
STAT3 Signaling in Breast Cancer: Multicellular Actions and Therapeutic Potential


Simple Summary
Abstract
1. Introduction
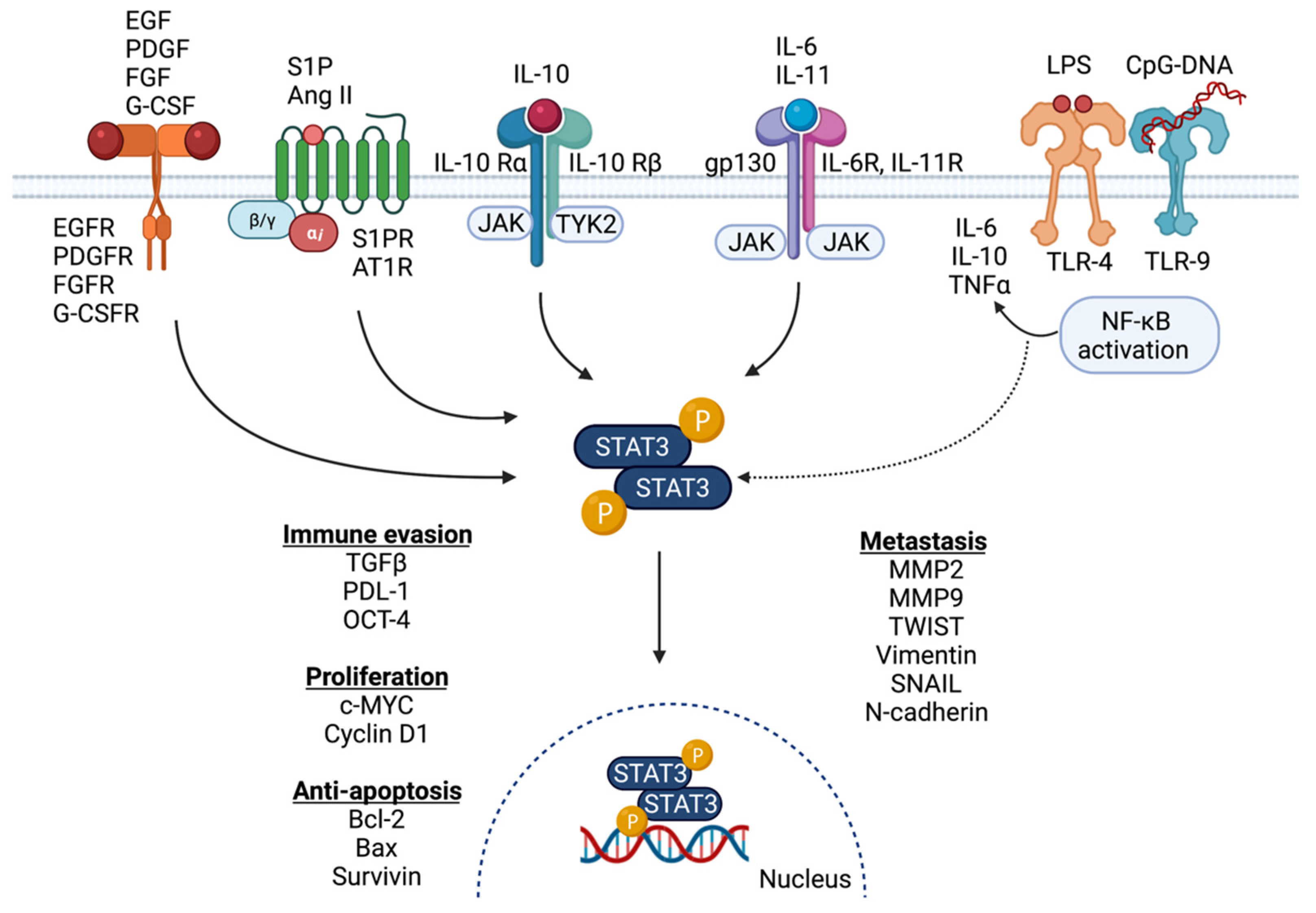
2. Activation of STAT3 in the Breast Cancer Microenvironment
2.1. Activation of STAT3 Signaling in Breast Cancer
2.2. Signaling Partners and Downstream Targets of STAT3 in Breast Cancer
2.3. STAT3 Signaling and Patient Outcome
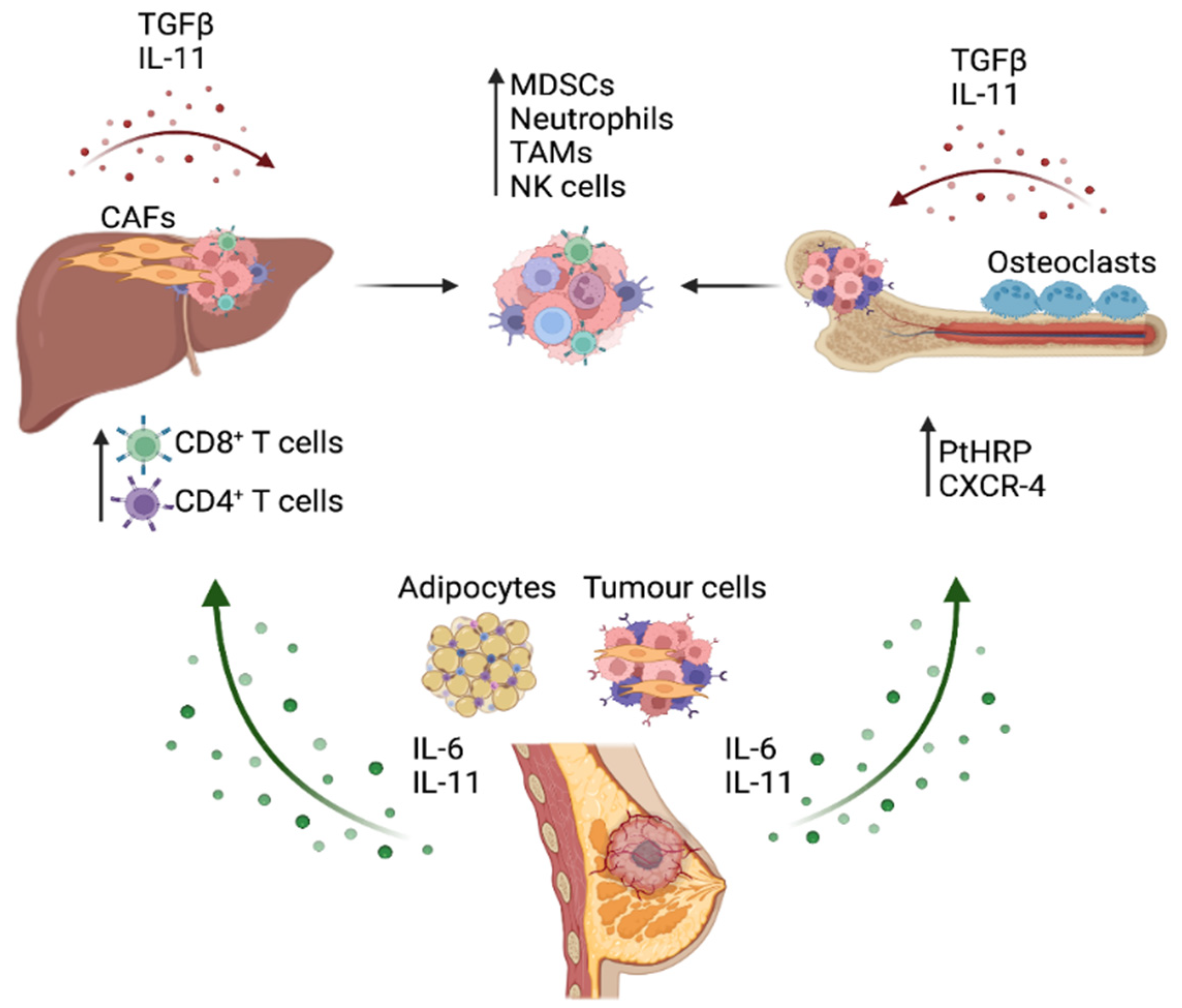
3. STAT3 Activation in the Breast Cancer Metastatic Process
4. Role of IL-6 Cytokine/STAT3 Signaling in Immune Evasion and Function in Metastatic Cancer
4.1. STAT3 Signaling and Immune Evasion
4.2. STAT3 and Macrophage Polarization
5. Therapeutically Targeting IL-6 Signaling and STAT3 Activation in Breast Cancer
5.1. Small-Molecule Inhibitor Therapies
5.2. Monoclonal Antibodies
5.3. Limitations to STAT3-Targeting Therapies
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Johnstone, C.N.; Chand, A.; Putoczki, T.L.; Ernst, M. Emerging roles for IL-11 signaling in cancer development and progression: Focus on breast cancer. Cytokine Growth Factor Rev. 2015, 26, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Thilakasiri, P.; Huynh, J.; Poh, A.; Tan, C.W.; Nero, T.; Tran, K.; Parslow, A.C.; Afshar-Sterle, S.; Baloyan, D.; Hannan, N.J.; et al. Repurposing the selective estrogen receptor modulator bazedoxifene to suppress gastrointestinal cancer growth. EMBO Mol. Med. 2019, 11, e9539. [Google Scholar] [CrossRef] [PubMed]
- Huynh, J.; Chand, A.; Gough, D.; Ernst, M. Therapeutically exploiting STAT3 activity in cancer-using tissue repair as a road map. Nat. Cancer 2019, 19, 82–96. [Google Scholar] [CrossRef]
- Ma, J.-H.; Qin, L.; Li, X. Role of STAT3 signaling pathway in breast cancer. Cell Commun. Signal. 2020, 18, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Pencik, J.; Pham, H.T.T.; Schmoellerl, J.; Javaheri, T.; Schlederer, M.; Culig, Z.; Merkel, O.; Moriggl, R.; Grebien, F.; Kenner, L. JAK-STAT signaling in cancer: From cytokines to non-coding genome. Cytokine 2016, 87, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Perugini, J.; Di Mercurio, E.; Tossetta, G.; Severi, I.; Monaco, F.; Reguzzoni, M.; Tomasetti, M.; Dani, C.; Cinti, S.; Giordano, A. Biological Effects of Ciliary Neurotrophic Factor on hMADS Adipocytes. Front. Endocrinol. 2019, 10, 768. [Google Scholar] [CrossRef] [PubMed]
- Eyking, A.; Ey, B.; Rünzi, M.; Roig, A.I.; Reis, H.; Schmid, K.W.; Gerken, G.; Podolsky, D.K.; Cario, E. Toll-like Receptor 4 Variant D299G Induces Features of Neoplastic Progression in Caco-2 Intestinal Cells and Is Associated with Advanced Human Colon Cancer. Gastroenterololgy 2011, 141, 2154–2165. [Google Scholar] [CrossRef] [PubMed]
- Hossain, D.M.S.; Dos Santos, C.; Zhang, Q.; Kozlowska, A.; Liu, H.; Gao, C.; Moreira, D.; Swiderski, P.; Jozwiak, A.; Kline, J.; et al. Leukemia cell–targeted STAT3 silencing and TLR9 triggering generate systemic antitumor immunity. Blood J. Am. Soc. Hematol. 2014, 123, 15–25. [Google Scholar] [CrossRef]
- Lee, H.; Deng, J.; Kujawski, M.; Yang, C.; Liu, Y.; Herrmann, A.; Kortylewski, M.; Horne, D.; Somlo, G.; Forman, S.; et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat. Med. 2010, 16, 1421–1428. [Google Scholar] [CrossRef]
- Xin, H.; Lu, R.; Lee, H.; Zhang, W.; Zhang, C.; Deng, J.; Liu, Y.; Shen, S.; Wagner, K.-U.; Forman, S. G-protein-coupled receptor agonist BV8/prokineticin-2 and STAT3 protein form a feed-forward loop in both normal and malignant myeloid cells. J. Biol. Chem. 2013, 288, 13842–13849. [Google Scholar] [CrossRef]
- Coulson, R.; Liew, S.H.; Connelly, A.A.; Yee, N.S.; Deb, S.; Kumar, B.; Vargas, A.C.; O’Toole, S.A.; Parslow, A.C.; Poh, A.; et al. The angiotensin receptor blocker, Losartan, inhibits mammary tumor development and progression to invasive carcinoma. Oncotarget 2017, 8, 18640–18656. [Google Scholar] [CrossRef]
- Perini, M.; Dmello, R.S.; Nero, T.L.; Chand, A.L. Evaluating the benefits of renin-angiotensin system inhibitors as cancer treatments. Pharmacol. Ther. 2020, 211, 107527. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, Z.; Moreira, D.; Su, Y.-L.; Won, H.; Adamus, T.; Dong, Z.; Liang, Y.; Yin, H.H.; Swiderski, P.; et al. B Cell Lymphoma Immunotherapy Using TLR9-Targeted Oligonucleotide STAT3 Inhibitors. Mol. Ther. 2018, 26, 695–707. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumor Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.; Liang, S.; Ghosh, S.; Hornsby, P.J.; Li, R. Interleukin 6 secreted from adipose stromal cells promotes migration and invasion of breast cancer cells. Oncogene 2009, 28, 2745–2755. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, I.S.; Karin, M. Inflammatory cytokines in cancer: Tumour necrosis factor and interleukin 6 take the stage. Ann. Rheum. Dis. 2011, 70, i104–i108. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Grivennikov, S.I.; Karin, M. The unholy trinity: Inflammation, cytokines, and STAT3 shape the cancer microenviron-ment. Cancer Cell 2011, 19, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Jostock, T.; Müllberg, J.; Özbek, S.; Atreya, R.; Blinn, G.; Voltz, N.; Fischer, M.; Neurath, M.F.; Rose-John, S. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. JBIC J. Biol. Inorg. Chem. 2001, 268, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Lamertz, L.; Rummel, F.; Polz, R.; Baran, P.; Hansen, S.; Waetzig, G.H.; Moll, J.M.; Floss, D.M.; Scheller, J. Soluble gp130 prevents interleukin-6 and interleukin-11 cluster signaling but not intracellular autocrine responses. Sci. Signal. 2018, 11, eaar7388. [Google Scholar] [CrossRef] [PubMed]
