
Targeting of RecQ Helicases as a Novel Therapeutic Strategy for Ovarian Cancer

Abstract
:Simple Summary
Abstract
1. Introduction
2. BLM Function, Its Interacting Partners, and Its Implication in Cancer
3. WRN Function, Its Interacting Partners, and Its Implication in Cancer
4. RECQL4 Function, Its Interacting Partners, and Its Implication in Cancer
5. Using Helicase Inhibitors in Combination Treatments with DNA Repair Inhibitors
6. Combination Therapy with a PARP Inhibitor
7. Combination Therapy with an ATR Inhibitor
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Abdel-Monem, M.; Durwald, H.; Hoffmann-Berling, H. Enzymic unwinding of DNA. 2. Chain separation by an ATP-dependent DNA unwinding enzyme. Eur. J. Biochem. 1976, 65, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Monem, M.; Hoffmann-Berling, H. Enzymic unwinding of DNA. 1. Purification and characterization of a DNA-dependent ATPase from Escherichia coli. Eur. J. Biochem. 1976, 65, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Mackay, V.; Linn, S. Selective inhibition of the dnase activity of the recBC enzyme by the DNA binding protein from Escherichia coli. J. Biol. Chem. 1976, 251, 3716–3719. [Google Scholar] [CrossRef]
- Singleton, M.R.; Dillingham, M.S.; Wigley, D.B. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007, 76, 23–50. [Google Scholar] [CrossRef]
- Umate, P.; Tuteja, N.; Tuteja, R. Genome-wide comprehensive analysis of human helicases. Commun. Integr. Biol. 2011, 4, 118–137. [Google Scholar] [CrossRef] [Green Version]
- Puranam, K.L.; Blackshear, P.J. Cloning and characterization of RECQL, a potential human homologue of the Escherichia coli DNA helicase RecQ. J. Biol. Chem. 1994, 269, 29838–29845. [Google Scholar] [CrossRef]
- Ellis, N.A.; Groden, J.; Ye, T.Z.; Straughen, J.; Lennon, D.J.; Ciocci, S.; Proytcheva, M.; German, J. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell 1995, 83, 655–666. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.E.; Oshima, J.; Fu, Y.H.; Wijsman, E.M.; Hisama, F.; Alisch, R.; Matthews, S.; Nakura, J.; Miki, T.; Ouais, S.; et al. Positional cloning of the Werner’s syndrome gene. Science 1996, 272, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Kitao, S.; Ohsugi, I.; Ichikawa, K.; Goto, M.; Furuichi, Y.; Shimamoto, A. Cloning of two new human helicase genes of the RecQ family: Biological significance of multiple species in higher eukaryotes. Genomics 1998, 54, 443–452. [Google Scholar] [CrossRef]
- Balajee, A.S. Human RecQL4 as a Novel Molecular Target for Cancer Therapy. Cytogenet. Genome Res. 2021, 161, 305–327. [Google Scholar] [CrossRef]
- German, J. Bloom syndrome: A mendelian prototype of somatic mutational disease. Medicine 1993, 72, 393–406. [Google Scholar] [CrossRef]
- Epstein, C.J.; Martin, G.M.; Schultz, A.L.; Motulsky, A.G. Werner’s syndrome a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine 1966, 45, 177–221. [Google Scholar] [CrossRef]
- Lauper, J.M.; Krause, A.; Vaughan, T.L.; Monnat, R.J., Jr. Spectrum and risk of neoplasia in Werner syndrome: A systematic review. PLoS ONE 2013, 8, e59709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitao, S.; Shimamoto, A.; Goto, M.; Miller, R.W.; Smithson, W.A.; Lindor, N.M.; Furuichi, Y. Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat. Genet. 1999, 22, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Siitonen, H.A.; Kopra, O.; Kaariainen, H.; Haravuori, H.; Winter, R.M.; Saamanen, A.M.; Peltonen, L.; Kestila, M. Molecular defect of RAPADILINO syndrome expands the phenotype spectrum of RECQL diseases. Hum. Mol. Genet. 2003, 12, 2837–2844. [Google Scholar] [CrossRef] [Green Version]
