

PITX1 Is a Regulator of TERT Expression in Prostate Cancer with Prognostic Power

 ,
,  , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Gene Expression Data
2.2. The Mixed Integer Linear Model of TERT Regulation
2.3. Regulatory TF-TF Network
2.4. Immunohistochemical Analysis of PITX1 and IRF1 in PCa Patients
2.5. Cell Lines
2.6. siRNA Knockdown
2.7. Chromatin Immunoprecipitation
2.8. Statistical Analysis
3. Results
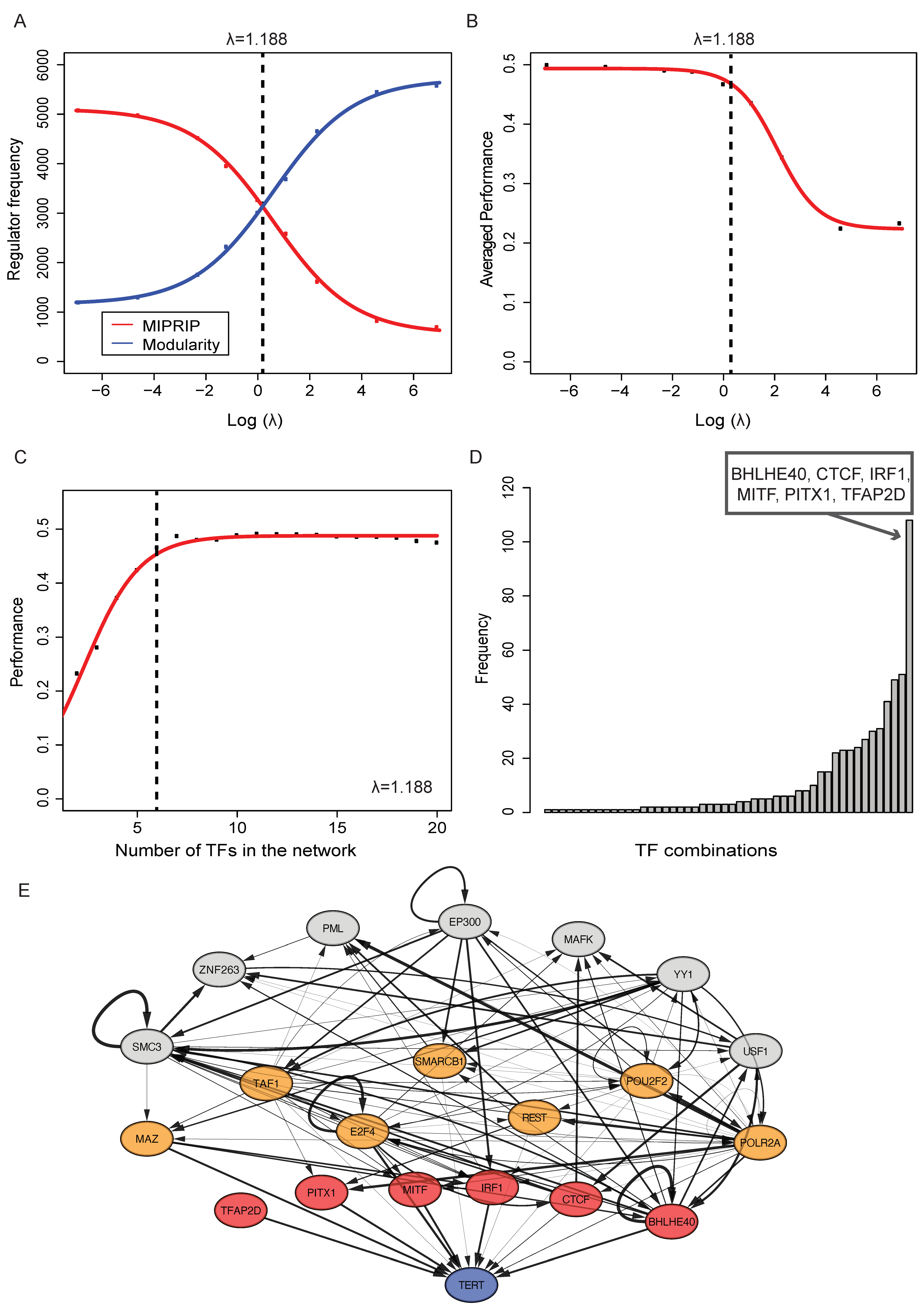
3.1. The Direct Model Predicts Specific Transcription Factors of TERT in PCa
3.2. Identifying a Regulatory Module for TERT Regulation in PCa
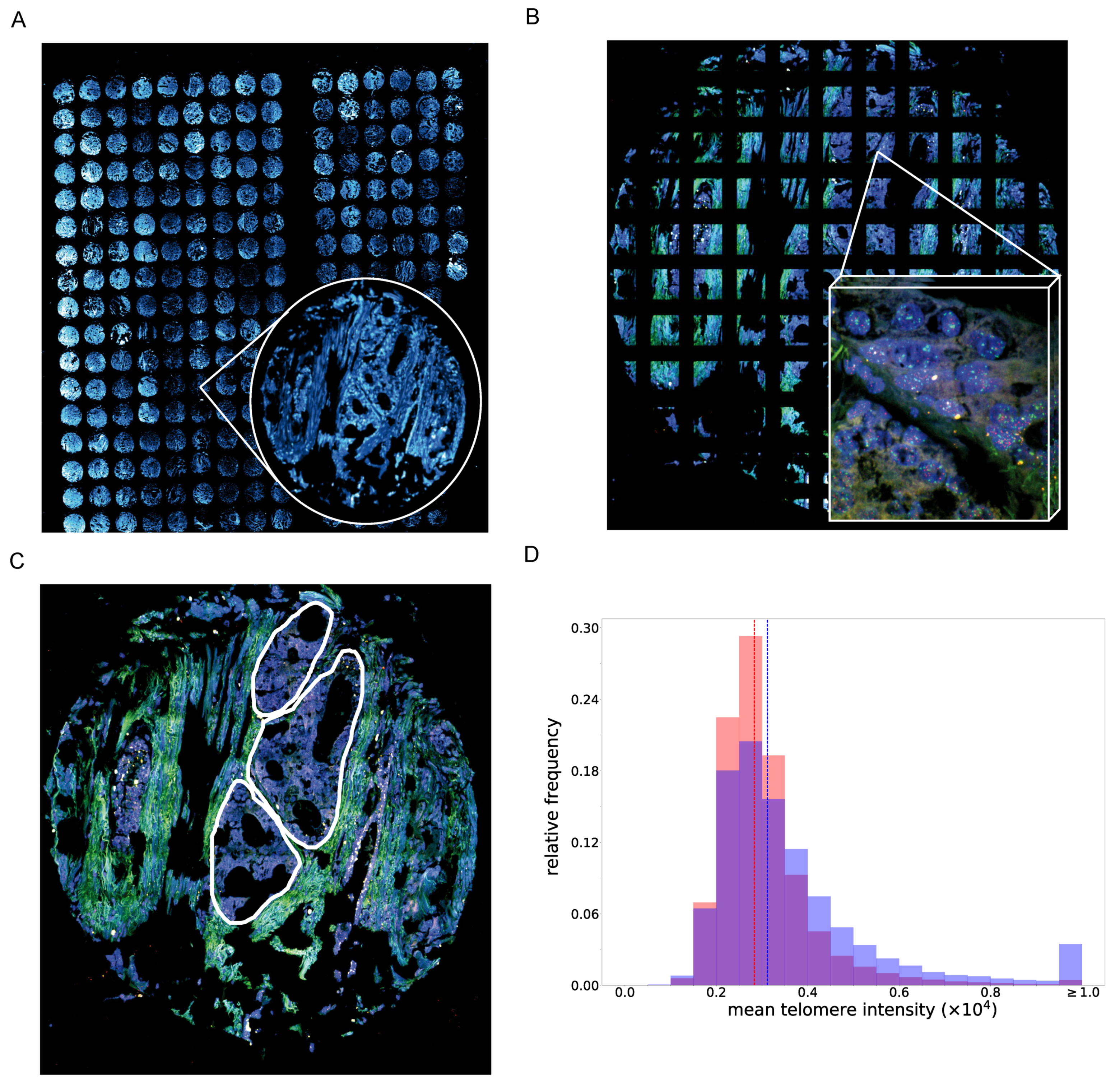
3.3. PCa Tissue Cells with A High PITX1 Protein Expression Show Higher Telomere Staining Intensity
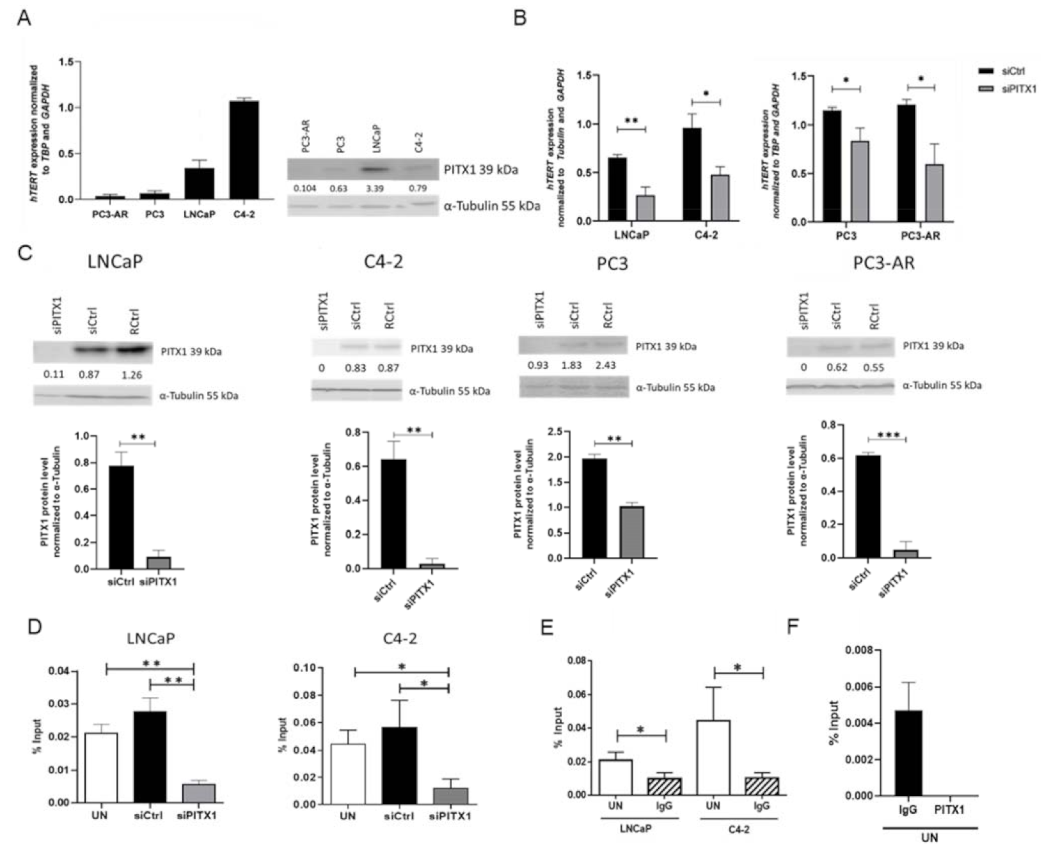
3.4. In Vitro Experiments Showed That PITX1 Binds to the Promoter of TERT and PITX1 Knockdown Reduces TERT Expression
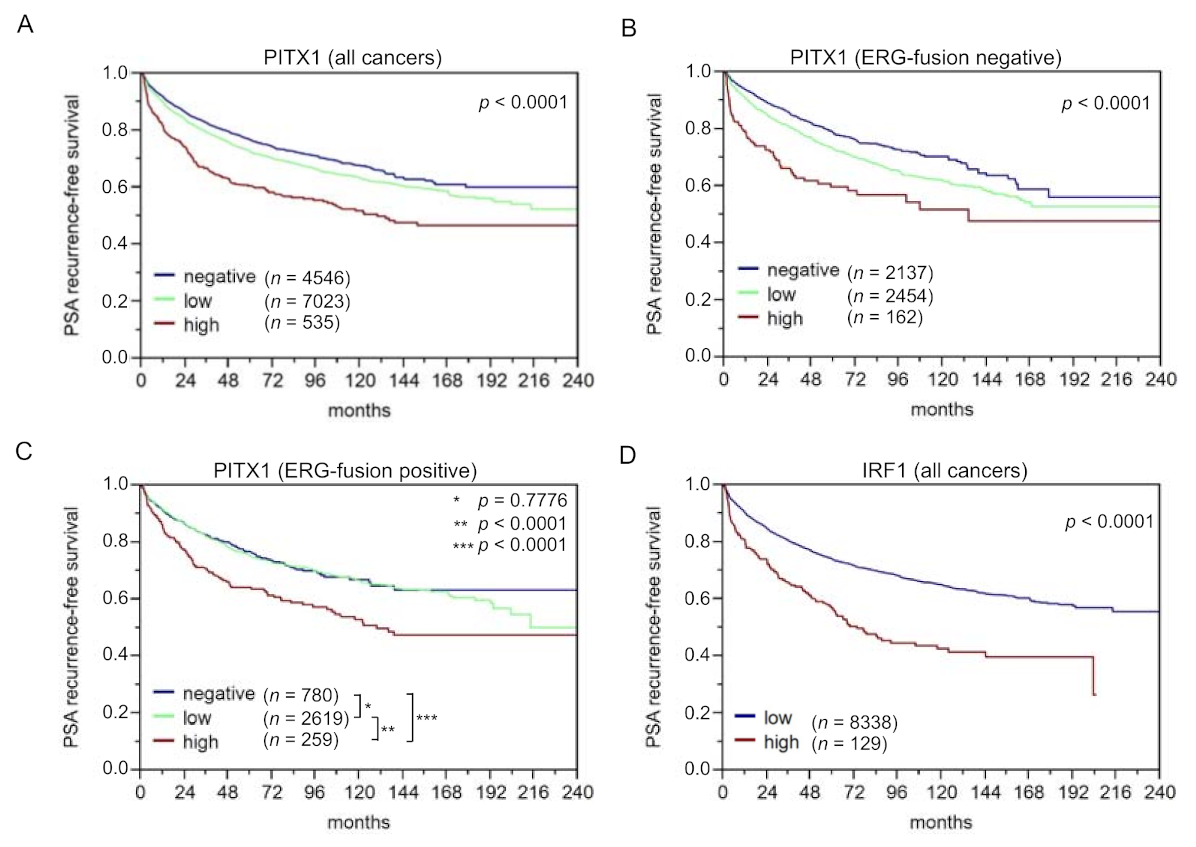
3.5. The Identified Transcription Factor PITX1 Suits as a Prognostic Marker
3.6. The Identified Transcription Factors IRF1, CTCF, and TFAP2D Also Suit as Prognostic Markers, Particularly when Combining them with PITX1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penney, K.L.; Stampfer, M.J.; Jahn, J.L.; Sinnott, J.A.; Flavin, R.; Rider, J.; Finn, S.; Giovannucci, E.; Sesso, H.D.; Loda, M.; et al. Gleason Grade Progression Is Uncommon. Cancer Res. 2013, 73, 5163–5168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Olama, A.A.; Kote-Jarai, Z.; Berndt, S.I.; Conti, D.V.; Schumacher, F.; Han, Y.; Benlloch, S.; Hazelett, D.; Wang, Z.; Saunders, E.; et al. A meta-analysis of 87,040 individuals identifies 23 new susceptibility loci for prostate cancer. Nat. Genet. 2014, 46, 1103–1109. [Google Scholar] [CrossRef] [Green Version]
- Board, P.A.T.E. Prostate Cancer Treatment (PDQ(R)): Patient Version. In PDQ Cancer Information Summaries; Bethesda: Rockville, MD, USA, 2002. [Google Scholar]
- Cooperberg, M.R.; Broering, J.M.; Carroll, P.R. Risk Assessment for Prostate Cancer Metastasis and Mortality at the Time of Diagnosis. J. Natl. Cancer Inst. 2009, 101, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.-W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent Fusion of TMPRSS2 and ETS Transcription Factor Genes in Prostate Cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef]
- Barbieri, C.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.-P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, E.H. Telomere states and cell fates. Nature 2000, 408, 53–56. [Google Scholar] [CrossRef]
- de Lange, T. How Shelterin Solves the Telomere End-Protection Problem. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Wright, W.E.; Tesmer, V.M.; Huffman, K.E.; Levene, S.D.; Shay, J.W. Normal human chromosomes have long G-rich telomeric overhangs at one end. Genes Dev. 1997, 11, 2801–2809. [Google Scholar] [CrossRef] [Green Version]
