
STAT1 and STAT3 Exhibit a Crosstalk and Are Associated with Increased Inflammation in Hepatocellular Carcinoma

 , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Tissue Microarrays
2.2. Tissue Microarray Construction
2.3. Immunohistochemistry
2.4. Tissue Microarray Evaluation
2.5. Cell Lines and siRNA-Mediated Knockdown
2.6. RNA Extraction, cDNA Synthesis and Semi-Quantitative Reverse-Transcription Polymerase Chain Reaction (RT-PCR)
2.7. Protein Isolation and Western Blot
2.8. Luciferase Reporter Assay
2.9. Statistical Analysis
3. Results
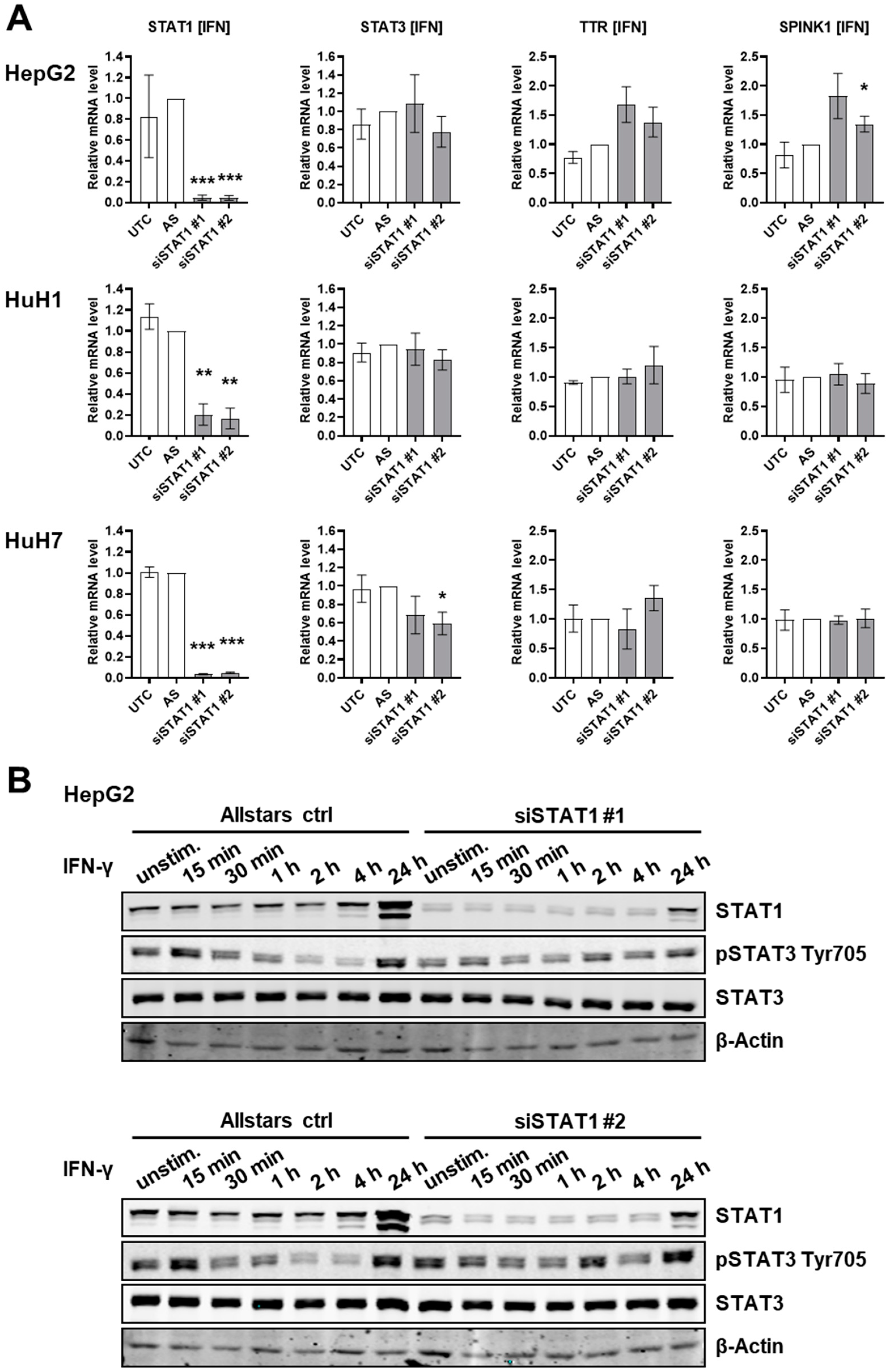
3.1. STAT1 Depletion Does Not Induce an IL-6-Like Response in HCC Cells
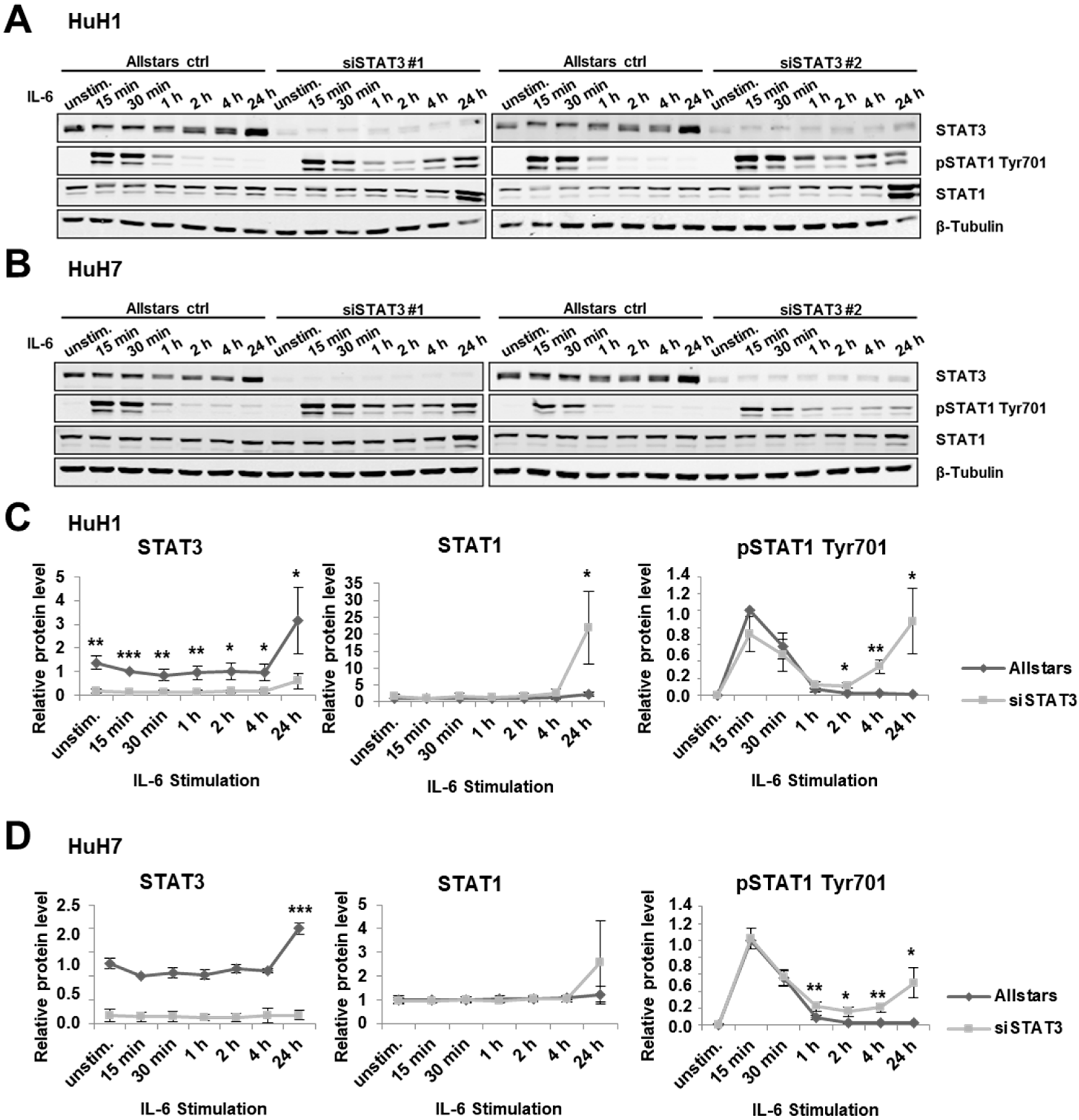
3.2. STAT3 Depletion Induces an IFN-γ-Like Response in HCC Cells
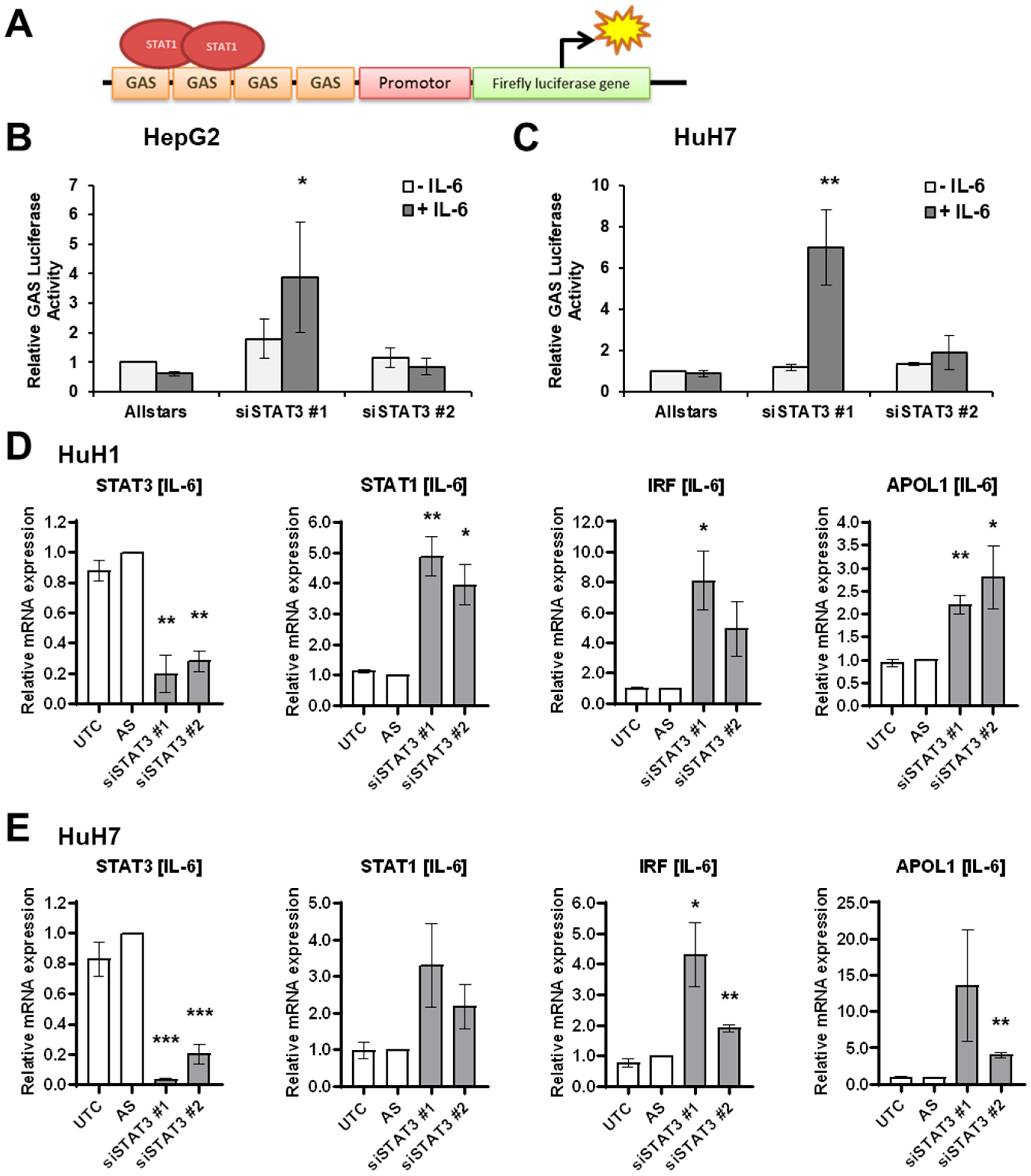
3.3. IL-6 Induces STAT1 Transcriptional Activity upon STAT3 Depletion
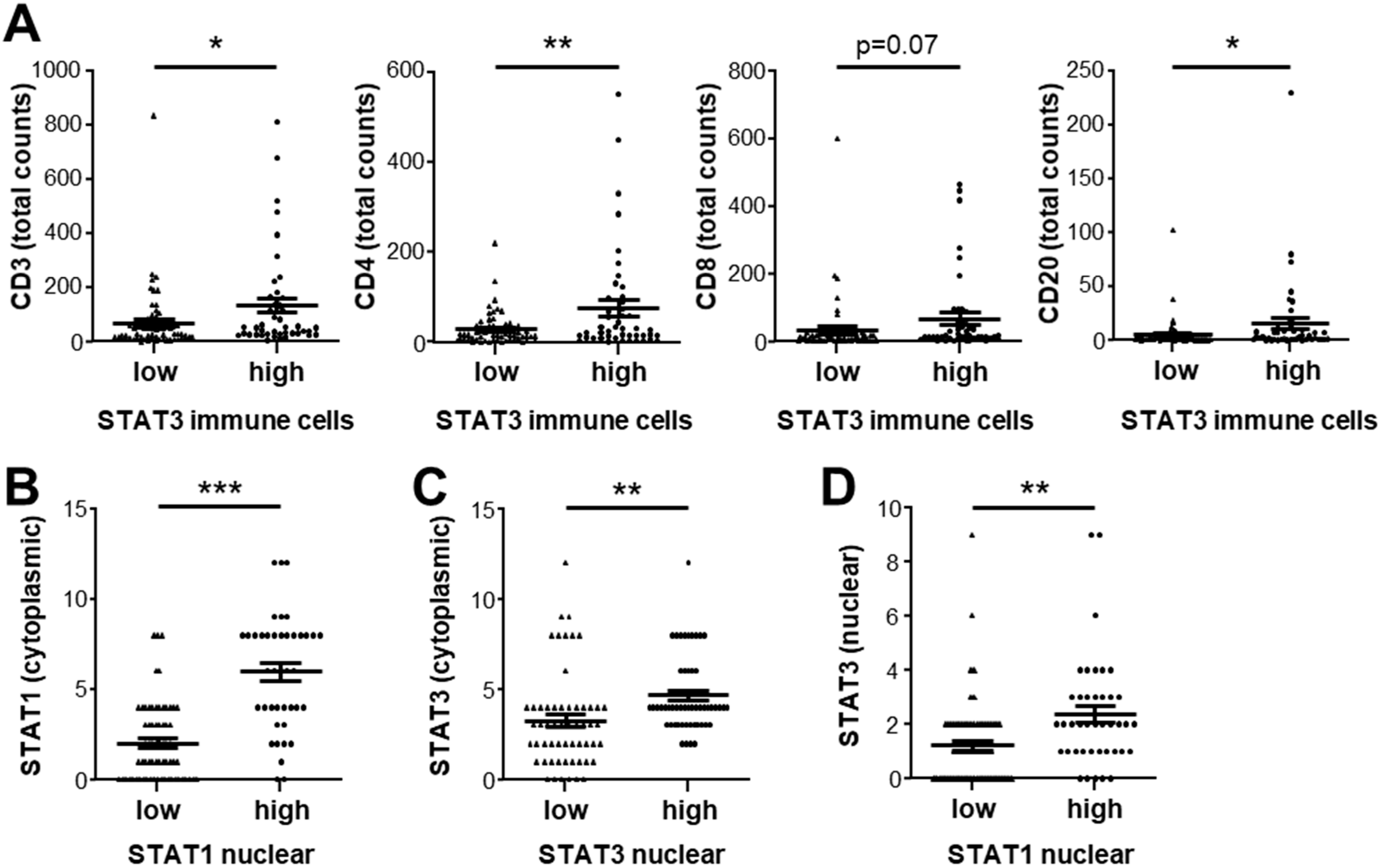
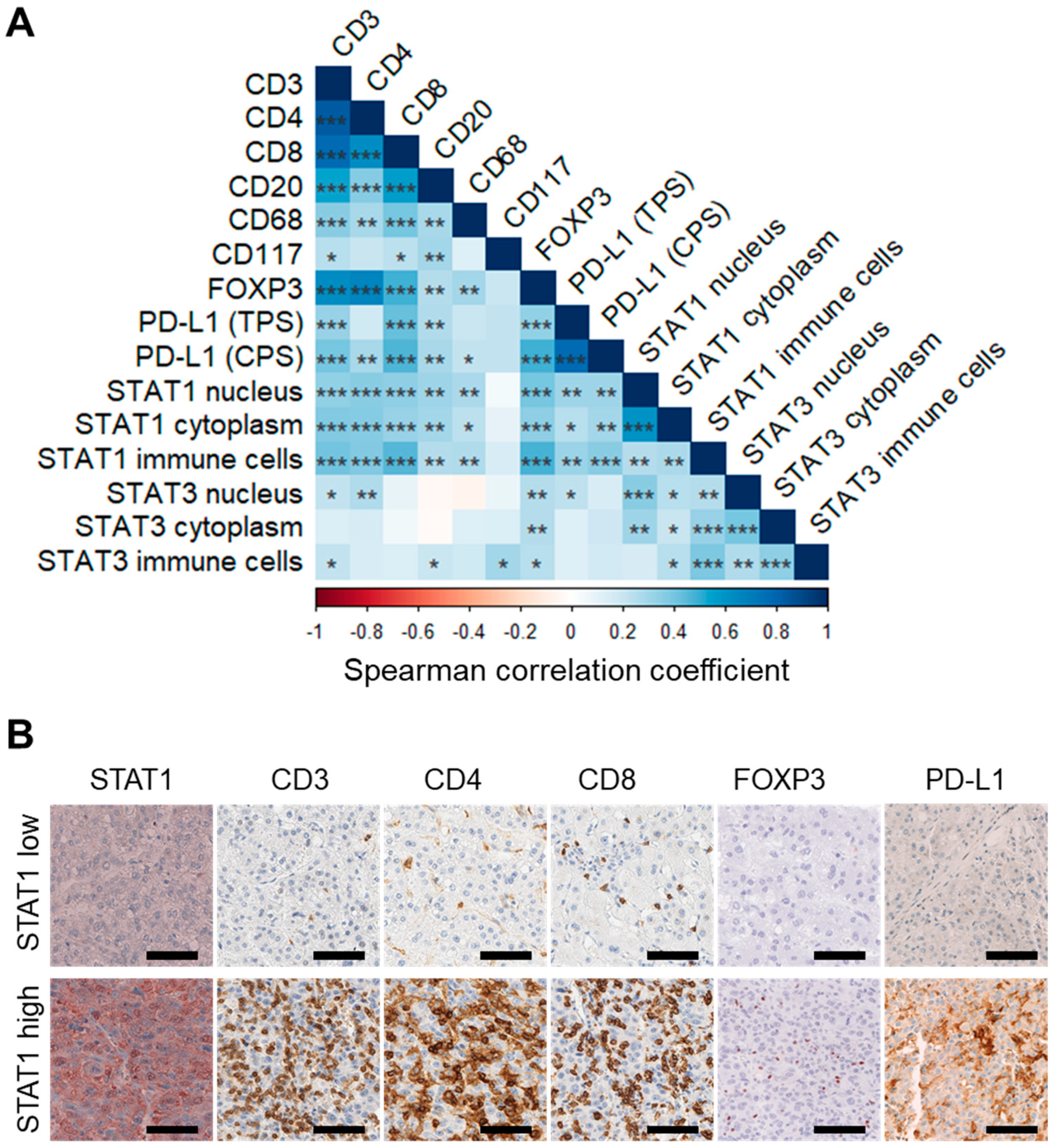
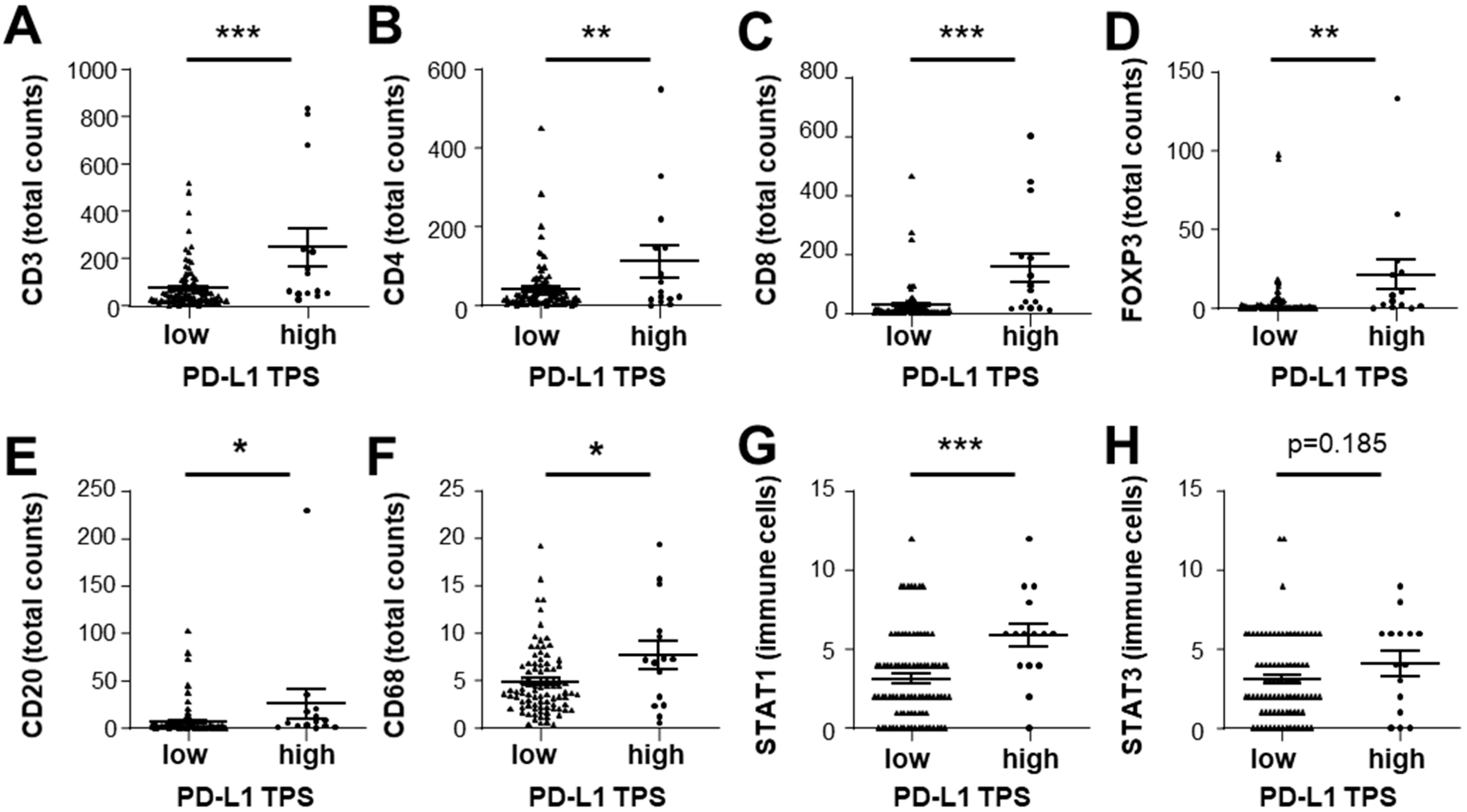
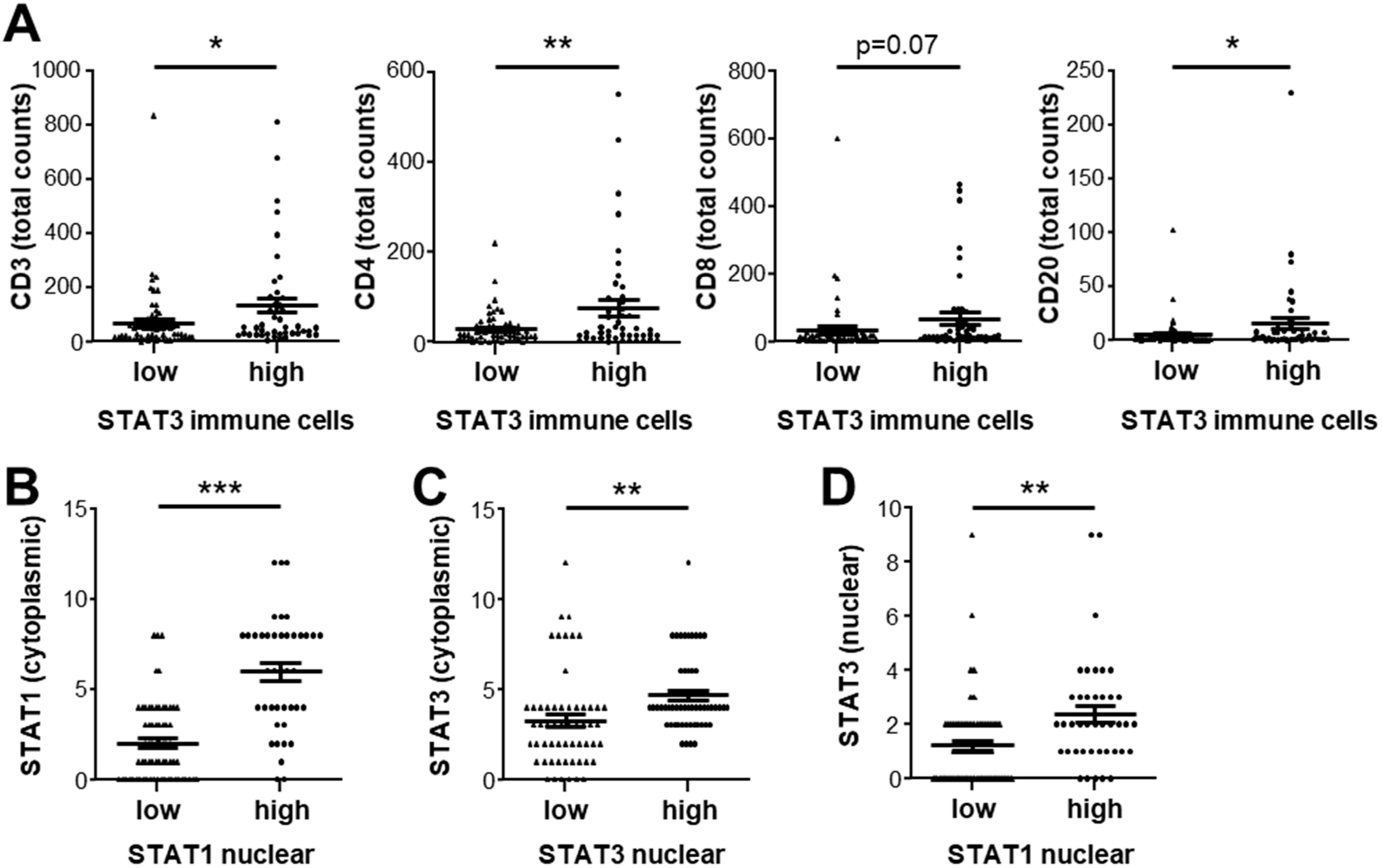
3.4. Expression of STAT1 and STAT3 Strongly Correlates with Infiltrating Immune Cell Profiles
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Li, X.; Ramadori, P.; Pfister, D.; Seehawer, M.; Zender, L.; Heikenwalder, M. The immunological and metabolic landscape in primary and metastatic liver cancer. Nat. Rev. Cancer 2021, 21, 541–557. [Google Scholar] [CrossRef] [PubMed]
- Refolo, M.G.; Messa, C.; Guerra, V.; Carr, B.I.; D’Alessandro, R. Inflammatory Mechanisms of HCC Development. Cancers 2020, 12, 641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e814. [Google Scholar] [CrossRef] [Green Version]
