
CHEK2p.I157T Mutation Is Associated with Increased Risk of Adult-Type Ovarian Granulosa Cell Tumors

 , ,
, , 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
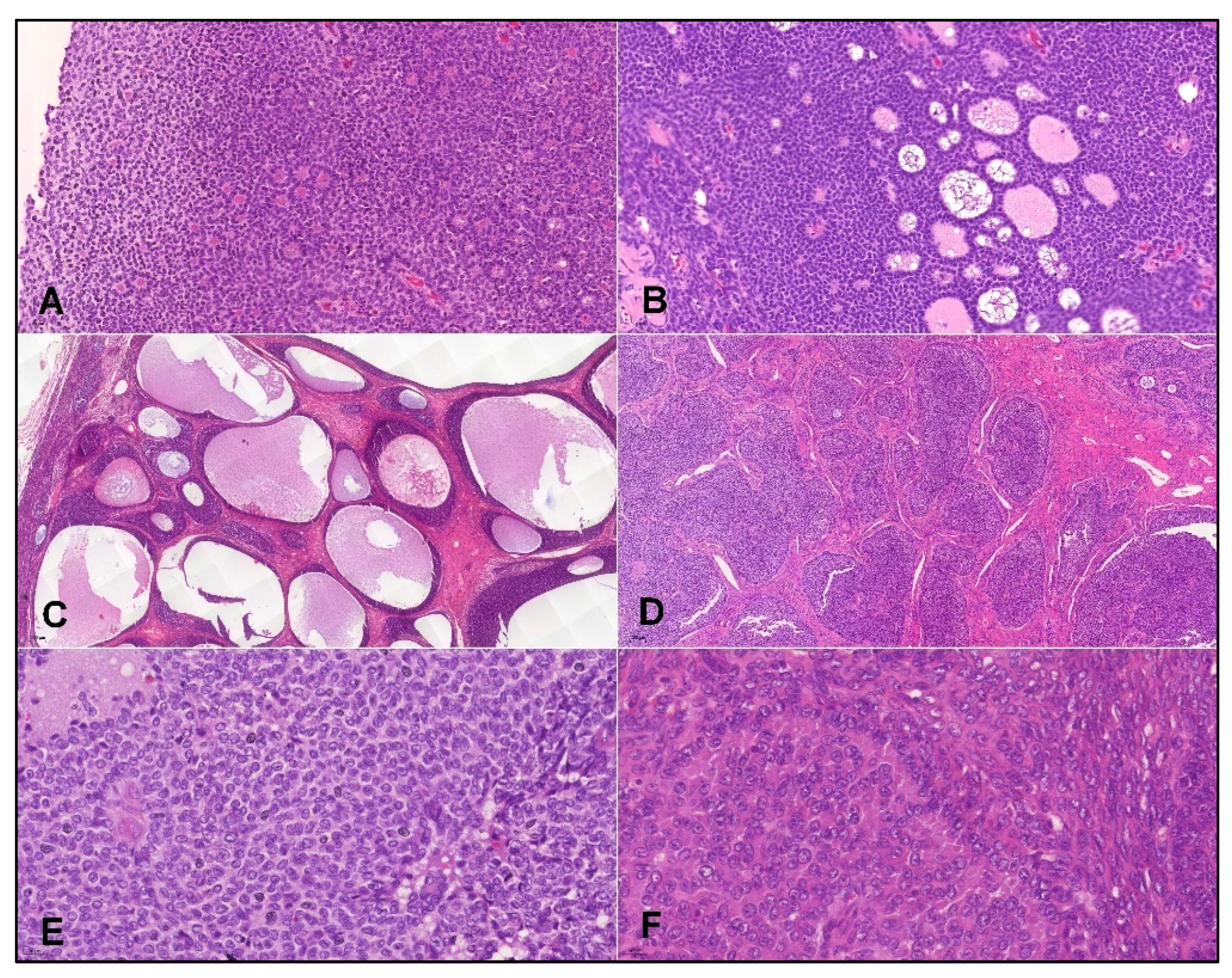
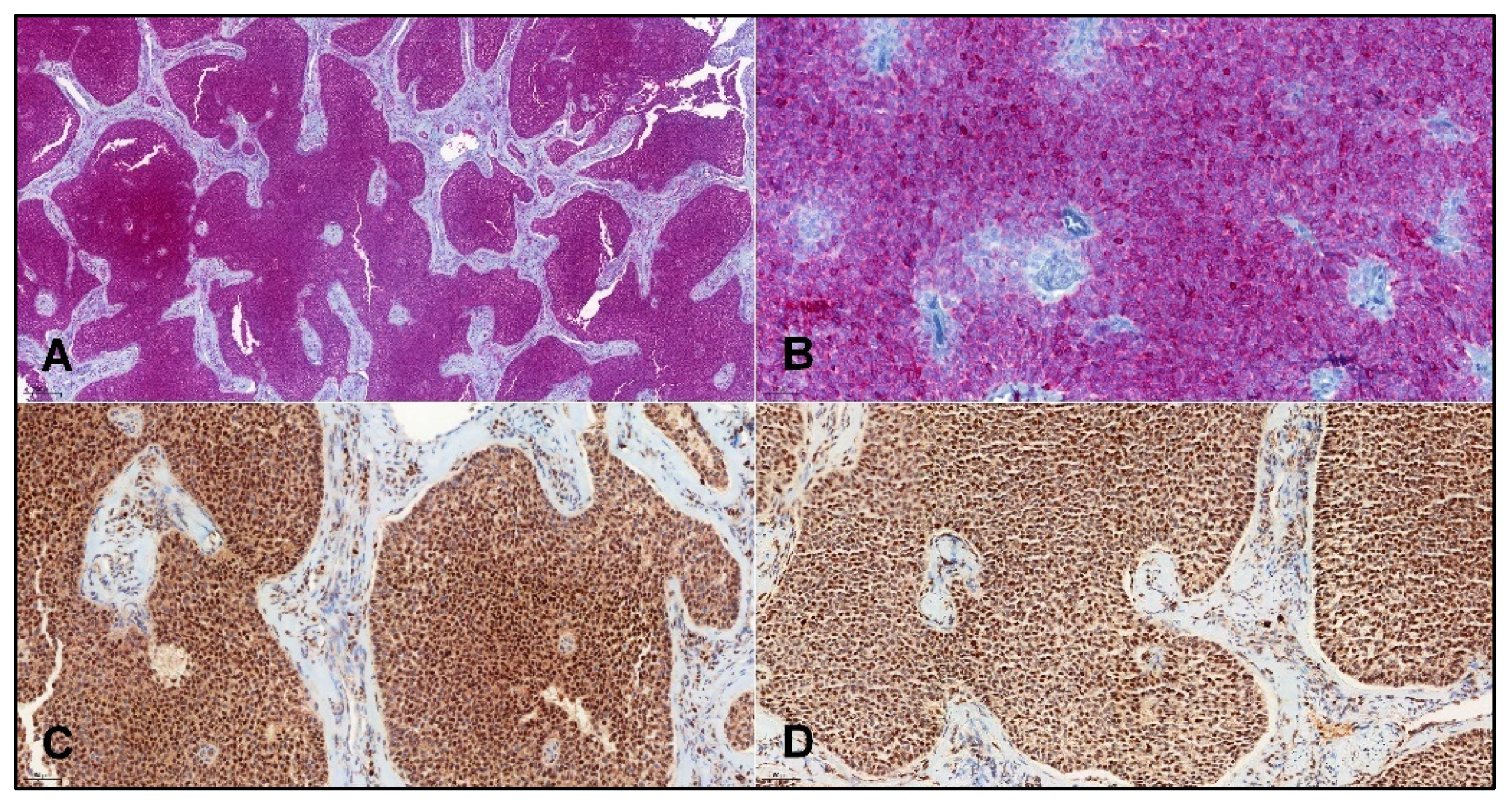
- Histopathology and IHC:
- DNA extraction
- Analysis of FOXL2 and CHEK2 mutations
- Data analysis and statistics
3. Results
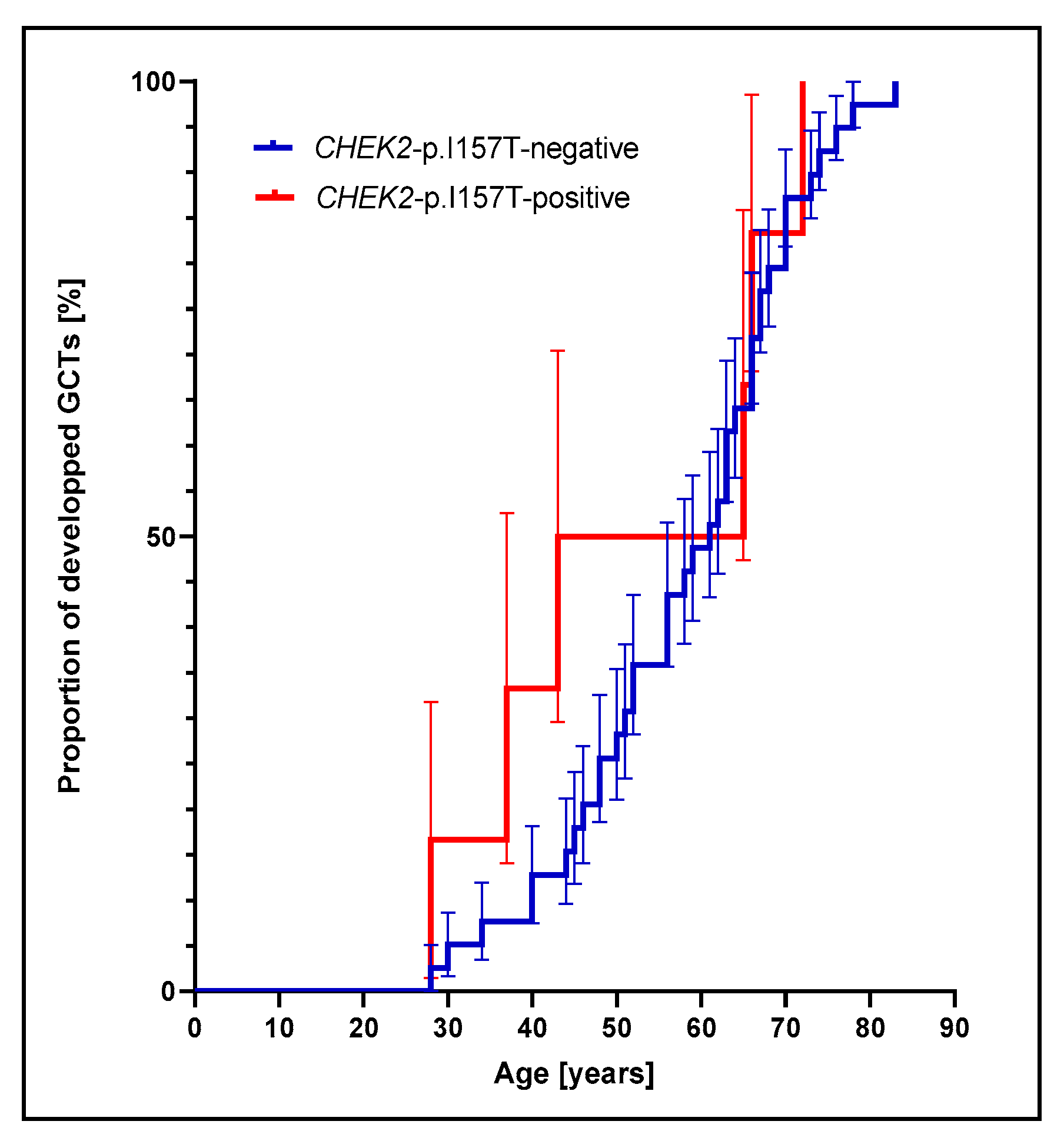
3.1. Prevalence of CHEK2p.I157T Mutation Is Increased among Adult GCT Patients
3.2. Performance of IHC Markers for Detection of Adult GCTs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence 5′-3′ |
|---|---|
| FOXL2-402-F1 | CCTCAACGAGTGCTTCATCA |
| FOXL2-402-R2 | GCCGGTAGTTGCCCTTCT |
| CHEK2 470 F2 | CTCTATTTTAGGAAGTGGGTCC |
| CHEK2 470 R2 | TAGTGACAGTGCAATTTCAGAA |
| CHEK2 1100F | TGTCTTCTTGGACTGGCAGA |
| CHEK2 1100R | GGGGTTCCACATAAGGTTCTC |
References
- Zannini, L.; Delia, D.; Buscemi, G. CHK2 kinase in the DNA damage response and beyond. J. Mol. Cell Biol. 2014, 6, 442–457. [Google Scholar] [CrossRef] [PubMed]