- Heaphy, C.M.; Subhawong, A.P.; Hong, S.-M.; Goggins, M.G.; Montgomery, E.A.; Gabrielson, E.; Netto, G.J.; Epstein, J.I.; Lotan, T.; Westra, W.H.; et al. Prevalence of the Alternative Lengthening of Telomeres Telomere Maintenance Mechanism in Human Cancer Subtypes. Am. J. Pathol. 2011, 179, 1608–1615. [Google Scholar] [CrossRef]
- Artandi, S.E.; DePinho, R. Telomeres and telomerase in cancer. Carcinogenesis 2009, 31, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Bacchetti, S. A survey of telomerase activity in human cancer. Eur. J. Cancer 1997, 33, 787–791. [Google Scholar] [CrossRef]
- Ramlee, M.K.; Wang, J.; Toh, W.X.; Li, S. Transcription Regulation of the Human Telomerase Reverse Transcriptase (hTERT) Gene. Genes 2016, 7, 50. [Google Scholar] [CrossRef]
- Poos, A.M.; Kordaß, T.; Kolte, A.; Ast, V.; Oswald, M.; Rippe, K.; König, R. Modelling TERT regulation across 19 different cancer types based on the MIPRIP 2.0 gene regulatory network approach. BMC Bioinform. 2019, 20, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Porter, M.A.; Onnela, J.P.; Mucha, P.J. Communities in networks. Notices Am. Math. Soc. 2009, 56, 1082. [Google Scholar]
- Fortunato, S. Community detection in graphs. Phys. Rep. 2009, 486, 75–174. [Google Scholar] [CrossRef] [Green Version]
- Newman, M.E.J. Communities, modules and large-scale structure in networks. Nat. Phys. 2011, 8, 25–31. [Google Scholar] [CrossRef]
- Fortunato, S.; Hric, D. Community detection in networks: A user guide. Phys. Rep. 2016, 659, 1–44. [Google Scholar] [CrossRef] [Green Version]
- Newman, M.E.J. Equivalence between modularity optimization and maximum likelihood methods for community detection. Phys. Rev. E 2016, 94, 052315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, M.E.J. Modularity and community structure in networks. Proc. Natl. Acad. Sci. USA 2006, 103, 8577–8582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poos, A.M.; Maicher, A.; Dieckmann, A.K.; Oswald, M.; Eils, R.; Kupiec, M.; Luke, B.; König, R. Mixed Integer Linear Programming based machine learning approach identifiesregulatorsof telomerase in yeast. Nucleic Acids Res. 2016, 44, e93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, M.J.; Shen, Y.; Giorgi, F.M.; Lachmann, A.; Ding, B.B.; Ye, B.H.; Califano, A. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 2016, 48, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Balwierz, P.J.; Pachkov, M.; Arnold, P.; Gruber, A.J.; Zavolan, M.; van Nimwegen, E. ISMARA: Automated modeling of genomic signals as a democracy of regulatory motifs. Genome Res. 2014, 24, 869–884. [Google Scholar] [CrossRef] [Green Version]
- Schacht, T.; Oswald, M.; Eils, R.; Eichmüller, S.; König, R. Estimating the activity of transcription factors by the effect on their target genes. Bioinformatics 2014, 30, i401–i407. [Google Scholar] [CrossRef] [Green Version]
- Lachmann, A.; Xu, H.; Krishnan, J.; Berger, S.; Mazloom, A.R.; Ma’Ayan, A. ChEA: Transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 2010, 26, 2438–2444. [Google Scholar] [CrossRef]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wu, G.; Ji, H. hmChIP: A database and web server for exploring publicly available human and mouse ChIP-seq and ChIP-chip data. Bioinformatics 2011, 27, 1447–1448. [Google Scholar] [CrossRef] [Green Version]