- Van Maldergem, L.; Siitonen, H.A.; Jalkh, N.; Chouery, E.; De Roy, M.; Delague, V.; Muenke, M.; Jabs, E.W.; Cai, J.; Wang, L.L.; et al. Revisiting the craniosynostosis-radial ray hypoplasia association: Baller-Gerold syndrome caused by mutations in the RECQL4 gene. J. Med. Genet. 2006, 43, 148–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vennos, E.M.; Collins, M.; James, W.D. Rothmund-Thomson syndrome: Review of the world literature. J. Am. Acad. Dermatol. 1992, 27, 750–762. [Google Scholar] [CrossRef]
- Wang, L.L.; Levy, M.L.; Lewis, R.A.; Chintagumpala, M.M.; Lev, D.; Rogers, M.; Plon, S.E. Clinical manifestations in a cohort of 41 Rothmund-Thomson syndrome patients. Am. J. Med. Genet. 2001, 102, 11–17. [Google Scholar] [CrossRef]
- Sun, H.; Karow, J.K.; Hickson, I.D.; Maizels, N. The Bloom’s syndrome helicase unwinds G4 DNA. J. Biol. Chem. 1998, 273, 27587–27592. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Hickson, I.D. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 2003, 426, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Robles, A.I.; Linke, S.P.; Sinogeeva, N.I.; Zhang, R.; Pedeux, R.; Ward, I.M.; Celeste, A.; Nussenzweig, A.; Chen, J.; et al. Functional interaction between BLM helicase and 53BP1 in a Chk1-mediated pathway during S-phase arrest. J. Cell Biol. 2004, 166, 801–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davalos, A.R.; Kaminker, P.; Hansen, R.K.; Campisi, J. ATR and ATM-dependent movement of BLM helicase during replication stress ensures optimal ATM activation and 53BP1 focus formation. Cell Cycle 2004, 3, 1579–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davalos, A.R.; Campisi, J. Bloom syndrome cells undergo p53-dependent apoptosis and delayed assembly of BRCA1 and NBS1 repair complexes at stalled replication forks. J. Cell Biol. 2003, 162, 1197–1209. [Google Scholar] [CrossRef]
- Franchitto, A.; Pichierri, P. Bloom’s syndrome protein is required for correct relocalization of RAD50/MRE11/NBS1 complex after replication fork arrest. J. Cell Biol. 2002, 157, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Michl, J.; Zimmer, J.; Tarsounas, M. Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. EMBO J. 2016, 35, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Beresten, S.F.; van Brabant, A.J.; Ye, T.Z.; Pandolfi, P.P.; Johnson, F.B.; Guarente, L.; Ellis, N.A. Evidence for BLM and Topoisomerase IIIalpha interaction in genomic stability. Hum. Mol. Genet. 2001, 10, 1287–1298. [Google Scholar] [CrossRef]
- Daley, J.M.; Chiba, T.; Xue, X.; Niu, H.; Sung, P. Multifaceted role of the Topo IIIalpha-RMI1-RMI2 complex and DNA2 in the BLM-dependent pathway of DNA break end resection. Nucleic Acids Res. 2014, 42, 11083–11091. [Google Scholar] [CrossRef] [Green Version]
- Xue, X.; Raynard, S.; Busygina, V.; Singh, A.K.; Sung, P. Role of replication protein A in double holliday junction dissolution mediated by the BLM-Topo IIIalpha-RMI1-RMI2 protein complex. J. Biol. Chem. 2013, 288, 14221–14227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shorrocks, A.K.; Jones, S.E.; Tsukada, K.; Morrow, C.A.; Belblidia, Z.; Shen, J.; Vendrell, I.; Fischer, R.; Kessler, B.M.; Blackford, A.N. The Bloom syndrome complex senses RPA-coated single-stranded DNA to restart stalled replication forks. Nat. Commun. 2021, 12, 585. [Google Scholar] [CrossRef]
- Qin, Z.; Bi, L.; Hou, X.M.; Zhang, S.; Zhang, X.; Lu, Y.; Li, M.; Modesti, M.; Xi, X.G.; Sun, B. Human RPA activates BLM’s bidirectional DNA unwinding from a nick. Elife 2020, 9, e54098. [Google Scholar] [CrossRef]