- Fisher, D.T.; Chen, Q.; Skitzki, J.J.; Muhitch, J.B.; Zhou, L.; Appenheimer, M.M.; Vardam, T.D.; Weis, E.L.; Passanese, J.; Wang, W.C.; et al. IL-6 trans-signaling licenses mouse and human tumor microvascular gateways for trafficking of cytotoxic T cells. J. Clin. Investig. 2011, 121, 3846–3859. [Google Scholar] [CrossRef] [Green Version]
- Yamada, D.; Rizvi, S.; Razumilava, N.; Bronk, S.F.; Davila, J.I.; Champion, M.D.; Borad, M.J.; Bezerra, J.A.; Chen, X.; Gores, G.J. IL-33 facilitates oncogene-induced cholangiocarcinoma in mice by an interleukin-6-sensitive mechanism. Hepatology 2015, 61, 1627–1642. [Google Scholar] [CrossRef]
- Li, R.; Wen, A.; Lin, J. Pro-Inflammatory Cytokines in the Formation of the Pre-Metastatic Niche. Cancers 2020, 12, 3752. [Google Scholar] [CrossRef] [PubMed]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, H.; Maeda, S.; Yoshida, H.; Tateishi, R.; Masuzaki, R.; Ohki, T.; Hayakawa, Y.; Kinoshita, H.; Yamakado, M.; Kato, N.; et al. Serum IL-6 levels and the risk for hepatocarcinogenesis in chronic hepatitis C patients: An analysis based on gender differences. Int. J. Cancer 2009, 125, 2264–2269. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Yu, J.; Cheng, A.S.; Wong, G.L.; Chan, H.Y.; Chu, E.S.; Ng, E.K.; Chan, F.K.; Sung, J.J.; Chan, H.L. High serum interleukin-6 level predicts future hepatocellular carcinoma development in patients with chronic hepatitis B. Int. J. Cancer 2009, 124, 2766–2770. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Wang, H.; Lafdil, F.; Feng, D. STAT proteins-key regulators of anti-viral responses, inflammation, and tumorigenesis in the liver. J. Hepatol. 2012, 57, 430–441. [Google Scholar] [CrossRef]