- Hunter, A.C.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef]
- Kim, G.; Ouzounova, M.; Quraishi, A.A.; Davis, A.K.; Tawakkol, N.; Clouthier, S.G.; Malik, F.; Paulson, A.; D’Angelo, R.C.; Korkaya, S.; et al. SOCS3-mediated regulation of inflammatory cytokines in PTEN and p53 inactivated triple negative breast cancer model. Oncogene 2015, 34, 671–680. [Google Scholar] [CrossRef]
- Kim, D.J.; Tremblay, M.L.; DiGiovanni, J. Protein Tyrosine Phosphatases, TC-PTP, SHP1, and SHP2, Cooperate in Rapid Dephosphorylation of Stat3 in Keratinocytes Following UVB Irradiation. PLoS ONE 2010, 5, e10290. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, Q.; Mi, S.; Liang, X.; Zhang, Z.; Su, X.; Liu, J.; Chen, Y.; Wang, M.; Zhang, Y.; et al. Both miR-17-5p and miR-20a Alleviate Suppressive Potential of Myeloid-Derived Suppressor Cells by Modulating STAT3 Expression. J. Immunol. 2011, 186, 4716–4724. [Google Scholar] [CrossRef]
- Kim, M.; Morales, L.D.; Jang, I.-S.; Cho, Y.-Y.; Kim, D.J. Protein Tyrosine Phosphatases as Potential Regulators of STAT3 Signaling. Int. J. Mol. Sci. 2018, 19, 2708. [Google Scholar] [CrossRef]
- Lee, H.; Zhang, P.; Herrmann, A.; Yang, C.; Xin, H.; Wang, Z.; Hoon, D.; Forman, S.J.; Jove, R.; Riggs, A.D.; et al. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc. Natl. Acad. Sci. USA 2012, 109, 7765–7769. [Google Scholar] [CrossRef]
- Zou, S.; Tong, Q.; Liu, B.; Huang, W.; Tian, Y.; Fu, X. Targeting STAT3 in Cancer Immunotherapy. Mol. Cancer 2020, 19, 1–19. [Google Scholar] [CrossRef]
- Nicholson, S.E.; DE Souza, D.; Fabri, L.; Corbin, J.; Willson, T.A.; Zhang, J.-G.; Silva, A.; Asimakis, M.; Farley, A.; Nash, A.D.; et al. Suppressor of cytokine signaling-3 preferentially binds to the SHP-2-binding site on the shared cytokine receptor subunit gp130. Proc. Natl. Acad. Sci. USA 2000, 97, 6493–6498. [Google Scholar] [CrossRef]
- Jiang, M.; Zhang, W.-W.; Liu, P.; Yu, W.; Liu, T.; Yu, J. Dysregulation of SOCS-Mediated Negative Feedback of Cytokine Signaling in Carcinogenesis and Its Significance in Cancer Treatment. Front. Immunol. 2017, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Fleming, J.D.; Giresi, P.G.; Lindahl-Allen, M.; Krall, E.B.; Lieb, J.D.; Struhl, K. STAT3 acts through pre-existing nucleosome-depleted regions bound by FOS during an epigenetic switch linking inflammation to cancer. Epigenet. Chromatin 2015, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Snyder, M.; Huang, J.; Huang, X.-Y.; Zhang, J.J. A Signal Transducer and Activator of Transcription 3·Nuclear Factor κB (Stat3·NFκB) Complex Is Necessary for the Expression of Fascin in Metastatic Breast Cancer Cells in Response to Interleukin (IL)-6 and Tumor Necrosis Factor (TNF)-α. J. Biol. Chem. 2014, 289, 30082–30089. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-Q.; Tang, C.-H.; Wang, Y.; Jin, L.; Wang, Q.; Li, X.; Hu, G.-N.; Huang, B.-F.; Zhao, Y.-M.; Su, C.-M. FSCN1 gene polymorphisms: Biomarkers for the development and progression of breast cancer. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dmello, R.; To, S.; Chand, A. Therapeutic Targeting of the Tumour Microenvironment in Metastatic Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 2067. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Shen, S.S.; Zhou, S.; Ni, J.; Chen, D.; Wang, G.; Li, Y. STAT3 activation and aberrant ligand-dependent sonic hedgehog signaling in human pulmonary adenocarcinoma. Exp. Mol. Pathol. 2012, 93, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Jasek-Gajda, E.; Jurkowska, H.; Jasińska, M.; Litwin, J.A.; Lis, G.J. Combination of ERK2 and STAT3 Inhibitors Promotes Anticancer Effects on Acute Lymphoblastic Leukemia Cells. Cancer Genom.-Proteom. 2020, 17, 517–527. [Google Scholar] [CrossRef]
- Tripathi, S.; Chen, Z.; Larjo, A.; Kanduri, K.; Nousiainen, K.; Äijö, T.; Ricaño-Ponce, I.; Hrdlickova, B.; Tuomela, S.; Laajala, E.; et al. Genome-wide Analysis of STAT3-Mediated Transcription during Early Human Th17 Cell Differentiation. Cell Rep. 2017, 19, 1888–1901. [Google Scholar] [CrossRef] [PubMed]
- Vallania, F.; Schiavone, D.; Dewilde, S.; Pupo, E.; Garbay, S.; Calogero, R.; Pontoglio, M.; Provero, P.; Poli, V. Genome-wide discovery of functional transcription factor binding sites by comparative genomics: The case of Stat 3. Proc. Natl. Acad. Sci. USA 2009, 106, 5117–5122. [Google Scholar] [CrossRef]