- Moder, M.; Velimezi, G.; Owusu, M.; Mazouzi, A.; Wiedner, M.; Ferreira da Silva, J.; Robinson-Garcia, L.; Schischlik, F.; Slavkovsky, R.; Kralovics, R.; et al. Parallel genome-wide screens identify synthetic viable interactions between the BLM helicase complex and Fanconi anemia. Nat. Commun. 2017, 8, 1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deans, A.J.; West, S.C. FANCM connects the genome instability disorders Bloom’s Syndrome and Fanconi Anemia. Mol. Cell 2009, 36, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.H.; Kusumoto, R.; Opresko, P.L.; Sui, X.; Huang, S.; Nicolette, M.L.; Paull, T.T.; Campisi, J.; Seidman, M.; Bohr, V.A. Collaboration of Werner syndrome protein and BRCA1 in cellular responses to DNA interstrand cross-links. Nucleic Acids Res. 2006, 34, 2751–2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groden, J.; Nakamura, Y.; German, J. Molecular evidence that homologous recombination occurs in proliferating human somatic cells. Proc. Natl. Acad. Sci. USA 1990, 87, 4315–4319. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Santoro, I.M.; McDaniel, L.D.; Nishijima, I.; Mills, M.; Youssoufian, H.; Vogel, H.; Schultz, R.A.; Bradley, A. Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nat. Genet. 2000, 26, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Kluzniak, W.; Wokolorczyk, D.; Rusak, B.; Huzarski, T.; Kashyap, A.; Stempa, K.; Rudnicka, H.; Jakubowska, A.; Szwiec, M.; Morawska, S.; et al. Inherited Variants in BLM and the Risk and Clinical Characteristics of Breast Cancer. Cancers 2019, 11, 1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunniff, C.; Bassetti, J.A.; Ellis, N.A. Bloom’s Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition. Mol. Syndromol. 2017, 8, 4–23. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Kaur, E.; Agrawal, R.; Sengupta, S. Functions of BLM Helicase in Cells: Is It Acting Like a Double-Edged Sword? Front. Genet. 2021, 12, 634789. [Google Scholar] [CrossRef]
- Alzahrani, F.A.; Ahmed, F.; Sharma, M.; Rehan, M.; Mahfuz, M.; Baeshen, M.N.; Hawsawi, Y.; Almatrafi, A.; Alsagaby, S.A.; Kamal, M.A.; et al. Investigating the pathogenic SNPs in BLM helicase and their biological consequences by computational approach. Sci. Rep. 2020, 10, 12377. [Google Scholar] [CrossRef]
- Norquist, B.M.; Brady, M.F.; Harrell, M.I.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Burger, R.A.; Tewari, K.S.; et al. Mutations in Homologous Recombination Genes and Outcomes in Ovarian Carcinoma Patients in GOG 218: An NRG Oncology/Gynecologic Oncology Group Study. Clin. Cancer Res. 2018, 24, 777–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birkbak, N.J.; Li, Y.; Pathania, S.; Greene-Colozzi, A.; Dreze, M.; Bowman-Colin, C.; Sztupinszki, Z.; Krzystanek, M.; Diossy, M.; Tung, N.; et al. Overexpression of BLM promotes DNA damage and increased sensitivity to platinum salts in triple-negative breast and serous ovarian cancers. Ann. Oncol. 2018, 29, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.D.; Shen, J.C.; Kamath-Loeb, A.S.; Blank, A.; Sopher, B.L.; Martin, G.M.; Oshima, J.; Loeb, L.A. The Werner syndrome protein is a DNA helicase. Nat. Genet. 1997, 17, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Mohaghegh, P.; Karow, J.K.; Brosh, R.M., Jr.; Bohr, V.A.; Hickson, I.D. The Bloom’s and Werner’s syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 2001, 29, 2843–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doherty, K.M.; Sommers, J.A.; Gray, M.D.; Lee, J.W.; von Kobbe, C.; Thoma, N.H.; Kureekattil, R.P.; Kenny, M.K.; Brosh, R.M., Jr. Physical and functional mapping of the replication protein a interaction domain of the werner and bloom syndrome helicases. J. Biol. Chem. 2005, 280, 29494–29505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.C.; Lao, Y.; Kamath-Loeb, A.; Wold, M.S.; Loeb, L.A. The N-terminal domain of the large subunit of human replication protein A binds to Werner syndrome protein and stimulates helicase activity. Mech. Ageing Dev. 2003, 124, 921–930. [Google Scholar] [CrossRef]