- Regis, G.; Pensa, S.; Boselli, D.; Novelli, F.; Poli, V. Ups and downs: The STAT1:STAT3 seesaw of Interferon and gp130 receptor signalling. Semin. Cell Dev. Biol. 2008, 19, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Avalle, L.; Pensa, S.; Regis, G.; Novelli, F.; Poli, V. STAT1 and STAT3 in tumorigenesis: A matter of balance. JAKSTAT 2012, 1, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Stephanou, A.; Latchman, D.S. STAT-1: A novel regulator of apoptosis. Int. J. Exp. Pathol. 2003, 84, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lafdil, F.; Wang, L.; Park, O.; Yin, S.; Niu, J.; Miller, A.M.; Sun, Z.; Gao, B. Hepatoprotective versus oncogenic functions of STAT3 in liver tumorigenesis. Am. J. Pathol. 2011, 179, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Ploeger, C.; Huth, T.; Sugiyanto, R.N.; Pusch, S.; Goeppert, B.; Singer, S.; Tabti, R.; Hausser, I.; Schirmacher, P.; Desaubry, L.; et al. Prohibitin, STAT3 and SH2D4A physically and functionally interact in tumor cell mitochondria. Cell Death Dis. 2020, 11, 1023. [Google Scholar] [CrossRef]
- Ploeger, C.; Waldburger, N.; Fraas, A.; Goeppert, B.; Pusch, S.; Breuhahn, K.; Wang, X.W.; Schirmacher, P.; Roessler, S. Chromosome 8p tumor suppressor genes SH2D4A and SORBS3 cooperate to inhibit interleukin-6 signaling in hepatocellular