- Bovolenta, L.A.; Acencio, M.L.; Lemke, N. HTRIdb: An open-access database for experimentally verified human transcriptional regulation interactions. BMC Genom. 2012, 13, 405. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.-H.; Li, J.-H.; Jiang, S.; Zhou, H.; Qu, L.-H. ChIPBase: A database for decoding the transcriptional regulation of long non-coding RNA and microRNA genes from ChIP-Seq data. Nucleic Acids Res. 2012, 41, D177–D187. [Google Scholar] [CrossRef]
- Molineris, I.; Grassi, E.; Ala, U.; Di Cunto, F.; Provero, P. Evolution of Promoter Affinity for Transcription Factors in the Human Lineage. Mol. Biol. Evol. 2011, 28, 2173–2183. [Google Scholar] [CrossRef] [Green Version]
- Grassi, E.; Zapparoli, E.; Molineris, I.; Provero, P. Total Binding Affinity Profiles of Regulatory Regions Predict Transcription Factor Binding and Gene Expression in Human Cells. PLoS ONE 2015, 10, e0143627. [Google Scholar] [CrossRef] [PubMed]
- Schlomm, T.; Iwers, L.; Kirstein, P.; Jessen, B.; Köllermann, J.; Minner, S.; Passow-Drolet, A.; Mirlacher, M.; Milde-Langosch, K.; Graefen, M.; et al. Clinical significance of p53 alterations in surgically treated prostate cancers. Mod. Pathol. 2008, 21, 1371–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weischenfeldt, J.; Simon, R.; Feuerbach, L.; Schlangen, K.; Weichenhan, D.; Minner, S.; Wuttig, D.; Warnatz, H.-J.; Stehr, H.; Rausch, T.; et al. Integrative Genomic Analyses Reveal an Androgen-Driven Somatic Alteration Landscape in Early-Onset Prostate Cancer. Cancer Cell 2013, 23, 159–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krohn, A.; Diedler, T.; Burkhardt, L.; Mayer, P.-S.; De Silva, C.; Meyer-Kornblum, M.; Kötschau, D.; Tennstedt, P.; Huang, J.; Gerhauser, C.; et al. Genomic Deletion of PTEN Is Associated with Tumor Progression and Early PSA Recurrence in ERG Fusion-Positive and Fusion-Negative Prostate Cancer. Am. J. Pathol. 2012, 181, 401–412. [Google Scholar] [CrossRef]
- Krohn, A.; Seidel, A.; Burkhardt, L.; Bachmann, F.; Mader, M.; Grupp, K.; Eichenauer, T.; Becker, A.; Adam, M.; Graefen, M.; et al. Recurrent deletion of 3p13 targets multiple tumour suppressor genes and defines a distinct subgroup of aggressive ERG fusion-positive prostate cancers. J. Pathol. 2013, 231, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Kluth, M.; Hesse, J.; Heinl, A.; Krohn, A.; Steurer, S.; Sirma, H.; Simon, R.; Mayer, P.-S.; Schumacher, U.; Grupp, K.; et al. Genomic deletion of MAP3K7 at 6q12-22 is associated with early PSA recurrence in prostate cancer and absence of TMPRSS2:ERG fusions. Mod. Pathol. 2013, 26, 975–983. [Google Scholar] [CrossRef] [Green Version]
- Burkhardt, L.; Fuchs, S.; Krohn, A.; Masser, S.; Mader, M.; Kluth, M.; Bachmann, F.; Huland, H.; Steuber, T.; Graefen, M.; et al. CHD1 Is a 5q21 Tumor Suppressor Required for ERG Rearrangement in Prostate Cancer. Cancer Res. 2013, 73, 2795–2805. [Google Scholar] [CrossRef] [Green Version]