- McDaniel, J.M.; Varley, K.; Gertz, J.; Savic, D.; Roberts, B.S.; Bailey, S.K.; Shevde, L.A.; Ramaker, R.C.; Lasseigne, B.; Kirby, M.K.; et al. Genomic regulation of invasion by STAT3 in triple negative breast cancer. Oncotarget 2016, 8, 8226–8238. [Google Scholar] [CrossRef]
- Sirkisoon, S.R.; Carpenter, R.; Rimkus, T.; Anderson, A.; Harrison, A.; Lange, A.M.; Jin, G.; Watabe, K.; Lo, H.-W. Interaction between STAT3 and GLI1/tGLI1 oncogenic transcription factors promotes the aggressiveness of triple-negative breast cancers and HER2-enriched breast cancer. Oncogene 2018, 37, 2502–2514. [Google Scholar] [CrossRef]
- Doheny, D.; Sirkisoon, S.; Carpenter, R.L.; Aguayo, N.R.; Regua, A.T.; Anguelov, M.; Manore, S.G.; Arrigo, A.; Abu Jalboush, S.; Wong, G.L.; et al. Combined inhibition of JAK2-STAT3 and SMO-GLI1/tGLI1 pathways suppresses breast cancer stem cells, tumor growth, and metastasis. Oncogene 2020, 39, 6589–6605. [Google Scholar] [CrossRef]
- Rozovski, U.; Harris, D.M.; Li, P.; Liu, Z.; Jain, P.; Manshouri, T.; Veletic, I.; Ferrajoli, A.; Bose, P.; Thompson, P.; et al. STAT3 induces the expression of GLI1 in chronic lymphocytic leukemia cells. Oncotarget 2021, 12, 401–411. [Google Scholar] [CrossRef]
- Larive, R.M.; Moriggi, G.; Menacho-Márquez, M.; Cañamero, M.; De Álava, E.; Alarcón, B.; Dosil, M.; Bustelo, X.R. Contribution of the R-Ras2 GTP-binding protein to primary breast tumorigenesis and late-stage metastatic disease. Nat. Commun. 2014, 5, 3881. [Google Scholar] [CrossRef]
- Xie, S.; Qin, J.; Liu, S.; Zhang, Y.; Wang, J.; Shi, X.; Li, D.; Zhou, J.; Liu, M. Cep70 overexpression stimulates pancreatic cancer by inducing centrosome abnormality and microtubule disorganization. Sci. Rep. 2016, 6, 21263. [Google Scholar] [CrossRef]
- Bao, X.; Huang, Y.; Xu, W.; Xiong, G. Functions and Clinical Significance of UPF3a Expression in Human Colorectal Cancer. Cancer Manag. Res. 2020, 12, 4271–4281. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.-Y.; Zeng, Y.; Lei, Z.; Wang, L.; Yang, H.; Liu, Z.; Zhao, J.; Zhang, H.-T. JAK/STAT3 signaling is required for TGF-β-induced epithelial-mesenchymal transition in lung cancer cells. Int. J. Oncol. 2014, 44, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D.; et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 2002, 21, 2000–2008. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-C.; Shi, L.-H.; Wang, X.-J.; Wang, S.-X.; Wan, X.-Q.; Liu, S.-R.; Wang, Y.-F.; Lu, Z.; Wang, L.-H.; Ding, Y. Stat3/Oct-4/c-Myc signal circuit for regulating stemness-mediated doxorubicin resistance of triple-negative breast cancer cells and inhibitory effects of WP1066. Int. J. Oncol. 2018, 53, 339–348. [Google Scholar] [CrossRef]
- Ashizawa, T.; Iizuka, A.; Maeda, C.; Tanaka, E.; Kondou, R.; Miyata, H.; Sugino, T.; Kawata, T.; Deguchi, S.; Mitsuya, K.; et al. Impact of combination therapy with anti-PD-1 blockade and a STAT3 inhibitor on the tumor-infiltrating lymphocyte status. Immunol. Lett. 2019, 216, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, T.; Yaguchi, T.; Kawakami, Y. Enhanced anti-tumor effects of the PD-1 blockade combined with a highly absorptive form of curcumin targeting STAT3. Cancer Sci. 2020, 111, 4326–4335. [Google Scholar] [CrossRef]
- Gupta, S.C.; Patchva, S.; Aggarwal, B.B. Therapeutic Roles of Curcumin: Lessons Learned from Clinical Trials. AAPS J. 2013, 15, 195–218. [Google Scholar] [CrossRef] [PubMed]
- Hanavadi, S.; Martin, T.A.; Watkins, G.; Mansel, R.E.; Jiang, W.G. Expression of Interleukin 11 and Its Receptor and Their Prognostic Value in Human Breast Cancer. Ann. Surg. Oncol. 2006, 13, 802–808. [Google Scholar] [CrossRef]
- Ren, L.; Wang, X.; Dong, Z.; Liu, J.; Zhang, S. Bone metastasis from breast cancer involves elevated IL-11 expression and the gp130/STAT3 pathway. Med Oncol. 2013, 30, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Ma, Q.; Ding, N.; Luo, F.; Bai, Y.; Kang, F.; Gong, X.; Dong, R.; Dai, J.; Dai, Q.; et al. IL-11 is essential in promoting osteolysis in breast cancer bone metastasis via RANKL-independent activation of osteoclastogenesis. Cell Death Dis. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Miller, C.P.; Thorpe, J.D.; Kortum, A.N.; Coy, C.M.; Cheng, W.-Y.; Yang, T.-H.O.; Anastassiou, D.; Beatty, J.D.; Urban, N.D.; Blau, C.A. JAK2 Expression Is Associated with Tumor-Infiltrating Lymphocytes and Improved Breast Cancer Outcomes: Implications for Evaluating JAK2 Inhibitors. Cancer Immunol. Res. 2014, 2, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Q.; Tang, C.-H.; Chen, H.-D.; Hu, G.-N.; Shao, J.-K.; Dong, X.-F.; Jin, L.-L.; Wang, C.-Q. p-STAT3 expression in breast cancer correlates negatively with tumor size and HER2 status. Medicine 2021, 100, e25124. [Google Scholar] [CrossRef]
- Zhang, N.; Ma, Z.P.; Wang, J.; Bai, H.L.; Li, Y.X.; Sun, Q.; Yang, L.; Tao, L.; Zhao, J.; Cao, Y.W.; et al. Human papillomavirus infection correlates with inflammatory Stat3 signaling activity and IL-17 expression in patients with breast cancer. Am. J. Transl. Res. 2016, 8, 3214–3226. [Google Scholar]