carcinoma. Hepatology 2016, 64, 828–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goeppert, B.; Frauenschuh, L.; Zucknick, M.; Stenzinger, A.; Andrulis, M.; Klauschen, F.; Joehrens, K.; Warth, A.; Renner, M.; Mehrabi, A.; et al. Prognostic impact of tumour-infiltrating immune cells on biliary tract cancer. Br. J. Cancer 2013, 109, 2665–2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albrecht, T.; Rausch, M.; Rossler, S.; Albrecht, M.; Braun, J.D.; Geissler, V.; Mehrabi, A.; Vogel, M.N.; Pathil-Warth, A.; Mechtersheimer, G.; et al. HER2 gene (ERBB2) amplification is a rare event in non-liver-fluke associated cholangiocarcinogenesis. BMC Cancer 2019, 19, 1191. [Google Scholar] [CrossRef] [Green Version]
- Goeppert, B.; Roessler, S.; Renner, M.; Singer, S.; Mehrabi, A.; Vogel, M.N.; Pathil, A.; Czink, E.; Kohler, B.; Springfeld, C.; et al. Mismatch repair deficiency is a rare but putative therapeutically relevant finding in non-liver fluke associated cholangiocarcinoma. Br. J. Cancer 2019, 120, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Kriegsmann, M.; Roessler, S.; Kriegsmann, K.; Renner, M.; Longuespee, R.; Albrecht, T.; Loeffler, M.; Singer, S.; Mehrabi, A.; Vogel, M.N.; et al. Programmed cell death ligand 1 (PD-L1, CD274) in cholangiocarcinoma-correlation with clinicopathological data and comparison of antibodies. BMC Cancer 2019, 19, 72. [Google Scholar] [CrossRef] [Green Version]