- Kononen, J.; Bubendorf, L.; Kallioniemi, O.; Bärlund, M.; Schraml, P.; Leighton, S.; Torhorst, J.; Mihatsch, M.J.; Sauter, G.; Kallionimeni, O.-P. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat. Med. 1998, 4, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Mirlacher, M.; Simon, R. Recipient Block TMA Technique. In Tissue Microarrays; Methods in Molecular Biology (Methods and Protocols); Simon, R., Ed.; Humana Press: Totowa, NJ, USA, 2010; Volume 664, pp. 37–44. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Fraune, C.; Harms, L.; Büscheck, F.; Höflmayer, D.; Tsourlakis, M.C.; Clauditz, T.S.; Simon, R.; Möller, K.; Luebke, A.M.; Möller-Koop, C.; et al. Upregulation of the transcription factor TFAP2D is associated with aggressive tumor phenotype in prostate cancer lacking the TMPRSS2:ERG fusion. Mol. Med. 2020, 26, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höflmayer, D.; Steinhoff, A.; Hube-Magg, C.; Kluth, M.; Simon, R.; Burandt, E.; Tsourlakis, M.C.; Minner, S.; Sauter, G.; Büscheck, F.; et al. Expression of CCCTC-binding factor (CTCF) is linked to poor prognosis in prostate cancer. Mol. Oncol. 2019, 14, 129–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protopopov, A.I.; Li, J.; Gizatullin, R.Z.; Kashuba, V.I.; Klein, G.; Zabarovsky, E.R.; Winberg, G. Human cell lines engineered for tetracycline-regulated expression of tumor suppressor candidate genes from a frequently affected chromosomal region, 3p21. J. Gene Med. 2002, 4, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Mirzakhani, K.; Kallenbach, J.; Rasa, S.M.M.; Ribaudo, F.; Ungelenk, M.; Ehsani, M.; Gong, W.; Gassler, N.; Leeder, M.; Grimm, M.-O.; et al. The androgen receptor—lncRNASAT1-AKT-p15 axis mediates androgen-induced cellular senescence in prostate cancer cells. Oncogene 2021, 41, 943–959. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, S.; Mirzakhani, K.; Neubert, L.; Stenzel, A.; Ehsani, M.; Esmaeili, M.; Pungsrinont, T.; Kacal, M.; Rasa, S.M.M.; Kallenbach, J.; et al. Antithetic hTERT Regulation by Androgens in Prostate Cancer Cells: hTERT Inhibition Is Mediated by the ING1 and ING2 Tumor Suppressors. Cancers 2021, 13, 4025. [Google Scholar] [CrossRef] [PubMed]
- Qi, D.-L.; Ohhira, T.; Fujisaki, C.; Inoue, T.; Ohta, T.; Osaki, M.; Ohshiro, E.; Seko, T.; Aoki, S.; Oshimura, M.; et al. Identification of PITX1 as a TERT Suppressor Gene Located on Human Chromosome 5. Mol. Cell. Biol. 2011, 31, 1624–1636. [Google Scholar] [CrossRef] [Green Version]
- Grove, C.A.; Walhout, A.J.M. Transcription factor functionality and transcription regulatory networks. Mol. BioSyst. 2008, 4, 309–314. [Google Scholar] [CrossRef]