- Lebel, M.; Spillare, E.A.; Harris, C.C.; Leder, P. The Werner syndrome gene product co-purifies with the DNA replication complex and interacts with PCNA and topoisomerase I. J. Biol. Chem. 1999, 274, 37795–37799. [Google Scholar] [CrossRef] [Green Version]
- Kamath-Loeb, A.S.; Johansson, E.; Burgers, P.M.; Loeb, L.A. Functional interaction between the Werner Syndrome protein and DNA polymerase delta. Proc. Natl. Acad. Sci. USA 2000, 97, 4603–4608. [Google Scholar] [CrossRef] [Green Version]
- Baynton, K.; Otterlei, M.; Bjoras, M.; von Kobbe, C.; Bohr, V.A.; Seeberg, E. WRN interacts physically and functionally with the recombination mediator protein RAD52. J. Biol. Chem. 2003, 278, 36476–36486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, W.H.; von Kobbe, C.; Opresko, P.L.; Arthur, L.M.; Komatsu, K.; Seidman, M.M.; Carney, J.P.; Bohr, V.A. Linkage between Werner syndrome protein and the Mre11 complex via Nbs1. J. Biol. Chem. 2004, 279, 21169–21176. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.H.; Muftic, D.; Muftuoglu, M.; Dawut, L.; Morris, C.; Helleday, T.; Shiloh, Y.; Bohr, V.A. WRN is required for ATM activation and the S-phase checkpoint in response to interstrand cross-link-induced DNA double-strand breaks. Mol. Biol. Cell 2008, 19, 3923–3933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Li, B.; Gray, M.D.; Oshima, J.; Mian, I.S.; Campisi, J. The premature ageing syndrome protein, WRN, is a 3’-->5’ exonuclease. Nat. Genet. 1998, 20, 114–116. [Google Scholar] [CrossRef]
- Kamath-Loeb, A.S.; Shen, J.C.; Schmitt, M.W.; Loeb, L.A. The Werner syndrome exonuclease facilitates DNA degradation and high fidelity DNA polymerization by human DNA polymerase delta. J. Biol. Chem. 2012, 287, 12480–12490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, E.M.; Shibue, T.; McFarland, J.M.; Gaeta, B.; Ghandi, M.; Dumont, N.; Gonzalez, A.; McPartlan, J.S.; Li, T.; Zhang, Y.; et al. WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature 2019, 568, 551–556. [Google Scholar] [CrossRef] [Green Version]
- Aaltonen, L.A.; Peltomaki, P.; Leach, F.S.; Sistonen, P.; Pylkkanen, L.; Mecklin, J.P.; Jarvinen, H.; Powell, S.M.; Jen, J.; Hamilton, S.R.; et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993, 260, 812–816. [Google Scholar] [CrossRef] [PubMed]
- Ionov, Y.; Peinado, M.A.; Malkhosyan, S.; Shibata, D.; Perucho, M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993, 363, 558–561. [Google Scholar] [CrossRef]
- Thibodeau, S.N.; Bren, G.; Schaid, D. Microsatellite instability in cancer of the proximal colon. Science 1993, 260, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Bonneville, R.; Krook, M.A.; Kautto, E.A.; Miya, J.; Wing, M.R.; Chen, H.Z.; Reeser, J.W.; Yu, L.; Roychowdhury, S. Landscape of Microsatellite Instability Across 39 Cancer Types. JCO Precis. Oncol. 2017, 1, 1–15. [Google Scholar] [CrossRef]
- Behan, F.M.; Iorio, F.; Picco, G.; Goncalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef]
- van Wietmarschen, N.; Sridharan, S.; Nathan, W.J.; Tubbs, A.; Chan, E.M.; Callen, E.; Wu, W.; Belinky, F.; Tripathi, V.; Wong, N.; et al. Repeat expansions confer WRN dependence in microsatellite-unstable cancers. Nature 2020, 586, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Sommers, J.A.; Shoemaker, R.H.; Brosh, R.M., Jr. Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc. Natl. Acad. Sci. USA 2011, 108, 1525–1530. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, M.; Banerjee, T.; Sommers, J.A.; Iannascoli, C.; Pichierri, P.; Shoemaker, R.H.; Brosh, R.M., Jr. Werner syndrome helicase has a critical role in DNA damage responses in the absence of a functional fanconi anemia pathway. Cancer Res. 2013, 73, 5497–5507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommers, J.A.; Kulikowicz, T.; Croteau, D.L.; Dexheimer, T.; Dorjsuren, D.; Jadhav, A.; Maloney, D.J.; Simeonov, A.; Bohr, V.A.; Brosh, R.M., Jr. A high-throughput screen to identify novel small molecule inhibitors of the Werner Syndrome Helicase-Nuclease (WRN). PLoS ONE 2019, 14, e0210525. [Google Scholar] [CrossRef] [Green Version]