- Goeppert, B.; Schmezer, P.; Dutruel, C.; Oakes, C.; Renner, M.; Breinig, M.; Warth, A.; Vogel, M.N.; Mittelbronn, M.; Mehrabi, A.; et al. Down-regulation of tumor suppressor A kinase anchor protein 12 in human hepatocarcinogenesis by epigenetic mechanisms. Hepatology 2010, 52, 2023–2033. [Google Scholar] [CrossRef]
- Goeppert, B.; Frauenschuh, L.; Zucknick, M.; Roessler, S.; Mehrabi, A.; Hafezi, M.; Stenzinger, A.; Warth, A.; Pathil, A.; Renner, M.; et al. Major histocompatibility complex class I expression impacts on patient survival and type and density of immune cells in biliary tract cancer. Br. J. Cancer 2015, 113, 1343–1349. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Qing, Y.; Stark, G.R. Alternative activation of STAT1 and STAT3 in response to interferon-gamma. J. Biol. Chem. 2004, 279, 41679–41685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil, M.P.; Bohn, E.; O’Guin, A.K.; Ramana, C.V.; Levine, B.; Stark, G.R.; Virgin, H.W.; Schreiber, R.D. Biologic consequences of Stat1-independent IFN signaling. Proc. Natl. Acad. Sci. USA 2001, 98, 6680–6685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Van der Leun, A.M.; Thommen, D.S.; Schumacher, T.N. CD8(+) T cell states in human cancer: Insights from single-cell analysis. Nat. Rev. Cancer 2020, 20, 218–232. [Google Scholar] [CrossRef]
- Gabrielson, A.; Wu, Y.; Wang, H.; Jiang, J.; Kallakury, B.; Gatalica, Z.; Reddy, S.; Kleiner, D.; Fishbein, T.; Johnson, L.; et al. Intratumoral CD3 and CD8 T-cell Densities Associated with Relapse-Free Survival in HCC. Cancer Immunol. Res. 2016, 4, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Bates, G.J.; Fox, S.B.; Han, C.; Leek, R.D.; Garcia, J.F.; Harris, A.L.; Banham, A.H. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J. Clin. Oncol. 2006, 24, 5373–5380. [Google Scholar] [CrossRef]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Zorn, E.; Nelson, E.A.; Mohseni, M.; Porcheray, F.; Kim, H.; Litsa, D.; Bellucci, R.; Raderschall, E.; Canning, C.; Soiffer, R.J.; et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood 2006, 108, 1571–1579. [Google Scholar] [CrossRef] [Green Version]
- Rebe, C.; Ghiringhelli, F. STAT3, a Master Regulator of Anti-Tumor Immune Response. Cancers 2019, 11, 1280. [Google Scholar] [CrossRef] [Green Version]
- Austin, J.W.; Lu, P.; Majumder, P.; Ahmed, R.; Boss, J.M. STAT3, STAT4, NFATc1, and CTCF regulate PD-1 through multiple novel regulatory regions in murine T cells. J. Immunol. 2014, 192, 4876–4886. [Google Scholar] [CrossRef] [PubMed]
- Owen, K.L.; Brockwell, N.K.; Parker, B.S. JAK-STAT Signaling: A Double-Edged Sword of Immune Regulation and Cancer Progression. Cancers 2019, 11, 2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Araujo, E.D.; Keseru, G.M.; Gunning, P.T.; Moriggl, R. Targeting STAT3 and STAT5 in Cancer. Cancers 2020, 12, 2002. [Google Scholar] [CrossRef] [PubMed]
- Hack, S.P.; Spahn, J.; Chen, M.; Cheng, A.L.; Kaseb, A.; Kudo, M.; Lee, H.C.; Yopp, A.; Chow, P.; Qin, S. IMbrave 050: A Phase III trial of atezolizumab plus bevacizumab in high-risk hepatocellular carcinoma after curative resection or ablation. Future Oncol. 2020, 16, 975–989. [Google Scholar] [CrossRef]