- Stolarova, L.; Kleiblova, P.; Janatova, M.; Soukupova, J.; Zemankova, P.; Macurek, L.; Kleibl, Z. CHEK2 germline variants in cancer predisposition: Stalemate rather than checkmate. Cells 2020, 9, 2675. [Google Scholar] [CrossRef]
- Cybulski, C.; Górski, B.; Huzarski, T.; Masojć, B.; Mierzejewski, M.; Dębniak, T.; Teodorczyk, U.; Byrski, T.; Gronwald, J.; Matyjasik, J.; et al. CHEK2 is a multiorgan cancer susceptibility gene. Am. J. Hum. Genet. 2004, 75, 1131–1135. [Google Scholar] [CrossRef]
- Leedom, T.P.; LaDuca, H.; McFarland, R.; Li, S.; Dolinsky, J.S.; Chao, E.C. Breast cancer risk is similar for CHEK2 founder and non-founder mutation carriers. Cancer Genet. 2016, 209, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Weischer, M.; Bojesen, S.E.; Ellervik, C.; Tybjærg-Hansen, A.; Nordestgaard, B.G. CHEK2*1100delC genotyping for clinical assessment of breast cancer risk: Meta-analyses of 26,000 patient cases and 27,000 controls. J. Clin. Oncol. 2008, 26, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Weischer, M.; Nordestgaard, B.G.; Pharoah, P.; Bolla, M.K.; Nevanlinna, H.; Veer, L.J.V.; Garcia-Closas, M.; Hopper, J.L.; Hall, P.; Andrulis, I.L.; et