- Arora, A.; Abdel-Fatah, T.M.; Agarwal, D.; Doherty, R.; Moseley, P.M.; Aleskandarany, M.A.; Green, A.R.; Ball, G.; Alshareeda, A.T.; Rakha, E.A.; et al. Transcriptomic and Protein Expression Analysis Reveals Clinicopathological Significance of Bloom Syndrome Helicase (BLM) in Breast Cancer. Mol. Cancer Ther. 2015, 14, 1057–1065. [Google Scholar] [CrossRef] [Green Version]
- Green, A.R.; Aleskandarany, M.A.; Ali, R.; Hodgson, E.G.; Atabani, S.; De Souza, K.; Rakha, E.A.; Ellis, I.O.; Madhusudan, S. Clinical Impact of Tumor DNA Repair Expression and T-cell Infiltration in Breast Cancers. Cancer Immunol. Res. 2017, 5, 292–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Chen, H.; Yang, Y.; Xu, C.; Zhou, J.; Zhou, J.; Chen, Y. Distinct prognosis of mRNA expression of the five RecQ DNA-helicase family members—RECQL, BLM, WRN, RECQL4, and RECQL5—In patients with breast cancer. Cancer Manag. Res. 2018, 10, 6649–6668. [Google Scholar] [CrossRef] [Green Version]
- Arora, A.; Agarwal, D.; Abdel-Fatah, T.M.; Lu, H.; Croteau, D.L.; Moseley, P.; Aleskandarany, M.A.; Green, A.R.; Ball, G.; Rakha, E.A.; et al. RECQL4 helicase has oncogenic potential in sporadic breast cancers. J. Pathol. 2016, 238, 495–501. [Google Scholar] [CrossRef]
- Thomassen, M.; Tan, Q.; Kruse, T.A. Gene expression meta-analysis identifies chromosomal regions and candidate genes involved in breast cancer metastasis. Breast Cancer Res. Treat. 2009, 113, 239–249. [Google Scholar] [CrossRef] [Green Version]
- Das, M.; Prasad, S.B.; Yadav, S.S.; Govardhan, H.B.; Pandey, L.K.; Singh, S.; Pradhan, S.; Narayan, G. Over expression of minichromosome maintenance genes is clinically correlated to cervical carcinogenesis. PLoS ONE 2013, 8, e69607. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Zeller, C.; Masrour, N.; Siddiqui, N.; Paul, J.; Brown, R. Promoter CpG island methylation of genes in key cancer pathways associates with clinical outcome in high-grade serous ovarian cancer. Clin. Cancer Res. 2013, 19, 5788–5797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Li, Y.; Zhao, C.; Peng, J.; Song, K.; Chen, L.; Zhang, P.; Ma, H.; Yuan, C.; Yan, S.; et al. RECQL4, Negatively Regulated by miR-10a-5p, Facilitates Cell Proliferation and Invasion via MAFB in Ovarian Cancer. Front. Oncol. 2020, 10, 524128. [Google Scholar] [CrossRef] [PubMed]
- Savva, C.; Sadiq, M.; Sheikh, O.; Karim, S.; Trivedi, S.; Green, A.R.; Rakha, E.A.; Madhusudan, S.; Arora, A. Werner Syndrome Protein Expression in Breast Cancer. Clin. Breast Cancer 2021, 21, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Croteau, D.L.; Rossi, M.L.; Canugovi, C.; Tian, J.; Sykora, P.; Ramamoorthy, M.; Wang, Z.M.; Singh, D.K.; Akbari, M.; Kasiviswanathan, R.; et al. RECQL4 localizes to mitochondria and preserves mitochondrial DNA integrity. Aging Cell 2012, 11, 456–466. [Google Scholar] [CrossRef] [Green Version]
- De, S.; Kumari, J.; Mudgal, R.; Modi, P.; Gupta, S.; Futami, K.; Goto, H.; Lindor, N.M.; Furuichi, Y.; Mohanty, D.; et al. RECQL4 is essential for the transport of p53 to mitochondria in normal human cells in the absence of exogenous stress. J. Cell Sci. 2012, 125, 2509–2522. [Google Scholar] [CrossRef] [Green Version]