- Li, Y.; Wang, Y.; Shi, Z.; Liu, J.; Zheng, S.; Yang, J.; Liu, Y.; Yang, Y.; Chang, F.; Yu, W. Clinicopathological and Prognostic Role of STAT3/p-STAT3 in Breast Cancer Patients in China: A Meta-Analysis. Sci. Rep. 2019, 9, 11243. [Google Scholar] [CrossRef]
- Nilsson, L.; Sandén, E.; Khazaei, S.; Tryggvadottir, H.; Nodin, B.; Jirström, K.; Borgquist, S.; Isaksson, K.; Jernström, H. Patient Characteristics Influence Activated Signal Transducer and Activator of Transcription 3 (STAT3) Levels in Primary Breast Cancer—Impact on Prognosis. Front. Oncol. 2020, 10, 1278. [Google Scholar] [CrossRef]
- Aleskandarany, M.; Agarwal, D.; Negm, O.H.; Ball, G.; Elmouna, A.; Ashankyty, I.; Nuglozeh, E.; Fazaludeen, F.; Diez-Rodriguez, M.; Nolan, C.C.; et al. The prognostic significance of STAT3 in invasive breast cancer: Analysis of protein and mRNA expressions in large cohorts. Breast Cancer Res. Treat. 2016, 156, 9–20. [Google Scholar] [CrossRef]
- Segatto, I.; Baldassarre, G.; Belletti, B. STAT3 in Breast Cancer Onset and Progression: A Matter of Time and Context. Int. J. Mol. Sci. 2018, 19, 2818. [Google Scholar] [CrossRef]
- Solakoglu, O.; Maierhofer, C.; Lahr, G.; Breit, E.; Scheunemann, P.; Heumos, I.; Pichlmeier, U.; Schlimok, G.; Oberneder, R.; Köllermann, M.W.; et al. Heterogeneous proliferative potential of occult metastatic cells in bone marrow of patients with solid epithelial tumors. Proc. Natl. Acad. Sci. USA 2002, 99, 2246–2251. [Google Scholar] [CrossRef] [PubMed]
- Salgado, R.; Junius, S.; Benoy, I.; Van Dam, P.; Vermeulen, P.; Van Marck, E.; Huget, P.; Dirix, L.Y. Circulating interleukin-6 predicts survival in patients with metastatic breast cancer. Int. J. Cancer 2003, 103, 642–646. [Google Scholar] [CrossRef]
- Maroni, P.; Bendinelli, P.; Ferraretto, A.; Lombardi, G. Interleukin 11 (IL-11): Role(s) in Breast Cancer Bone Metastases. Biomedicines 2021, 9, 659. [Google Scholar] [CrossRef] [PubMed]
- Horwood, N.J.; Elliott, J.; Martin, T.J.; Gillespie, M.T. Osteotropic Agents Regulate the Expression of Osteoclast Differentiation Factor and Osteoprotegerin in Osteoblastic Stromal Cells. Endocrinology 1998, 139, 4743. [Google Scholar] [CrossRef]
- Sims, N.A.; Jenkins, B.J.; Nakamura, A.; Quinn, J.M.; Li, R.; Gillespie, M.T.; Ernst, M.; Robb, L.; Martin, T.J. Interleukin-11 Receptor Signaling Is Required for Normal Bone Remodeling. J. Bone Miner. Res. 2005, 20, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Kozlow, W.; Guise, T.A. Breast Cancer Metastasis to Bone: Mechanisms of Osteolysis and Implications for Therapy. J. Mammary Gland. Biol. Neoplasia 2005, 10, 169–180. [Google Scholar] [CrossRef]
- Kang, Y.; He, W.; Tulley, S.; Gupta, G.P.; Serganova, I.; Chen, C.-R.; Manova-Todorova, K.; Blasberg, R.; Gerald, W.L.; Massagué, J. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 13909–13914. [Google Scholar] [CrossRef]
- Kim, Y.K.; Bae, G.-U.; Kang, J.K.; Park, J.W.; Lee, E.K.; Lee, H.Y.; Choi, W.S.; Lee, H.W.; Han, J.-W. Cooperation of H2O2-mediated ERK activation with Smad pathway in TGF-β1 induction of p21WAF1/Cip 1. Cell. Signal. 2006, 18, 236–243. [Google Scholar] [CrossRef]
- Yuan, J.-H.; Yang, F.; Wang, F.; Ma, J.-Z.; Guo, Y.-J.; Tao, Q.-F.; Liu, F.; Pan, W.; Wang, T.-T.; Zhou, C.-C.; et al. A Long Noncoding RNA Activated by TGF-β Promotes the Invasion-Metastasis Cascade in Hepatocellular Carcinoma. Cancer Cell 2014, 25, 666–681. [Google Scholar] [CrossRef]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordón-Cardo, C.; Guise, T.A.; Massagué, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef]
- Pollari, S.; Leivonen, S.-K.; Perälä, M.; Fey, V.; Käkönen, S.-M.; Kallioniemi, O. Identification of MicroRNAs Inhibiting TGF-β-Induced IL-11 Production in Bone Metastatic Breast Cancer Cells. PLoS ONE 2012, 7, e37361. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.-L.; Huang, W.-D.; Li, B.; Chen, T.-R.; Li, Z.-X.; Zhao, C.-L.; Li, H.-Y.; Wu, Y.-M.; Yan, W.-J.; Xiao, J.-R. microRNA-124 inhibits bone metastasis of breast cancer by repressing Interleukin-11. Mol. Cancer 2018, 17, 1–14. [Google Scholar] [CrossRef]
- Helbig, G.; Christopherson, K.W.; Bhat-Nakshatri, P.; Kumar, S.; Kishimoto, H.; Miller, K.D.; Broxmeyer, H.E.; Nakshatri, H. NF-κB Promotes Breast Cancer Cell Migration and Metastasis by Inducing the Expression of the Chemokine Receptor CXCR. J. Biol. Chem. 2003, 278, 21631–21638. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, C.; Pan, J.; Chen, L.; Qi, S.-T. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human adamantinomatous craniopharyngioma cells and promotes tumor cell migration. Mol. Med. Rep. 2017, 15, 4123–4131. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.-W.; Liu, L.-J.; Huang, J. Interleukin-6-induced epithelial-mesenchymal transition through signal transducer and activator of transcription 3 in human cervical carcinoma. Int. J. Oncol. 2014, 45, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Ebbing, E.A.; van der Zalm, A.P.; Steins, A.; Creemers, A.; Hermsen, S.; Rentenaar, R.; Klein, M.; Waasdorp, C.; Hooijer, G.K.J.; Meijer, S.L.; et al. Stromal-derived interleukin 6 drives epithelial-to-mesenchymal transition and therapy resistance in esophageal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2019, 116, 2237–2242. [Google Scholar] [CrossRef]