al. CHEK2*1100delC heterozygosity in women with breast cancer associated with early death, breast cancer–specific death, and increased risk of a second breast cancer. J. Clin. Oncol. 2012, 30, 4308–4316. [Google Scholar] [CrossRef]
- Fletcher, O.; Johnson, N.; dos Santos Silva, I.; Kilpivaara, O.; Aittomäki, K.; Blomqvist, C.; Nevanlinna, H.; Wasielewski, M.; Meijers-Heijerboer, H.; Broeks, A.; et al. Family history, genetic testing, and clinical risk prediction: Pooled analysis of CHEK2*1100delC in 1828 bilateral breast cancers and 7030 controls. Cancer Epidemiol. Biomark. Prev. 2009, 18, 230–234. [Google Scholar] [CrossRef]
- Muranen, T.A.; Blomqvist, C.; Dörk, T.; Jakubowska, A.; Heikkilä, P.; Fagerholm, R.; Greco, D.; Aittomäki, K.; Bojesen, S.E.; Shah, M.; et al. Patient survival and tumor characteristics associated with CHEK2: p.I157T—Findings from the Breast Cancer Association Consortium. Breast Cancer Res. 2016, 18, 98. [Google Scholar] [CrossRef]
- Sutcliffe, E.G.; Stettner, A.R.; Miller, S.A.; Solomon, S.R.; Marshall, M.L.; Roberts, M.E.; Susswein, L.R.; Arvai, K.J.; Klein, R.T.; Murphy, P.D.; et al. Differences in cancer prevalence among CHEK2 carriers identified via multi-gene panel testing. Cancer Genet. 2020, 246–247, 12–17. [Google Scholar] [CrossRef]
- Alexiadis, M.; Rowley, S.M.; Chu, S.; Leung, D.T.; Stewart, C.J.; Amarasinghe, K.C.; Campbell, I.G.; Fuller, P.J. Mutational landscape of ovarian adult granulosa cell tumors from whole exome and targeted TERT promoter sequencing. Mol. Cancer Res. 2018, 17, 177–185. [Google Scholar] [CrossRef]
- Van Dongen, J.J.M.; Langerak, A.; Brüggemann, M.; Evans, P.A.S.; Hummel, M.; Lavender, F.; Delabesse, E.; Davi, F.; Schuuring, E.; Garcia-Sanz, R.; et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: Report of the BIOMED-2 concerted action BMH4-CT98-3936. Leukemia 2003, 17, 2257–2317. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. Basic Local Alignment Search Tool (BLAST); National Center for Biotechnology Information: Bethesda, MD, USA, 1990. [Google Scholar]
- The Genome Aggregation Database. Available online: https://gnomad.broadinstitute.org/about (accessed on 22 October 2021).
