.; Malka, D.; Trégouët, D.A.; Bouché, O.; Le Corre, D.; et al. Pharmacogenetic assessment of toxicity and outcome in patients with metastatic colorectal cancer treated with LV5FU2, FOLFOX, and FOLFIRI: FFCD 2000-05. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 2556–2564. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Satoh, M.; Kinouchi, M.; Yamamoto, K.; Hasegawa, Y.; Kakugawa, Y.; Kawai, M.; Uchimi, K.; Aizawa, H.; Ohnuma, S.; et al. The use of natural products in colorectal cancer drug discovery. Expert Opin. Drug Discov. 2015, 10, 411–426. [Google Scholar] [CrossRef]
- Ravindranathan, P.; Pasham, D.; Balaji, U.; Cardenas, J.; Gu, J.; Toden, S.; Goel, A. A combination of curcumin and oligomeric proanthocyanidins offer superior anti-tumorigenic properties in colorectal cancer. Sci. Rep. 2018, 8, 13869. [Google Scholar] [CrossRef]
- Lee, S.H.; Koo, B.S.; Kim, J.M.; Huang, S.; Rho, Y.S.; Bae, W.J.; Kang, H.J.; Kim, Y.S.; Moon, J.H.; Lim, Y.C. Wnt/β-catenin signalling maintains self-renewal and tumourigenicity of head and neck squamous cell carcinoma stem-like cells by activating Oct4. J. Pathol. 2014, 234, 99–107. [Google Scholar] [CrossRef]
- He, L.; Zhu, H.; Zhou, S.; Wu, T.; Wu, H.; Yang, H.; Mao, H.; SekharKathera, C.; Janardhan, A.; Edick, A.M.; et al. Wnt pathway is involved in 5-FU drug resistance of colorectal cancer cells. Exp. Mol. Med. 2018, 50, 1–12. [Google Scholar] [CrossRef]
- Albini, A.; Bruno, A.; Gallo, C.; Pajardi, G.; Noonan, D.M.; Dallaglio, K. Cancer stem cells and the tumor microenvironment: Interplay in tumor heterogeneity. Connect. Tissue Res. 2015, 56, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yang, Z.; Dong, D.-L.; Jang, T.-S.; Knowles, J.C.; Kim, H.-W.; Jin, G.-Z.; Xuan, Y. 3D culture technologies of cancer stem cells: Promising ex vivo tumor models. J. Tissue Eng. 2020, 11, 2041731420933407. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, H.; Zheng, P.; Zheng, Y.; Luo, Q.; Xie, G.; Ma, Y.; Shen, L. ICG-001 suppresses growth of gastric cancer cells and reduces chemoresistance of cancer stem cell-like population. J. Exp. Clin. Cancer Res. CR 2017, 36, 125. [Google Scholar] [CrossRef]
- Emami, K.H.; Nguyen, C.; Ma, H.; Kim, D.H.; Jeong, K.W.; Eguchi, M.; Moon, R.T.; Teo, J.-L.; Oh, S.W.; Kim, H.Y.; et al. A small molecule inhibitor of β-catenin/cyclic AMP response element-binding protein transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 12682–12687. [Google Scholar] [CrossRef]
- Ghadimi, B.M.; Grade, M.; Difilippantonio, M.J.; Varma, S.; Simon, R.; Montagna, C.; Füzesi, L.; Langer, C.; Becker, H.; Liersch, T.; et al. Effectiveness of gene expression profiling for response prediction of rectal adenocarcinomas to preoperative chemoradiotherapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 1826–1838. [Google Scholar] [CrossRef]
- Xie, J.; Xiang, D.-B.; Wang, H.; Zhao, C.; Chen, J.; Xiong, F.; Li, T.-Y.; Wang, X.-L. Inhibition of Tcf-4 Induces Apoptosis and Enhances Chemosensitivity of Colon Cancer Cells. PLoS ONE 2012, 7, e45617. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Jung, S.; Jeong, H.; Yu, S.-W. Autophagy as a decisive process for cell death. Exp. Mol. Med. 2020, 52, 921–930. [Google Scholar] [CrossRef]