- Goulet, C.R.; Champagne, A.; Bernard, G.; Vandal, D.; Chabaud, S.; Pouliot, F.; Bolduc, S. Cancer-associated fibroblasts induce epithelial–mesenchymal transition of bladder cancer cells through paracrine IL-6 signalling. BMC Cancer 2019, 19, 1–13. [Google Scholar] [CrossRef]
- Yadav, A.; Kumar, B.; Datta, J.; Teknos, T.N.; Kumar, P. IL-6 Promotes Head and Neck Tumor Metastasis by Inducing Epithelial–Mesenchymal Transition via the JAK-STAT3-SNAIL Signaling Pathway. Mol. Cancer Res. 2011, 9, 1658–1667. [Google Scholar] [CrossRef]
- Takebe, N.; Warren, R.Q.; Ivy, S.P. Breast cancer growth and metastasis: Interplay between cancer stem cells, embryonic signaling pathways and epithelial-to-mesenchymal transition. Breast Cancer Res. 2011, 13, 211. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, N.J.; Sasser, A.K.; Axel, E.A.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial–mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef]
- Sansone, P.; Storci, G.; Tavolari, S.; Guarnieri, T.; Giovannini, C.; Taffurelli, M.; Ceccarelli, C.; Santini, D.; Paterini, P.; Marcu, K.B.; et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J. Clin. Investig. 2007, 117, 3988–4002. [Google Scholar] [CrossRef]
- Gyamfi, J.; Lee, Y.-H.; Eom, M.; Choi, J. Interleukin-6/STAT3 signalling regulates adipocyte induced epithelial-mesenchymal transition in breast cancer cells. Sci. Rep. 2018, 8, 8859. [Google Scholar] [CrossRef]
- He, J.-Y.; Wei, X.-H.; Li, S.-J.; Liu, Y.; Hu, H.-L.; Li, Z.-Z.; Kuang, X.-H.; Wang, L.; Shi, X.; Yuan, S.-T.; et al. Adipocyte-derived IL-6 and leptin promote breast Cancer metastasis via upregulation of Lysyl Hydroxylase-2 expression. Cell Commun. Signal. 2018, 16, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Lee, W.S.; Jeong, J.; Kim, S.-J.; Jin, W. Induction of metastatic potential by TrkB via activation of IL6/JAK2/STAT3 and PI3K/AKT signaling in breast cancer. Oncotarget 2015, 6, 40158–40171. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.; Lee, O.-Y.; Shon, S.Y.; Nam, O.; Ryu, P.D.; Seo, M.W.; Lee, D.-S. A mutual activation loop between breast cancer cells and myeloid-derived suppressor cells facilitates spontaneous metastasis through IL-6 trans-signaling in a murine model. Breast Cancer Res. 2013, 15, R79. [Google Scholar] [CrossRef]
- Xiao, Y.; Cong, M.; Li, J.; He, D.; Wu, Q.; Tian, P.; Wang, Y.; Yang, S.; Liang, C.; Liang, Y.; et al. Cathepsin C promotes breast cancer lung metastasis by modulating neutrophil infiltration and neutrophil extracellular trap formation. Cancer Cell 2021, 39, 423–437.e7. [Google Scholar] [CrossRef]
- Weng, Y.-S.; Tseng, H.-Y.; Chen, Y.-A.; Shen, P.-C.; Al Haq, A.T.; Chen, L.-M.; Tung, Y.-C.; Hsu, H.-L. MCT-1/miR-34a/IL-6/IL-6R signaling axis promotes EMT progression, cancer stemness and M2 macrophage polarization in triple-negative breast cancer. Mol. Cancer 2019, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Siersbæk, R.; Scabia, V.; Nagarajan, S.; Chernukhin, I.; Papachristou, E.K.; Broome, R.; Johnston, S.J.; Joosten, S.E.; Green, A.R.; Kumar, S.; et al. IL6/STAT3 Signaling Hijacks Estrogen Receptor α Enhancers to Drive Breast Cancer Metastasis. Cancer Cell 2020, 38, 412–423.e9. [Google Scholar] [CrossRef] [PubMed]
- Sansone, P.; Ceccarelli, C.; Berishaj, M.; Chang, Q.; Rajasekhar, V.K.; Perna, F.; Bowman, R.L.; Vidone, M.; Daly, L.; Nnoli, J.; et al. Self-renewal of CD133hi cells by IL6/Notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nat. Commun. 2016, 7, 10442. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.F.; Iglesias, M.; Céspedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.-F.; et al. Dependency of Colorectal Cancer on a TGF-β-Driven Program in Stromal Cells for Metastasis Initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef]
- Huynh, J.; Baloyan, D.; Chisanga, D.; Shi, W.; O’Brien, M.; Afshar-Sterle, S.; Alorro, M.; Pang, L.; Williams, D.S.; Parslow, A.C.; et al. Host IL11 Signaling Suppresses CD4+ T cell–Mediated Antitumor Responses to Colon Cancer in Mice. Cancer Immunol. Res. 2021, 9, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.-A.; Kim, M.; Kang, M.-C.; Kong, J.-S.; Kim, K.-M.; Lee, S.; Hong, B.-K.; Jeong, G.H.; Lee, J.; Shin, M.-G.; et al. Placental growth factor regulates the generation of TH17 cells to link angiogenesis with autoimmunity. Nat. Immunol. 2019, 20, 1348–1359. [Google Scholar] [CrossRef]