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- VassarStats. Website for Statistical Computation. Available online: http://vassarstats.net/ (accessed on 22 October 2021).
- Sullivan, K.M.; Dean, A.; Soe, M.M. OpenEpi: A web-based epidemiologic and statistical calculator for public health. Public Health Rep. 2009, 124, 471–474. [Google Scholar] [CrossRef]
- OpenEpi. Open-Source Epidemiologic Statistics for Public Health, Version 3.01, Updated. 6 April 2013. Available online: https://www.openepi.com/Menu/OE_Menu.htm (accessed on 22 October 2021).
- StatPages. The Interactive Statistical Pages. Available online: https://statpages.info/index.html (accessed on 22 October 2021).
- Shah, S.P.; Köbel, M.; Senz, J.; Morin, R.D.; Clarke, B.A.; Wiegand, K.C.; Leung, G.; Zayed, A.; Mehl, E.; Kalloger, S.E.; et al. Mutation of FOXL2 in granulosa-cell tumors of the ovary. N. Engl. J. Med. 2009, 360, 2719–2729. [Google Scholar] [CrossRef]
- Kottarathil, V.D.; Antony, M.A.; Nair, I.R.; Pavithran, K. Recent advances in granulosa cell tumor ovary: A review. Indian J. Surg. Oncol. 2012, 4, 37–47. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, E.; Mozos, A.; Nakayama, D.; Espinosa, I.; Catasus, L.; Muñoz, J.; Prat, J.; Mu, J. Prognostic significance of FOXL2 mutation and mRNA expression in adult and juvenile granulosa cell tumors of the ovary. Mod. Pathol. 2011, 24, 1360–1367. [Google Scholar] [CrossRef]
- Schumer, S.T.; Cannistra, S.A. Granulosa cell tumor of the ovary. J. Clin. Oncol. 2003, 21, 1180–1189. [Google Scholar] [CrossRef]
- Young, R.H.; Dickersin, G.R.; Scully, R.E. Juvenile granulosa cell tumor of the ovary: A clinicopathological analysis of 125 cases. Am. J. Surg. Pathol. 1984, 8, 575–596. [Google Scholar] [CrossRef]
- Shim, S.-H.; Lee, S.J.; Kim, D.-Y.; Kim, J.; Kim, S.-N.; Kang, S.-B.; Kim, J.-H.; Kim, Y.-M.; Kim, Y.-T.; Nam, J.-H. A long-term follow-up study of 91 cases with ovarian granulosa cell tumors. Anticancer Res. 2014, 34, 1001–1010. [Google Scholar]
- McConechy, M.K.; Färkkilä, A.; Horlings, H.M.; Talhouk, A.; Unkila-Kallio, L.; van Meurs, H.S.; Yang, W.; Rozenberg, N.; Andersson, N.; Zaby, K.; et al. Molecularly defined adult granulosa cell tumor of the ovary: The clinical phenotype. JNCI J. Natl. Cancer Inst. 2016, 108, djw134. [Google Scholar] [CrossRef]
- Jamieson, S.; Fuller, P.J. Molecular pathogenesis of granulosa cell tumors of the ovary. Endocr. Rev. 2012, 33, 109–144. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Hur, S.Y.; Yoo, N.J.; Lee, S.H. Mutational analysis of FOXL2 codon 134 in granulosa cell tumour of ovary and other human cancers. J. Pathol. 2010, 221, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Berman, J.J. Neoplasms: Principles of Development and Diversity; Jones & Bartlett Learning: Burlington, MA, USA, 2009. [Google Scholar]
- Bryk, S.; Pukkala, E.; Martinsen, J.I.; Unkila-Kallio, L.; Tryggvadottir, L.; Sparén, P.; Kjærheim, K.; Weiderpass, E.; Riska, A. Incidence and occupational variation of ovarian granulosa cell tumours in Finland, Iceland, Norway and Sweden during 1953–2012: A longitudinal cohort study. BJOG 2016, 124, 143–149. [Google Scholar] [CrossRef]
- SEER Program. Incidence—SEER Research Data, 13 Registries, November 2020 Sub (1992–2018)—Linked to County Attributes—Time Dependent (1990–2018) Income/Rurality, 1969–2019 Counties, National Cancer Institute, DCCPS, Surveillance Research Program, Released April 2021, Based on the November 2020 Submission. Available online: www.seer.cancer.gov (accessed on 18 January 2022).