- Croteau, D.L.; Singh, D.K.; Hoh Ferrarelli, L.; Lu, H.; Bohr, V.A. RECQL4 in genomic instability and aging. Trends Genet. 2012, 28, 624–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, M.D.; Duquette, M.L.; Shiels, J.C.; Maizels, N. A conserved G4 DNA binding domain in RecQ family helicases. J. Mol. Biol. 2006, 358, 1071–1080. [Google Scholar] [CrossRef]
- Xu, X.; Liu, Y. Dual DNA unwinding activities of the Rothmund-Thomson syndrome protein, RECQ4. EMBO J. 2009, 28, 568–577. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Shamanna, R.A.; Keijzers, G.; Anand, R.; Rasmussen, L.J.; Cejka, P.; Croteau, D.L.; Bohr, V.A. RECQL4 Promotes DNA End Resection in Repair of DNA Double-Strand Breaks. Cell Rep. 2016, 16, 161–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, H.; Nie, L.; Chi, Z.; Liu, J.; Guo, D.; Lu, X.; Hei, T.K.; Balajee, A.S.; Zhao, Y. RecQL4 helicase amplification is involved in human breast tumorigenesis. PLoS ONE 2013, 8, e69600. [Google Scholar] [CrossRef] [Green Version]
- Datta, A.; Brosh, R.M., Jr. New Insights Into DNA Helicases as Druggable Targets for Cancer Therapy. Front. Mol. Biosci. 2018, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Dziadkowiec, K.N.; Gasiorowska, E.; Nowak-Markwitz, E.; Jankowska, A. PARP inhibitors: Review of mechanisms of action and BRCA1/2 mutation targeting. Prz. Menopauzalny 2016, 15, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Tew, W.P.; Lacchetti, C.; Ellis, A.; Maxian, K.; Banerjee, S.; Bookman, M.; Jones, M.B.; Lee, J.M.; Lheureux, S.; Liu, J.F.; et al. PARP Inhibitors in the Management of Ovarian Cancer: ASCO Guideline. J. Clin. Oncol. 2020, 38, 3468–3493. [Google Scholar] [CrossRef]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(ADP)-ribose polymerase inhibition: Frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol. 2010, 28, 2512–2519. [Google Scholar] [CrossRef]
- Dias, M.P.; Moser, S.C.; Ganesan, S.; Jonkers, J. Understanding and overcoming resistance to PARP inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 773–791. [Google Scholar] [CrossRef]
- Haynes, B.; Murai, J.; Lee, J.M. Restored replication fork stabilization, a mechanism of PARP inhibitor resistance, can be overcome by cell cycle checkpoint inhibition. Cancer Treat. Rev. 2018, 71, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bol, G.M.; Vesuna, F.; Xie, M.; Zeng, J.; Aziz, K.; Gandhi, N.; Levine, A.; Irving, A.; Korz, D.; Tantravedi, S.; et al. Targeting DDX3 with a small molecule inhibitor for lung cancer therapy. EMBO Mol. Med. 2015, 7, 648–669. [Google Scholar] [CrossRef]
- Heerma van Voss, M.R.; Brilliant, J.D.; Vesuna, F.; Bol, G.M.; van der Wall, E.; van Diest, P.J.; Raman, V. Combination treatment using DDX3 and PARP inhibitors induces synthetic lethality in BRCA1-proficient breast cancer. Med. Oncol. 2017, 34, 33. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Biswas, K.; Sommers, J.A.; Thompson, H.; Awate, S.; Nicolae, C.M.; Thakar, T.; Moldovan, G.L.; Shoemaker, R.H.; Sharan, S.K.; et al. WRN helicase safeguards deprotected replication forks in BRCA2-mutated cancer cells. Nat. Commun. 2021, 12, 6561. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Minasian, L.; Kohn, E.C. New strategies in ovarian cancer treatment. Cancer 2019, 125 (Suppl 24), 4623–4629. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [Green Version]
- Wagner, J.M.; Kaufmann, S.H. Prospects for the Use of ATR Inhibitors to Treat Cancer. Pharmaceuticals 2010, 3, 1311–1334. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; George, E.; Ragland, R.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.; Herlyn, M.; Brown, E.; Simpkins, F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef] [Green Version]

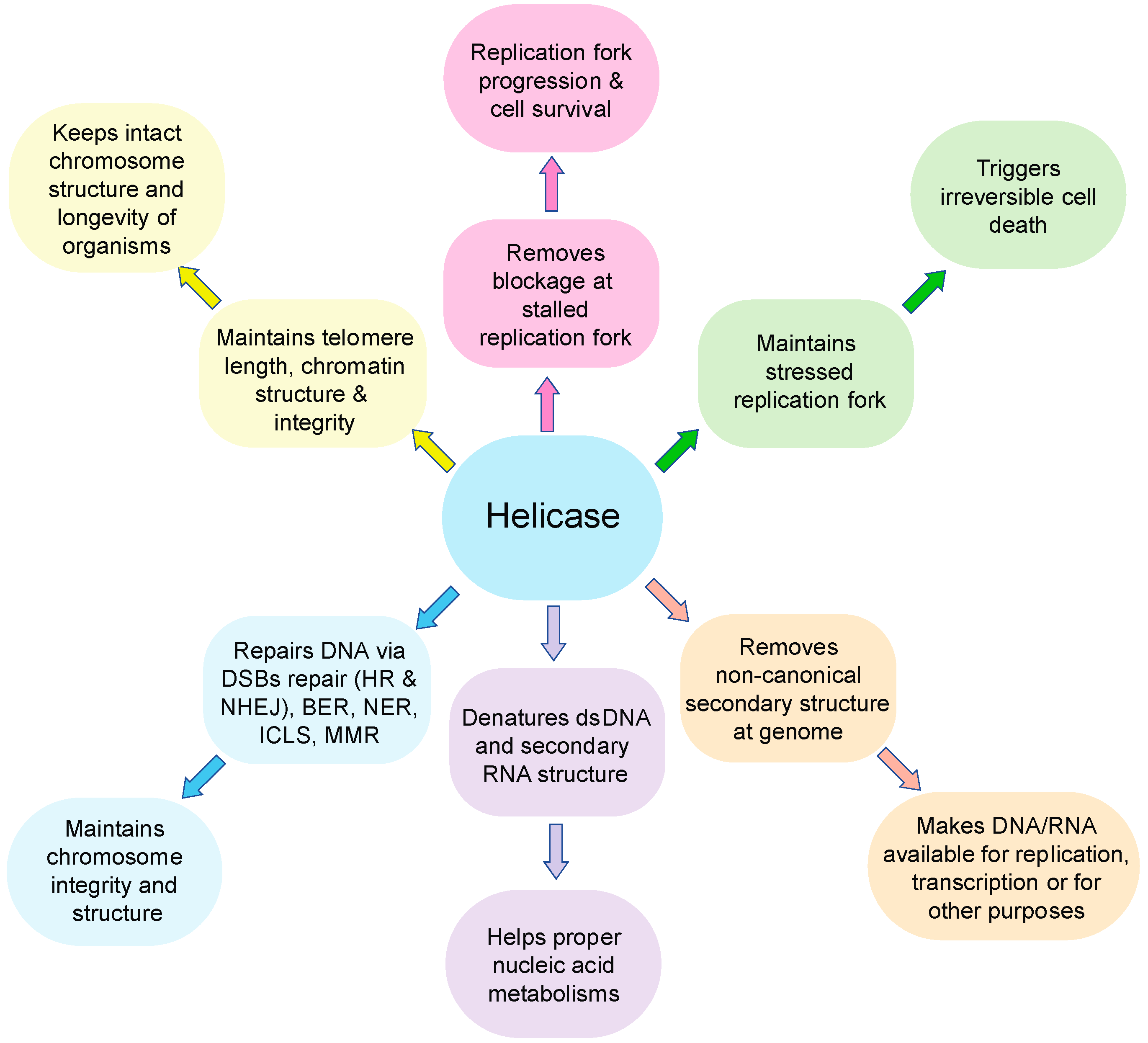
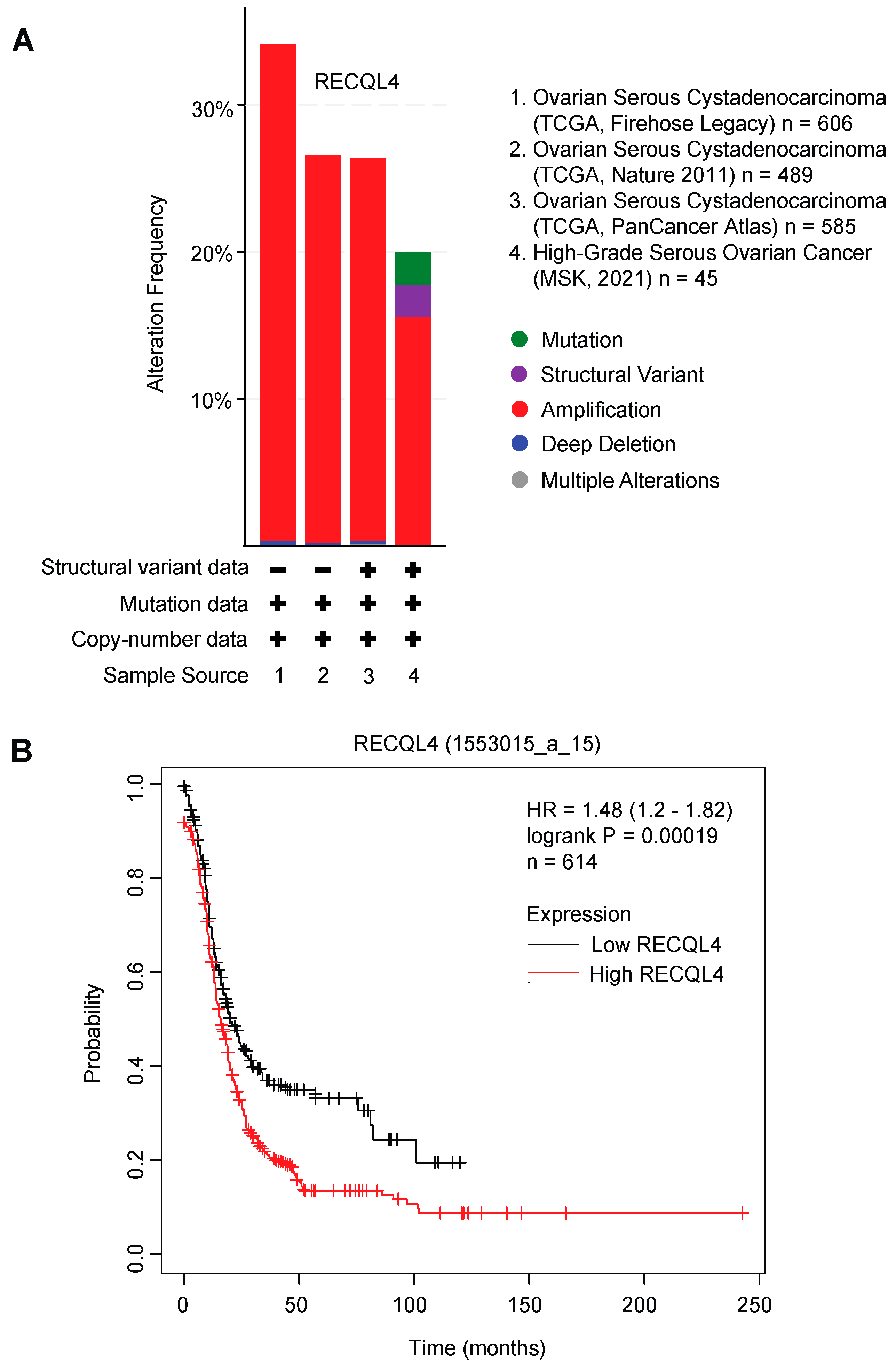

{kind=link}