- Braun, D.M.; Chung, I.; Kepper, N.; Deeg, K.I.; Rippe, K. TelNet - a database for human and yeast genes involved in telomere maintenance. BMC Genet. 2018, 19, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Gunkel, M.; Chung, I.; Wörz, S.; Deeg, K.I.; Simon, R.; Sauter, G.; Jones, D.T.; Korshunov, A.; Rohr, K.; Erfle, H.; et al. Quantification of telomere features in tumor tissue sections by an automated 3D imaging-based workflow. Methods 2016, 114, 60–73. [Google Scholar] [CrossRef]
- Akincilar, S.C.; Unal, B.; Tergaonkar, V. Reactivation of telomerase in cancer. Cell. Mol. Life Sci. 2016, 73, 1659–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wollmann, T.I.; Ivanova, J.; Gunkel, M.; Chung, I.; Erfle, H.; Rippe, K.; Rohr, K. Multi-channel deep transfer learning for nuclei segmentation in glioblastoma cell tissue images. In Bildverarbeitung für die Medizin, Informatik Aktuell; Springer: Berlin/Heidelberg, Germany, 2018; pp. 316–321. [Google Scholar]
- Ulman, V.; Maska, M.; Magnusson, K.E.G.; Ronneberger, O.; Haubold, C.; Harder, N.; Matula, P.; Matula, P.; Svoboda, D.; Radojevic, M.; et al. An objective comparison of cell-tracking algorithms. Nat. Methods 2017, 14, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Worz, S.; Sander, P.; Pfannmoller, M.; Rieker, R.J.; Joos, S.; Mechtersheimer, G.; Boukamp, P.; Lichter, P.; Rohr, K. 3d geometry-based quantification of colocalizations in multichannel 3d microscopy images of human soft tissue tumors. IEEE Trans Med. Imaging 2010, 29, 1474–1484. [Google Scholar] [CrossRef]
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Mangino, M.; Aviv, A.; Spector, T.; Durbin, R.; Consortium, U.K. Estimating telomere length from whole genome sequence data. Nucleic Acids Res 2014, 42, e75. [Google Scholar] [CrossRef] [Green Version]
- De Castro, M.F.; Calle, P.F.; Viloria, A.; Larrocha, C.; Jiménez, M.C. Evaluation of a totally automated alternative system for determining the rate of erythrocyte sedimentation. Sangre 1989, 34, 4–9. [Google Scholar]
- Meyerson, M.; Counter, C.M.; Eaton, E.N.; Ellisen, L.W.; Steiner, P.; Caddle, S.D.; Ziaugra, L.; Beijersbergen, R.; Davidoff, M.J.; Liu, Q.; et al. hEST2, the Putative Human Telomerase Catalytic Subunit Gene, Is Up-Regulated in Tumor Cells and during Immortalization. Cell 1997, 90, 785–795. [Google Scholar] [CrossRef] [Green Version]
- Epstein, J.I.; Feng, Z.; Trock, B.J.; Pierorazio, P.M. Upgrading and Downgrading of Prostate Cancer from Biopsy to Radical Prostatectomy: Incidence and Predictive Factors Using the Modified Gleason Grading System and Factoring in Tertiary Grades. Eur. Urol. 2012, 61, 1019–1024. [Google Scholar] [CrossRef] [Green Version]
- Ohira, T.; Kojima, H.; Kuroda, Y.; Aoki, S.; Inaoka, D.; Osaki, M.; Wanibuchi, H.; Okada, F.; Oshimura, M.; Kugoh, H. PITX1 protein interacts with ZCCHC10 to regulate hTERT mRNA transcription. PLoS ONE 2019, 14, e0217605. [Google Scholar] [CrossRef] [Green Version]
- Qiao, F.; Gong, P.; Song, Y.; Shen, X.; Su, X.; Li, Y.; Wu, H.; Zhao, Z.; Fan, H. Downregulated PITX1 Modulated by MiR-19a-3p Promotes Cell Malignancy and Predicts a Poor Prognosis of Gastric Cancer by Affecting Transcriptionally Activated PDCD5. Cell. Physiol. Biochem. 2018, 46, 2215–2231. [Google Scholar] [CrossRef] [Green Version]