- Howe, K.L.; Achuthan, P.; Allen, J.; Alvarez-Jarreta, J.; Ridwan Amode, M.; Armean, I.M.; Azov, A.G.; Bennet, R.; Bhai, J.; Billis, K.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef] [PubMed]
- Boyce, E.; Costaggini, I.; Vitonis, A.; Feltmate, C.; Muto, M.; Berkowitz, R.; Cramer, D.; Horowitz, N. The epidemiology of ovarian granulosa cell tumors: A case-control study. Gynecol. Oncol. 2009, 115, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Nizialek, E.; Lotan, T.L.; Isaacs, W.B.; Yegnasubramanian, S.; Paller, C.J.; Antonarakis, E.S. The somatic mutation landscape of germline CHEK2-altered prostate cancer. J. Clin. Oncol. 2021, 39, 5084. [Google Scholar] [CrossRef]
- Benayoun, B.A.; Georges, A.B.; L’Hôte, D.; Andersson, N.; Dipietromaria, A.; Todeschini, A.-L.; Caburet, S.; Bazin, C.; Anttonen, M.; Veitia, R.A. Transcription factor FOXL2 protects granulosa cells from stress and delays cell cycle: Role of its regulation by the SIRT1 deacetylase. Hum. Mol. Genet. 2011, 20, 1673–1686. [Google Scholar] [CrossRef]



| FOXL2 Status | ||||
|---|---|---|---|---|
| FOXL2p.C134W | FOXL2 Wild-Type | Unknown | ALL | |
| Number of cases | 58 | 11 | 24 | 93 |
| Age at diagnosis (years) | ||||
| Median (range) | 59 (28–83) | 52 (18–81) | 59.5 (32–75) | - |
| CHEK2 mutation p.I157T | ||||
| Positive | 6 | 1 | 0 | 7 |
| Negative | 40 | 6 | 0 | 46 |
| Unknown | 12 | 4 | 24 | 40 |
| CHEK2 mutation c.1100delC | ||||
| Positive | 1 | 0 | 0 | 1 |
| Negative | 39 | 8 | 8 | 55 |
| Unknown | 18 | 3 | 16 | 37 |
| FOXL2 by IHC | ||||
| Positive | 55 | 8 | 20 | 83 |
| Negative | 3 | 3 | 4 | 10 |
| Inhibin by IHC | ||||
| Positive | 50 | 6 | 20 | 76 |
| Negative | 8 | 5 | 4 | 17 |
| Calretinin by IHC | ||||
| Positive | 50 | 6 | 20 | 76 |
| Negative | 8 | 5 | 4 | 17 |
| SF1 by IHC | ||||
| Positive | 49 | 8 | 20 | 77 |
| Negative | 9 | 3 | 4 | 16 |
| FOXL2 | Inhibin | Calretinin | SF1 | |
|---|---|---|---|---|
| Sensitivity (CI95) | 94.8% | 86.2% | 86.2% | 84.5% |
| (85.9–98.6%) | (75.1–92.8%) | (75.1–92.8%) | (73.1–91.6%) | |
| Specificity (CI95) | 27.3% | 45.5% | 45.5% | 27.3% |
| (9.7–56.6%) | (21.3–72.0%) | (21.3–72.0%) | (9.7–56.6%) | |
| Youden’s J (CI95) | 0.221 | 0.317 | 0.317 | 0.118 |
| (−0.009–0.448) | (0.008–0.631) | (0.008–0.631) | (−0.116–0.455) |
| Model Variables (Model 1 and Model 2) | ||||||||
|---|---|---|---|---|---|---|---|---|
| B * | SE | Wald | Df | p-Value | OR | 95% CI for OR | ||
| Lower | Upper | |||||||
| Constant | ||||||||
| Model 1 | −2.24 | 1.22 | 3.38 | 1 | 0.066 | - | - | - |
| Model 2 | −2.41 | 1.21 | 3.97 | 1 | 0.046 | - | - | - |