- Kumar, V.; Cheng, P.; Condamine, T.; Mony, S.; Languino, L.; McCaffrey, J.C.; Hockstein, N.; Guarino, M.; Masters, G.; Penman, E.; et al. CD45 Phosphatase Inhibits STAT3 Transcription Factor Activity in Myeloid Cells and Promotes Tumor-Associated Macrophage Differentiation. Immunity 2016, 44, 303–315. [Google Scholar] [CrossRef]
- Kortylewski, M.; Kujawski, M.; Wang, T.; Wei, S.; Zhang, S.; Pilon-Thomas, S.; Niu, G.; Kay, H.; Mulé, J.; Kerr, W.; et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 2005, 11, 1314–1321. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Coussens, L.M. Macrophages and Therapeutic Resistance in Cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Wang, H.-W.; Bowman, R.L.; Joyce, J.A. STAT3 and STAT6 Signaling Pathways Synergize to Promote Cathepsin Secretion from Macrophages via IRE1α Activation. Cell Rep. 2016, 16, 2914–2927. [Google Scholar] [CrossRef]
- Irey, E.A.; Lassiter, C.M.; Brady, N.J.; Chuntova, P.; Wang, Y.; Knutson, T.P.; Henzler, C.; Chaffee, T.S.; Vogel, R.; Nelson, A.C.; et al. JAK/STAT inhibition in macrophages promotes therapeutic resistance by inducing expression of protumorigenic factors. Proc. Natl. Acad. Sci. USA 2019, 116, 12442–12451. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Qu, Z.; Sun, F.; Han, L.; Li, L.; Yan, S.; Stabile, L.P.; Chen, L.-F.; Siegfried, J.M.; Xiao, G. Myeloid STAT3 Promotes Lung Tumorigenesis by Transforming Tumor Immunosurveillance into Tumor-Promoting Inflammation. Cancer Immunol. Res. 2017, 5, 257–268. [Google Scholar] [CrossRef]
- Pathria, P.; Gotthardt, D.; Prchal-Murphy, M.; Putz, E.-M.; Holcmann, M.; Schlederer, M.; Grabner, B.; Crncec, I.; Svinka, J.; Musteanu, M.; et al. MyeloidSTAT3promotes formation of colitis-associated colorectal cancer in mice. OncoImmunology 2015, 4, e998529. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xiao, H.; Lin, L.; Jou, D.; Kumari, V.; Lin, J.; Li, C. Drug Design Targeting Protein–Protein Interactions (PPIs) Using Multiple Ligand Simultaneous Docking (MLSD) and Drug Repositioning: Discovery of Raloxifene and Bazedoxifene as Novel Inhibitors of IL-6/GP130 Interface. J. Med. Chem. 2014, 57, 632–641. [Google Scholar] [CrossRef]
- Thilakasiri, P.S.; Dmello, R.S.; Nero, T.L.; Parker, M.; Ernst, M.; Chand, A.L. Repurposing of drugs as STAT3 inhibitors for cancer therapy. Semin. Cancer Biol. 2021, 68, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Chen, X.; Fu, S.; Zhang, R.; Pan, L.; Cao, Y.; Wu, X.; Xiao, H.; Lin, H.-J.; Lo, H.-W.; et al. Bazedoxifene is a novel IL-6/GP130 inhibitor for treating triple-negative breast cancer. Breast Cancer Res. Treat. 2019, 175, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Santen, R.J.; Wang, J.-P.; Yue, W. Inhibitory Effects of a Bazedoxifene/Conjugated Equine Estrogen Combination on Human Breast Cancer Cells In Vitro. Endocrinology 2012, 154, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Chen, X.; Lo, H.-W.; Lin, J. Combined bazedoxifene and paclitaxel treatments inhibit cell viability, cell migration, colony formation, and tumor growth and induce apoptosis in breast cancer. Cancer Lett. 2019, 448, 11–19. [Google Scholar] [CrossRef]
- Balko, J.M.; Schwarz, L.J.; Luo, N.; Estrada, M.V.; Giltnane, J.M.; Dávila-González, D.; Wang, K.; Sánchez, V.; Dean, P.T.; Combs, S.E.; et al. Triple-negative breast cancers with amplification of JAK2 at the 9p24 locus demonstrate JAK2-specific dependence. Sci. Transl. Med. 2016, 8, 334ra53. [Google Scholar] [CrossRef]
- Gadina, M.; Johnson, C.; Schwartz, D.; Bonelli, M.; Hasni, S.; Kanno, Y.; Changelian, P.; Laurence, A.; O’Shea, J.J. Translational and clinical advances in JAK-STAT biology: The present and future of jakinibs. J. Leukoc. Biol. 2018, 104, 499–514. [Google Scholar] [CrossRef]
- Stover, D.G.; Gil Del Alcazar, C.R.; Brock, J.; Guo, H.; Overmoyer, B.; Balko, J.; Xu, Q.; Bardia, A.; Tolaney, S.M.; Gelman, R.; et al. Phase II study of ruxolitinib, a selective JAK1/2 inhibitor, in patients with metastatic triple-negative breast cancer. NPJ Breast Cancer 2018, 4, 10. [Google Scholar] [CrossRef]
- Pan, L.; Chen, X.; Fu, S.; Yu, W.; Li, C.; Wang, T.; Lo, H.-W.; Lin, J. LLY17, a novel small molecule STAT3 inhibitor induces apoptosis and suppresses cell migration and tumor growth in triple-negative breast cancer. Breast Cancer Res. Treat. 2020, 181, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.W.; Saadalla, A.; Ewida, A.H.; Al-Katranji, K.; Al-Saoudi, G.; Giaccone, Z.T.; Gounari, F.; Zhang, M.; Frank, D.A.; Khazaie, K. The STAT3 inhibitor pyrimethamine displays anti-cancer and immune stimulatory effects in murine models of breast cancer. Cancer Immunol. Immunother. 2018, 67, 13–23. [Google Scholar] [CrossRef]
- Nair, V.S.; Toor, S.M.; Ali, B.R.; Elkord, E. Dual inhibition of STAT1 and STAT3 activation downregulates expression of PD-L1 in human breast cancer cells. Expert Opin. Ther. Targets 2018, 22, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Kim, S.-J.; Hahn, Y.-I.; Yoon, H.-J.; Han, B.; Kim, K.; Lee, S.; Kim, K.P.; Suh, Y.G.; Na, H.-K.; et al. 15-Keto prostaglandin E2 suppresses STAT3 signaling and inhibits breast cancer cell growth and progression. Redox Biol. 2019, 23, 101175. [Google Scholar] [CrossRef]