- Su, M.; Mei, Y.; Sinha, S. Role of the Crosstalk between Autophagy and Apoptosis in Cancer. J. Oncol. 2013, 2013, 102735. [Google Scholar] [CrossRef]
- Liu, Y.; Levine, B. Autosis and autophagic cell death: The dark side of autophagy. Cell Death Differ. 2015, 22, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef]
- Sena, P.; Mancini, S.; Benincasa, M.; Mariani, F.; Palumbo, C.; Roncucci, L. Metformin Induces Apoptosis and Alters Cellular Responses to Oxidative Stress in Ht29 Colon Cancer Cells: Preliminary Findings. Int. J. Mol. Sci. 2018, 19, 1478. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Li, Y.; Duan, H.; Yuan, L. Metformin Suppresses the Proliferation and Promotes the Apoptosis of Colon Cancer Cells Through Inhibiting the Expression of Long Noncoding RNA-UCA1. OncoTargets Ther. 2020, 13, 4169–4181. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Plasencia, C.; López-Urrutia, E.; García-Castillo, V.; Trujano-Camacho, S.; López-Camarillo, C.; Campos-Parra, A.D. Interplay between Autophagy and Wnt/β-Catenin Signaling in Cancer: Therapeutic Potential Through Drug Repositioning. Front. Oncol. 2020, 10, 1037. [Google Scholar] [CrossRef]
- Cicchini, M.; Chakrabarti, R.; Kongara, S.; Price, S.; Nahar, R.; Lozy, F.; Zhong, H.; Vazquez, A.; Kang, Y.; Karantza, V. Autophagy regulator BECN1 suppresses mammary tumorigenesis driven by WNT1 activation and following parity. Autophagy 2014, 10, 2036–2052. [Google Scholar] [CrossRef]
- Nàger, M.; Sallán, M.C.; Visa, A.; Pushparaj, C.; Santacana, M.; Macià, A.; Yeramian, A.; Cantí, C.; Herreros, J. Inhibition of WNT-CTNNB1 signaling upregulates SQSTM1 and sensitizes glioblastoma cells to autophagy blockers. Autophagy 2018, 14, 619–636. [Google Scholar] [CrossRef]
- Gunti, S.; Hoke, A.T.K.; Vu, K.P.; London, N.R., Jr. Organoid and Spheroid Tumor Models: Techniques and Applications. Cancers 2021, 13, 874. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, S.; Zhao, Y.; Yuan, Y.-C.; Goel, A. Metformin and ICG-001 Act Synergistically to Abrogate Cancer Stem Cells-Mediated Chemoresistance in Colorectal Cancer by Promoting Apoptosis and Autophagy. Cancers 2022, 14, 1281. https://doi.org/10.3390/cancers14051281
Roy S, Zhao Y, Yuan Y-C, Goel A. Metformin and ICG-001 Act Synergistically to Abrogate Cancer Stem Cells-Mediated Chemoresistance in Colorectal Cancer by Promoting Apoptosis and Autophagy. Cancers. 2022; 14(5):1281. https://doi.org/10.3390/cancers14051281
Chicago/Turabian StyleRoy, Souvick, Yinghui Zhao, Yate-Ching Yuan, and Ajay Goel. 2022. "Metformin and ICG-001 Act Synergistically to Abrogate Cancer Stem Cells-Mediated Chemoresistance in Colorectal Cancer by Promoting Apoptosis and Autophagy" Cancers 14, no. 5: 1281. https://doi.org/10.3390/cancers14051281
APA StyleRoy, S., Zhao, Y., Yuan, Y.-C., & Goel, A. (2022). Metformin and ICG-001 Act Synergistically to Abrogate Cancer Stem Cells-Mediated Chemoresistance in Colorectal Cancer by Promoting Apoptosis and Autophagy. Cancers, 14(5), 1281. https://doi.org/10.3390/cancers14051281