| Inhibin | ||||||||
| Model 1 | 1.61 | 0.86 | 3.47 | 1 | 0.062 | 5.00 | 0.92 | 27.13 |
| Model 2 | 1.44 | 0.82 | 3.03 | 1 | 0.082 | 4.20 | 0.84 | 21.15 |
| FOXL2 | ||||||||
| Model 1 | 2.52 | 1.02 | 6.17 | 1 | 0.013 | 12.44 | 1.70 | 90.94 |
| Model 2 | 2.36 | 1.00 | 5.63 | 1 | 0.018 | 10.61 | 1.51 | 74.63 |
| SF1 | ||||||||
| Model 1 | −0.67 | 1.05 | 0.42 | 1 | 0.519 | 0.510 | 0.066 | 3.956 |
| Model 2 | NA | NA | NA | NA | NA | NA | NA | NA |
| Calretinin | ||||||||
| Model 1 | 1.61 | 0.86 | 3.47 | 1 | 0.062 | 5.00 | 0.920 | 27.131 |
| Model 2 | 1.44 | 0.82 | 3.03 | 1 | 0.082 | 4.20 | 0.835 | 21.149 |
| Model fit and classification performance | ||||||||
| Omnibus test of model coefficients | Chi-squared | Df | p-value | |||||
| Model 1 | 13.51 | 4 | 0.009 | |||||
| Model 2 | 13.07 | 3 | 0.004 | |||||
| HL test | Chi-squared | Df | p-value | |||||
| Model 1 | 1.13 | 3 | 0.771 | |||||
| Model 2 | 1.74 | 2 | 0.419 | |||||
| -2 Log likelihood | Chi-squared | Df | p-value | |||||
| Model 1 | 47.03 | NA | NA | |||||
| Model 2 | 47.47 | NA | NA | |||||
| Classification results | FOXL2p.C134W | FOXL2wt | FOXL2p.C134W/FOXL2wt | |||||
| Model 1 | Correct | 56 (96.6%) | 3 (27.3%) | 59 (85.5%) | ||||
| Incorrect | 2 | 8 | 10 | |||||
| Youden’s J | 0.238 (CI95: 0.010–0.414) | |||||||
| Model 2 | Correct | 55 (94.8%) | 5 (45.5%) | 60 (87.0%) | ||||
| Incorrect | 3 | 6 | 9 | |||||
| Youden’s J | 0.403 (CI95: 0.104–0.629) | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Švajdler, P.; Vasovčák, P.; Švajdler, M.; Šedivcová, M.; Urbán, V.; Michal, M.; Mezencev, R. CHEK2p.I157T Mutation Is Associated with Increased Risk of Adult-Type Ovarian Granulosa Cell Tumors. Cancers 2022, 14, 1208. https://doi.org/10.3390/cancers14051208
Švajdler P, Vasovčák P, Švajdler M, Šedivcová M, Urbán V, Michal M, Mezencev R. CHEK2p.I157T Mutation Is Associated with Increased Risk of Adult-Type Ovarian Granulosa Cell Tumors. Cancers. 2022; 14(5):1208. https://doi.org/10.3390/cancers14051208
Chicago/Turabian StyleŠvajdler, Peter, Peter Vasovčák, Marián Švajdler, Monika Šedivcová, Veronika Urbán, Michal Michal, and Roman Mezencev. 2022. "CHEK2p.I157T Mutation Is Associated with Increased Risk of Adult-Type Ovarian Granulosa Cell Tumors" Cancers 14, no. 5: 1208. https://doi.org/10.3390/cancers14051208
APA StyleŠvajdler, P., Vasovčák, P., Švajdler, M., Šedivcová, M., Urbán, V., Michal, M., & Mezencev, R. (2022). CHEK2p.I157T Mutation Is Associated with Increased Risk of Adult-Type Ovarian Granulosa Cell Tumors. Cancers, 14(5), 1208. https://doi.org/10.3390/cancers14051208