- Ruf, B.; Heinrich, B.; Greten, T.F. Immunobiology and immunotherapy of HCC: Spotlight on innate and innate-like immune cells. Cell. Mol. Immunol. 2021, 18, 112–127. [Google Scholar] [CrossRef]
- Fletcher, J.S.; Springer, M.G.; Choi, K.; Jousma, E.; Rizvi, T.A.; Dombi, E.; Kim, M.O.; Wu, J.; Ratner, N. STAT3 inhibition reduces macrophage number and tumor growth in neurofibroma. Oncogene 2019, 38, 2876–2884. [Google Scholar] [CrossRef]
- Jahangiri, A.; Dadmanesh, M.; Ghorban, K. STAT3 inhibition reduced PD-L1 expression and enhanced antitumor immune responses. J. Cell. Physiol. 2020, 235, 9457–9463. [Google Scholar] [CrossRef]
- Liu, H.; Shen, J.; Lu, K. IL-6 and PD-L1 blockade combination inhibits hepatocellular carcinoma cancer development in mouse model. Biochem. Biophys. Res. Commun. 2017, 486, 239–244. [Google Scholar] [CrossRef]
- Pore, N.; Wu, S.; Standifer, N.; Jure-Kunkel, M.; de Los Reyes, M.; Shrestha, Y.; Halpin, R.; Rothstein, R.; Mulgrew, K.; Blackmore, S.; et al. Resistance to Durvalumab and Durvalumab plus Tremelimumab Is Associated with Functional STK11 Mutations in Patients with Non-Small Cell Lung Cancer and Is Reversed by STAT3 Knockdown. Cancer Discov. 2021, 11, 2828–2845. [Google Scholar] [CrossRef]
- Wang, H.; Li, S.; Wang, Q.; Jin, Z.; Shao, W.; Gao, Y.; Li, L.; Lin, K.; Zhu, L.; Wang, H.; et al. Tumor immunological phenotype signature-based high-throughput screening for the discovery of combination immunotherapy compounds. Sci. Adv. 2021, 7, eabd7851. [Google Scholar] [CrossRef]