{kind=link}
{kind=link}
| Cancer Subtypes | Study Setting | Key Findings Related to BLM | Ref. |
|---|---|---|---|
| Newly diagnosed, stage III or IV ovarian, fallopian tube, or primary peritoneal carcinoma | Post-hoc exploratory biomarker analysis from GOG-0218 (NCT00262847), a phase III trial. | Homologous recombination repair (HRR) mutations were found in 25.7% of tissue samples. BLM mutation was found in 1.6% of these patients. | [41] |
| Arm 1: carboplatin plus paclitaxel Arm 2: carboplatin plus paclitaxel and concurrent bevacizumab Arm 3: carboplatin plus paclitaxel and concurrent bevacizumab followed by bevacizumab maintenance | BRCA wild type, HRR mutations were associated with increased progression-free survival (PFS) and overall survival (OS) independent of treatment. However, no difference for PFS was identified by addition of bevacizumab between patients with or without HRR mutation. | ||
| Stage II or III triple negative breast cancer (TNBC) | Post-hoc exploratory biomarker analyses of two, single arm, neoadjuvant phase II trials: | BLM copy number gain was found in 33% of platinum-sensitive and 0% in resistant tumors in trial 1 and 44% of combination therapy-sensitive and 12% in resistant tumors in trial 2. | [42] |
| Trial 1 (NCT00148694) evaluated cisplatin Trial 2 (NCT00580333) evaluated cisplatin and bevacizumab | BLM mRNA levels were higher in cisplatin-sensitive tumors compared to resistant tumors. Stratification of results by bevacizumab was not given in this article. |
| Biomarker | Cancer Subtype | Study Setting | Key Findings Related to BLM, WRN, or RECQL4 | Ref. |
|---|---|---|---|---|
| BLM | Breast Cancer (BC) | Retrospective study of BLM mRNA expression in BC (n= 1950) and in publicly available external BC dataset (n = 2413). BLM protein level was also evaluated in another BC dataset (n = 1650) and 20 normal breast tissues. | High BLM mRNA expression was associated with aggressive clinicopathological features and poor survival. | [65] |
| At a protein level, high cytoplasmic BLM (53% of tumors) and low nuclear BLM (54% of tumors) were associated with aggressive phenotypes. Strong nuclear BLM expression was found in 95% of normal breast tissues. | ||||
| BLM, WRN, RECQL4 | Breast cancer (BC) | Retrospective study of 1269 invasive BC. Of which, 1032 were positive for tumor-infiltrating CD8+ T lymphocytes (TILs), and 237 cases were negative for CD8+ TILs. Independent ER- BC cohort was used for validation (n = 279). | BLM and RECQL4 protein expressions were not associated with survival in CD8+ TIL+ or CD8+ TIL- BCs. | [66] |
| Low WRN protein expression was associated with poor survival in CD8+ TIL- BCs, but not in CD8+ TIL+ BCs. | ||||
| BLM, WRN, RECQL4 | BC | Retrospective study of gene expression data and clinical outcomes from a publicly available dataset on BC with relapse-free survival (RFS) (n = 3955), overall survival (OS) (n = 1402), distant metastasis-free survival (DMFS) (n = 1747), and post-progression survival (PPS) (n = 414). Additional BC samples (n = 160) were used for IHC staining. | High BLM mRNA levels were associated with worse DMFS but were not correlated with OS, RFS, or PPS. | [67] |
| High WRN mRNA levels were associated with better OS and better but were not correlated with DMFS or PPS. | ||||
| High RECQL4 mRNA levels were associated with worse OS, DMFS and RFS, and moderately correlated with poor PPS. | ||||
| Since WRN and RECQL4 mRNA expressions were associated with OS, WRN and RECQL4 protein expressions were tested for association with OS. High levels of WRN protein level were associated with increased OS while high RECQL4 protein level was associated with reduced OS. | ||||