- Dai, X.; Yin, C.; Zhang, Y.; Guo, G.; Zhao, C.; Wang, O.; Xiang, Y.; Zhang, X.; Liang, G. Osthole inhibits triple negative breast cancer cells by suppressing STAT3. J. Exp. Clin. Cancer Res. 2018, 37, 1–11. [Google Scholar] [CrossRef]
- Zeng, A.-Q.; Yu, Y.; Yao, Y.-Q.; Yang, F.-F.; Liao, M.; Song, L.-J.; Li, Y.-L.; Li, Y.-J.; Deng, Y.-L.; Yang, S.-P.; et al. Betulinic acid impairs metastasis and reduces immunosuppressive cells in breast cancer models. Oncotarget 2017, 9, 3794–3804. [Google Scholar] [CrossRef]
- Kasembeli, M.M.; Singhmar, P.; Ma, J.; Edralin, J.; Tang, Y.; Adams, C.; Heijnen, C.J.; Kavelaars, A.; Tweardy, D.J. TTI-101: A competitive inhibitor of STAT3 that spares oxidative phosphorylation and reverses mechanical allodynia in mouse models of neuropathic pain. Biochem. Pharmacol. 2021, 192, 114688. [Google Scholar] [CrossRef]
- Doberer, K.; Duerr, M.; Halloran, P.F.; Eskandary, F.; Budde, K.; Regele, H.; Reeve, J.; Borski, A.; Kozakowski, N.; Reindl-Schwaighofer, R.; et al. A Randomized Clinical Trial of Anti–IL-6 Antibody Clazakizumab in Late Antibody-Mediated Kidney Transplant Rejection. J. Am. Soc. Nephrol. 2021, 32, 708–722. [Google Scholar] [CrossRef]
- Weinblatt, M.E.; Mease, P.; Mysler, E.; Takeuchi, T.; Drescher, E.; Berman, A.; Xing, J.; Zilberstein, M.; Banerjee, S.; Emery, P. The Efficacy and Safety of Subcutaneous Clazakizumab in Patients with Moderate-to-Severe Rheumatoid Arthritis and an Inadequate Response to Methotrexate: Results from a Multinational, Phase IIb, Randomized, Double-Blind, Placebo/Active-Controlled, Dose-Ra. Arthritis Rheumatol. 2015, 67, 2591–2600. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, G.; Czer, L.S.; Kobashigawa, J.; Kittleson, M.; Patel, J.; Chang, D.; Kransdorf, E.; Shikhare, A.; Tran, H.; Vo, A.; et al. Successful Treatment of Severe COVID-19 Pneumonia with Clazakizumab in a Heart Transplant Recipient: A Case Report. Transplant. Proc. 2020, 52, 2711–2714. [Google Scholar] [CrossRef]
- Wang, D.; Fu, B.; Peng, Z.; Yang, D.; Han, M.; Li, M.; Yang, Y.; Yang, T.; Sun, L.; Li, W.; et al. Tocilizumab in patients with moderate or severe COVID-19: A randomized, controlled, open-label, multicenter trial. Front. Med. 2021, 15, 486–494. [Google Scholar] [CrossRef]
- Hagi, T.; Nakamura, T.; Kita, K.; Iino, T.; Asanuma, K.; Sudo, A. Anti-tumour effect of tocilizumab for osteosarcoma cell lines. Bone Jt. Res. 2020, 9, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.-H.; Kim, S.-K.; Kim, D.-S.; Zhang, D.; Park, J.-A.; Yi, H.; Kim, J.-S.; Shin, H.-C. Anti-proliferative action of IL-6R-targeted antibody tocilizumab for non-small cell lung cancer cells. Oncol. Lett. 2015, 9, 2283–2288. [Google Scholar] [CrossRef][Green Version]
- Zhong, H.; Davis, A.; Ouzounova, M.; Carrasco, R.A.; Chen, C.; Breen, S.; Chang, Y.S.; Huang, J.; Liu, Z.; Yao, Y.; et al. A Novel IL6 Antibody Sensitizes Multiple Tumor Types to Chemotherapy Including Trastuzumab-Resistant Tumors. Cancer Res. 2016, 76, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Alraouji, N.N.; Aboussekhra, A. Tocilizumab inhibits IL-8 and the proangiogenic potential of triple negative breast cancer cells. Mol. Carcinog. 2021, 60, 51–59. [Google Scholar] [CrossRef]
- Rossi, J.-F.; Négrier, S.; James, N.D.; Kocak, I.; Hawkins, R.; Davis, H.; Prabhakar, U.; Qin, X.; Mulders, P.; Berns, B. A phase I/II study of siltuximab (CNTO 328), an anti-interleukin-6 monoclonal antibody, in metastatic renal cell cancer. Br. J. Cancer 2010, 103, 1154–1162. [Google Scholar] [CrossRef]
- Karkera, J.; Steiner, H.; Li, W.; Skradski, V.; Moser, P.L.; Riethdorf, S.; Reddy, M.; Puchalski, T.; Safer, K.; Prabhakar, U.; et al. The anti-interleukin-6 antibody siltuximab down-regulates genes implicated in tumorigenesis in prostate cancer patients from a phase I study. Prostate 2011, 71, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- van Rhee, F.; Fayad, L.; Voorhees, P.; Furman, R.; Lonial, S.; Borghaei, H.; Sokol, L.; Crawford, J.; Cornfeld, M.; Qi, M.; et al. Siltuximab, a Novel Anti–Interleukin-6 Monoclonal Antibody, for Castleman’s Disease. J. Clin. Oncol. 2010, 28, 3701–3708. [Google Scholar] [CrossRef] [PubMed]
- Long, K.B.; Tooker, G.; Tooker, E.; Luque, S.L.; Lee, J.; Pan, X.; Beatty, G.L. IL6 Receptor Blockade Enhances Chemotherapy Efficacy in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2017, 16, 1898–1908. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Che, X.; Liu, C.; Fan, Y.; Bai, M.; Hou, K.; Shi, X.; Zhang, X.; Liu, B.; Zheng, C.; et al. Cancer-associated fibroblasts-stimulated interleukin-11 promotes metastasis of gastric cancer cells mediated by upregulation of MUC 1. Exp. Cell Res. 2018, 368, 184–193. [Google Scholar] [CrossRef]
- Winship, A.; Van Sinderen, M.; Rainczuk, K.; Dimitriadis, E. Therapeutically blocking interleukin-11 receptor-α enhances doxorubicin cytotoxicity in high grade type I endometrioid tumours. Oncotarget 2017, 8, 22716–22729. [Google Scholar] [CrossRef]