- Ohira, T.; Naohiro, S.; Nakayama, Y.; Osaki, M.; Okada, F.; Oshimura, M.; Kugoh, H. miR-19b regulates hTERT mRNA expression through targeting PITX1 mRNA in melanoma cells. Sci. Rep. 2015, 5, 8201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kragesteen, B.K.; Spielmann, M.; Paliou, C.; Heinrich, V.; Schöpflin, R.; Esposito, A.; Annunziatella, C.; Bianco, S.; Chiariello, A.M.; Jerković, I.; et al. Dynamic 3D chromatin architecture contributes to enhancer specificity and limb morphogenesis. Nat. Genet. 2018, 50, 1463–1473. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, X.; Chen, Y.; Cao, M.; Tang, J.; Zhong, B.; He, M. The PITX gene family as potential biomarkers and therapeutic targets in lung adenocarcinoma. Medicine 2021, 100, e23936. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zhao, C.; Jiang, L.; Lin, S.; Bi, J.; Wei, Q.; Yu, L.; Zhao, L.; Wei, M. High PITX1 expression in lung adenocarcinoma patients is associated with DNA methylation and poor prognosis. Pathology-Res. Pr. 2018, 214, 2046–2053. [Google Scholar] [CrossRef]
- Sailer, V.; Charpentier, A.; Dietrich, J.; Vogt, T.J.; Franzen, A.; Bootz, F.; Dietrich, D.; Schroeck, A. Intragenic DNA methylation of PITX1 and the adjacent long non-coding RNA C5orf66-AS1 are prognostic biomarkers in patients with head and neck squamous cell carcinomas. PLoS ONE 2018, 13, e0192742. [Google Scholar] [CrossRef]
- Kong, G.; Liu, Z.; Wu, K.; Zhang, Y.; Deng, Z.; Feng, W.; Chen, S.; Wang, H. Strong expression of paired-like homeodomain transcription factor 1 (PITX1) is associated with a favorable outcome in human osteosarcoma. Tumor Biol. 2015, 36, 7735–7741. [Google Scholar] [CrossRef]
- Knösel, T.; Chen, Y.; Hotovy, S.; Settmacher, U.; Altendorf-Hofmann, A.; Petersen, I. Loss of desmocollin 1-3 and homeobox genes PITX1 and CDX2 are associated with tumor progression and survival in colorectal carcinoma. Int. J. Color. Dis. 2012, 27, 1391–1399. [Google Scholar] [CrossRef]
- Otsubo, T.; Yamada, K.; Hagiwara, T.; Oshima, K.; Iida, K.; Nishikata, K.; Toyoda, T.; Igari, T.; Nohara, K.; Yamashita, S.; et al. DNA hypermethyation and silencing ofPITX1correlated with advanced stage and poor postoperative prognosis of esophageal squamous cell carcinoma. Oncotarget 2017, 8, 84434–84448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolfschoten, I.G.; van Leeuwen, B.; Berns, K.; Mullenders, J.; Beijersbergen, R.L.; Bernards, R.; Voorhoeve, P.M.; Agami, R. A Genetic Screen Identifies PITX1 as a Suppressor of RAS Activity and Tumorigenicity. Cell 2005, 121, 849–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.X.; Lobie, P.E. Transcriptional activation of p53 by Pitx1. Cell Death Differ. 2007, 14, 1893–1907. [Google Scholar] [CrossRef]
- Dejardin, J.; Kingston, R.E. Purification of Proteins Associated with Specific Genomic Loci. Cell 2009, 136, 175–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovejoy, C.A.; Li, W.; Reisenweber, S.; Thongthip, S.; Bruno, J.; De Lange, T.; De, S.; Petrini, J.; Sung, P.A.; Jasin, M.; et al. Loss of ATRX, Genome Instability, and an Altered DNA Damage Response Are Hallmarks of the Alternative Lengthening of Telomeres Pathway. PLoS Genet. 2012, 8, e1002772. [Google Scholar] [CrossRef] [PubMed]