| RECQL4 | BC | Retrospective study of independent cohorts of general BC to study copy number changes (n = 1970), mRNA expression (n = 1977), protein levels (n = 1902), and BC incidence in type II Rothmund-Thomson syndrome (RTS) (n = 58). | RECQL4 copy number gain and amplification were found in 27.6% and 3% of tumors, respectively. ER- tumors showed higher likelihood of gain or amplification of RECQL4 compared to ER+ tumors. | [68] |
| RECQL4 mRNA expressions were high in 51% of tumors. ER- tumors had higher RECQL4 mRNA expressions compared to ER+ tumors. | ||||
| RECQL4 protein expression had complex subcellular localizations in BC. RECQL4 staining were exclusively found in 17.6% of nucleus, 23.4% in cytoplasm, 24.8% in both nucleus and cytoplasm, or 34.2% with absence of staining. | ||||
| No increased incidence of BC was found in type II RTS patients. | ||||
| RECQL4 | BC | Meta-analysis of gene expression data from eight public datasets of BC patients (n = 1366 total with some possible overlap between datasets) | Differential expression of genes analysis showed that RECQL4 was differentially upregulated in metastatic versus non-metastatic tumors. | [69] |
| RECQL4 | Cervical cancer | Cross-sectional comparative study of primary tumor biopsy (n = 60) and hysterectomized control patients (n = 30) | RECQL4 mRNA levels were higher in tumor samples (n = 30) than in control samples (n = 60), but they did not correlate with tumor stage. | [70] |
| RECQL4 | Stage III or IV High-grade serous ovarian cancer (HGSOC) | Prespecified post-hoc exploratory biomarker study of newly diagnosed HGSOC patients who either received platinum alone (n = 42), combination of platinum and taxane (n = 85), or other platinum-based treatment (n = 16). A public dataset (TCGA) of HGSOC DNA containing methylation data (n = 311) was used for validation. | Hypermethylation of RECQL4 promoter was associated with increased hazard of disease progression in the prospective cohorts and in the TCGA dataset of HGSOC. | [71] |
| RECQL4 | Ovarian cancer (OC) | Retrospective cohort study of OC patients (n = 157) and fallopian tube (FT) tissue (n = 54) from benign tumors of patients undergoing hysterectomy and adnexectomy for tissue microarray study. Fresh-frozen OC (n = 40) and normal FT tissues (n = 20) were used to measure RECQL4 mRNA and protein expressions. | RECQL4 mRNA levels were about 10-fold higher in OCs compared to normal FT tissues. | [72] |
| 60.5% of patients had high nuclear RECQL4 expression. | ||||
| High RECQL4 protein expression was associated with poor OS, cisplatin resistance status, serum CA125 level, and omental metastasis. | ||||
| WRN | BRCA-mutant and sporadic BC | Retrospective study of BRCA-mutant (n = 75) and sporadic (n = 1650) invasive BC patients | Low nuclear or cytoplasmic WRN protein expression was associated with poor overall BC-specific survival. | [73] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maity, J.; Horibata, S.; Zurcher, G.; Lee, J.-M. Targeting of RecQ Helicases as a Novel Therapeutic Strategy for Ovarian Cancer. Cancers 2022, 14, 1219. https://doi.org/10.3390/cancers14051219
Maity J, Horibata S, Zurcher G, Lee J-M. Targeting of RecQ Helicases as a Novel Therapeutic Strategy for Ovarian Cancer. Cancers. 2022; 14(5):1219. https://doi.org/10.3390/cancers14051219
Chicago/Turabian StyleMaity, Jyotirindra, Sachi Horibata, Grant Zurcher, and Jung-Min Lee. 2022. "Targeting of RecQ Helicases as a Novel Therapeutic Strategy for Ovarian Cancer" Cancers 14, no. 5: 1219. https://doi.org/10.3390/cancers14051219
APA StyleMaity, J., Horibata, S., Zurcher, G., & Lee, J.-M. (2022). Targeting of RecQ Helicases as a Novel Therapeutic Strategy for Ovarian Cancer. Cancers, 14(5), 1219. https://doi.org/10.3390/cancers14051219