- Verhoeven, Y.; Tilborghs, S.; Jacobs, J.; De Waele, J.; Quatannens, D.; Deben, C.; Prenen, H.; Pauwels, P.; Trinh, X.B.; Wouters, A.; et al. The potential and controversy of targeting STAT family members in cancer. Semin. Cancer Biol. 2020, 60, 41–56. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ploeger, C.; Schreck, J.; Huth, T.; Fraas, A.; Albrecht, T.; Charbel, A.; Ji, J.; Singer, S.; Breuhahn, K.; Pusch, S.; et al. STAT1 and STAT3 Exhibit a Crosstalk and Are Associated with Increased Inflammation in Hepatocellular Carcinoma. Cancers 2022, 14, 1154. https://doi.org/10.3390/cancers14051154
Ploeger C, Schreck J, Huth T, Fraas A, Albrecht T, Charbel A, Ji J, Singer S, Breuhahn K, Pusch S, et al. STAT1 and STAT3 Exhibit a Crosstalk and Are Associated with Increased Inflammation in Hepatocellular Carcinoma. Cancers. 2022; 14(5):1154. https://doi.org/10.3390/cancers14051154
Chicago/Turabian StylePloeger, Carolin, Johannes Schreck, Thorben Huth, Angelika Fraas, Thomas Albrecht, Alphonse Charbel, Junfang Ji, Stephan Singer, Kai Breuhahn, Stefan Pusch, and et al. 2022. "STAT1 and STAT3 Exhibit a Crosstalk and Are Associated with Increased Inflammation in Hepatocellular Carcinoma" Cancers 14, no. 5: 1154. https://doi.org/10.3390/cancers14051154
APA StylePloeger, C., Schreck, J., Huth, T., Fraas, A., Albrecht, T., Charbel, A., Ji, J., Singer, S., Breuhahn, K., Pusch, S., Köhler, B. C., Springfeld, C., Schirmacher, P., Mehrabi, A., Goeppert, B., & Roessler, S. (2022). STAT1 and STAT3 Exhibit a Crosstalk and Are Associated with Increased Inflammation in Hepatocellular Carcinoma. Cancers, 14(5), 1154. https://doi.org/10.3390/cancers14051154