- Yeager, T.R.; Neumann, A.A.; Englezou, A.; Huschtscha, L.I.; Noble, J.R.; Reddel, R.R. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999, 59, 4175–4179. [Google Scholar]
- Draskovic, I.; Arnoult, N.; Steiner, V.; Bacchetti, S.; Lomonte, P.; Londoño-Vallejo, A. Probing PML body function in ALT cells reveals spatiotemporal requirements for telomere recombination. Proc. Natl. Acad. Sci. USA 2009, 106, 15726–15731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauter, G.; Clauditz, T.; Steurer, S.; Wittmer, C.; Büscheck, F.; Krech, T.; Lutz, F.; Lennartz, M.; Harms, L.; Lawrenz, L.; et al. Integrating Tertiary Gleason 5 Patterns into Quantitative Gleason Grading in Prostate Biopsies and Prostatectomy Specimens. Eur. Urol. 2017, 73, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Schramm, G.; Wiesberg, S.; Diessl, N.; Kranz, A.-L.; Sagulenko, V.; Oswald, M.; Reinelt, G.; Westermann, F.; Eils, R.; König, R. PathWave: Discovering patterns of differentially regulated enzymes in metabolic pathways. Bioinformatics 2010, 26, 1225–1231. [Google Scholar] [CrossRef] [Green Version]
- Dittrich, M.T.; Klau, G.W.; Rosenwald, A.; Dandekar, T.; Müller, T. Identifying functional modules in protein-protein interaction networks: An integrated exact approach. Bioinformatics 2008, 24, i223–i231. [Google Scholar] [CrossRef] [Green Version]
- Saraiva, J.P.; Zubiria-Barrera, C.; Klassert, T.E.; Lautenbach, M.J.; Blaess, M.; Claus, R.A.; Slevogt, H.; König, R. Combination of Classifiers Identifies Fungal-Specific Activation of Lysosome Genes in Human Monocytes. Front. Microbiol. 2017, 8, 2366. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regulators Tumor | Frequency Tumor (n = 300 Models) | Frequency Normal (n = 300 Models) | p-Value ** |
|---|---|---|---|
| PITX1 * | 186 (62%) | 35 (12%) | 1.56 × 10−37 |
| MITF * | 119 (40%) | 28 (9%) | 5.97 × 10−17 |
| AR * | 92 (31%) | 21 (7%) | 1.26 × 10−12 |
| TFAP2C * | 72 (24%) | 11 (4%) | 1.67 × 10−12 |
| E2F2 * | 92 (31%) | 24 (8%) | 1.31 × 10−11 |
| NR2F2 * | 97 (32%) | 27 (9%) | 1.31 × 10−11 |
| SMARCB1 | 88 (29%) | 24 (8%) | 1.15 × 10−10 |
| CEBPA * | 65 (22%) | 20 (7%) | 6.08 × 10−7 |
| BHLHE40 * | 53 (18%) | 16 (5%) | 8.26 × 10−6 |
| CTCF * | 48 (16%) | 15 (5%) | 4.13 × 10−5 |
| ETS1 * | 63 (21%) | 26 (9%) | 7.43 × 10−5 |
| MXI1 | 27 (9%) | 5 (2%) | 1.75 × 10−4 |
| POLR2A | 34 (11%) | 9 (3%) | 2.23 × 10−4 |
| RAD21 | 32 (11%) | 11 (4%) | 2.37 × 10−3 |
| IRF1 * | 31 (10%) | 12 (4%) | 6.38 × 10−3 |
| TFAP2D * | 34 (11%) | 18 (6%) | 3.91 × 10−2 |
| MAX | 36 (12%) | 20 (7%) | 4.62 × 10−2 |
| TERT Regulator | Regulators Used in At Least 20% of the Models | Number of Direct Regulators | Number of TERT Regulators |
|---|---|---|---|
| PITX1 | SMARCC1, TAF1 *, HEY1 *, POLR2A *, FOXO1, HNF4A, ESR1 *, RBBP5, SMAD1, SMARCB1 * | 10 | 5 |
| AR | MAFF, MAFK, ZBTB17, CREB3, GATA2, TCF4, CTCF *, EGR1 * | 8 | 2 |
| MITF | MXI1 *, ZNF263, SMC3, TAL1 *, MYC *, EP300, MAX * | 7 | 4 |
| CTCF | MAX *, PRDM16, YY1, RBBP5, REST *, POU2F2 *, FOXP2, EP300 | 8 | 3 |
| BHLHE40 | ARNTL, HIF1A *, SIN3AK20 *, EGR1 *, NCOR1, AR *, CEBPB, GABPA, ZNF143 | 9 | 4 |
| ETS1 | ETV2, PAX5 *, FOS, CEBPB, USF1, FOXA1, TCF7L2, IRF4, GATA2 | 9 | 1 |
| CEBPA | SP1, CLOCK, IKZF1 *, MYC *, NCOR1, FOXP2, JUN, SREBF1, MAZ * | 9 | 3 |
| E2F2 | E2F4 *, PML, E2F7, MAFK, ELF1, HEY1 *, EBF1, E2F6 *, MAFF, TCF12 * | 10 | 4 |
| NR2F2 | MXI1 *, TP53 *, USF1, E2F4 *, SF1, FOXP2, SIN3AK20 *, ZNF263 | 8 | 4 |
| IRF1 | NFKB.P50.P65 *, IRF2, SPI1, EGR1 *, MYB * | 5 | 3 |
| TFAP2C | TP63, MAX *, RAD21 *, RBPJ, SP1, POU5F1, ZFP36L1, MTA1, E2F1 *, EZH2, SETDB1 | 11 | 3 |
| PITX1 | |||||
|---|---|---|---|---|---|
| Parameter | n Evaluable | Negative (%) | Low (%) | High (%) | p Value |
| All cancers | 15,011 | 38.3 | 57.7 | 4.0 | |
| Tumor stage | <0.0001 | ||||
| pT2 | 9555 | 41.5 | 55.5 | 3.0 | |
| pT3a | 3366 | 34.6 | 60.4 | 5.0 | |
| pT3b-pT4 | 2030 | 30.0 | 63.0 | 7.0 | |
| Gleason grade | <0.0001 | ||||
| ≤3 + 3 | 2794 | 41.8 | 55.3 | 2.8 | |
| 3 + 4 | 7971 | 40.2 | 56.5 | 3.3 | |
| 3 + 4 Tert.5 | 720 | 38.9 | 57.6 | 3.5 | |
| 4 + 3 | 1479 | 30.6 | 62.8 | 6.6 | |
| 4 + 3 Tert.5 | 1056 | 31.3 | 63.5 | 5.2 | |
| ≥4 + 4 | 867 | 28.7 | 61.5 | 9.8 | |
| Lymph node metastasis | <0.0001 | ||||
| N0 | 9067 | 37.7 | 58.0 | 4.3 | |
| N+ | 1121 | 30.2 | 63.2 | 6.6 | |
| Surgical margin | <0.0001 | ||||
| negative | 11,973 | 39.2 | 57.1 | 3.7 | |
| positive | 2985 | 35.1 | 59.8 | 5.1 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poos, A.M.; Schroeder, C.; Jaishankar, N.; Röll, D.; Oswald, M.; Meiners, J.; Braun, D.M.; Knotz, C.; Frank, L.; Gunkel, M.; et al. PITX1 Is a Regulator of TERT Expression in Prostate Cancer with Prognostic Power. Cancers 2022, 14, 1267. https://doi.org/10.3390/cancers14051267
Poos AM, Schroeder C, Jaishankar N, Röll D, Oswald M, Meiners J, Braun DM, Knotz C, Frank L, Gunkel M, et al. PITX1 Is a Regulator of TERT Expression in Prostate Cancer with Prognostic Power. Cancers. 2022; 14(5):1267. https://doi.org/10.3390/cancers14051267
Chicago/Turabian StylePoos, Alexandra M., Cornelia Schroeder, Neeraja Jaishankar, Daniela Röll, Marcus Oswald, Jan Meiners, Delia M. Braun, Caroline Knotz, Lukas Frank, Manuel Gunkel, and et al. 2022. "PITX1 Is a Regulator of TERT Expression in Prostate Cancer with Prognostic Power" Cancers 14, no. 5: 1267. https://doi.org/10.3390/cancers14051267
APA StylePoos, A. M., Schroeder, C., Jaishankar, N., Röll, D., Oswald, M., Meiners, J., Braun, D. M., Knotz, C., Frank, L., Gunkel, M., Spilger, R., Wollmann, T., Polonski, A., Makrypidi-Fraune, G., Fraune, C., Graefen, M., Chung, I., Stenzel, A., Erfle, H., ... Koenig, R. (2022). PITX1 Is a Regulator of TERT Expression in Prostate Cancer with Prognostic Power. Cancers, 14(5), 1267. https://doi.org/10.3390/cancers14051267