- Winship, A.; Van Sinderen, M.; Donoghue, J.; Rainczuk, K.; Dimitriadis, E. Targeting Interleukin-11 Receptor-α Impairs Human Endometrial Cancer Cell Proliferation and Invasion In Vitro and Reduces Tumor Growth and Metastasis In Vivo. Mol. Cancer Ther. 2016, 15, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.-H. Inhibition of the Interleukin-11-STAT3 Axis Attenuates Hypoxia-Induced Migration and Invasion in MDA-MB-231 Breast Cancer Cells. Kor. J. Physiol. Pharmacol. 2014, 18, 391–396. [Google Scholar] [CrossRef]
- Schmidt, S.; Schumacher, N.; Schwarz, J.; Tangermann, S.; Kenner, L.; Schlederer, M.; Sibilia, M.; Linder, M.; Altendorf-Hofmann, A.; Knösel, T.; et al. ADAM17 is required for EGF-R–induced intestinal tumors via IL-6 trans-signaling. J. Exp. Med. 2018, 215, 1205–1225. [Google Scholar] [CrossRef]
- Brooks, G.D.; McLeod, L.; Alhayyani, S.; Miller, A.; Russell, P.A.; Ferlin, W.; Rose-John, S.; Ruwanpura, S.; Jenkins, B.J. IL6 Trans-signaling Promotes KRAS-Driven Lung Carcinogenesis. Cancer Res. 2016, 76, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Putoczki, T.L.; Thiem, S.; Loving, A.; Busuttil, R.A.; Wilson, N.J.; Ziegler, P.K.; Nguyen, P.M.; Preaudet, A.; Farid, R.; Edwards, K.M.; et al. Interleukin-11 Is the Dominant IL-6 Family Cytokine during Gastrointestinal Tumorigenesis and Can Be Targeted Therapeutically. Cancer Cell 2013, 24, 257–271. [Google Scholar] [CrossRef]
- Qin, J.-J.; Yan, L.; Zhang, J.; Zhang, W.-D. STAT3 as a potential therapeutic target in triple negative breast cancer: A systematic review. J. Exp. Clin. Cancer Res. 2019, 38, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, Y.; Tilborghs, S.; Jacobs, J.; De Waele, J.; Quatannens, D.; Deben, C.; Prenen, H.; Pauwels, P.; Trinh, X.B.; Wouters, A.; et al. The potential and controversy of targeting STAT family members in cancer. Semin. Cancer Biol. 2020, 60, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Z. STAT1 in cancer: Friend or foe? Discov. Med. 2017, 24, 19–29. [Google Scholar] [PubMed]
- Bendell, J.C.; Hong, D.S.; Burris, H.A.; Naing, A.; Jones, S.F.; Falchook, G.; Bricmont, P.; Elekes, A.; Rock, E.P.; Kurzrock, R. Phase 1, open-label, dose-escalation, and pharmacokinetic study of STAT3 inhibitor OPB-31121 in subjects with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 74, 125–130. [Google Scholar] [CrossRef]
- Jonker, D.J.; Nott, L.; Yoshino, T.; Gill, S.; Shapiro, J.; Ohtsu, A.; Zalcberg, J.; Vickers, M.M.; Wei, A.C.; Gao, Y.; et al. Napabucasin versus placebo in refractory advanced colorectal cancer: A randomised phase 3 trial. Lancet Gastroenterol. Hepatol. 2018, 3, 263–270. [Google Scholar] [CrossRef]
- Balic, J.J.; Albargy, H.; Luu, K.; Kirby, F.J.; Jayasekara, W.S.N.; Mansell, F.; Garama, D.J.; De Nardo, D.; Baschuk, N.; Louis, C.; et al. STAT3 serine phosphorylation is required for TLR4 metabolic reprogramming and IL-1β expression. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Belo, Y.; Mielko, Z.; Nudelman, H.; Afek, A.; Ben-David, O.; Shahar, A.; Zarivach, R.; Gordan, R.; Arbely, E. Unexpected implications of STAT3 acetylation revealed by genetic encoding of acetyl-lysine. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1343–1350. [Google Scholar] [CrossRef]
- Hirahara, K.; Onodera, A.; Villarino, A.; Bonelli, M.; Sciume, G.; Laurence, A.; Sun, H.-W.; Brooks, S.R.; Vahedi, G.; Shih, H.-Y.; et al. Asymmetric Action of STAT Transcription Factors Drives Transcriptional Outputs and Cytokine Specificity. Immunity 2015, 42, 877–889. [Google Scholar] [CrossRef]
- Wingelhofer, B.; Neubauer, H.A.; Valent, P.; Han, X.; Constantinescu, S.N.; Gunning, P.T.; Müller, M.; Moriggl, R. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia 2018, 32, 1713–1726. [Google Scholar] [CrossRef]
- Bai, L.; Zhou, H.; Xu, R.; Zhao, Y.; Chinnaswamy, K.; McEachern, D.; Chen, J.; Yang, C.-Y.; Liu, Z.; Wang, M.; et al. A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo. Cancer Cell 2019, 36, 498–511.e17. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Therapeutic | Target | Mechanism | FDA Approval |
|---|---|---|---|
| Bazedoxifene | gp130 receptor | Selective estrogen receptor modulator (SERM) which disrupts interaction between IL6 and gp130 | Menopause osteoporosis and moderate to severe hot flushes |
| Baricitinib | JAK1 and JAK2 | Small molecule inhibition of JAK1 and JAK2 | Rheumatoid arthritis |
| Ruxolitinib | JAK1 and JAK2 | Small molecule inhibition of JAK1 and JAK2 | Myelofibrosis and polycythemia vera |
| LLY17 | STAT3 | Inhibits STAT3 phosphorylation and dimerization | N/A |
| Pyrimethamine | STAT3 | Inhibition of STAT3 Phosphorylation | N/A |
| TTI-01 | STAT3 | Inhibits recruitment of STAT3 to activated cytokine receptor complexes and dimerization | N/A |
| BBI608 | STAT3 | Inhibition of STAT3 Phosphorylation | N/A |
| SD-36 | STAT3 | Targets STAT3 for ubiquitination and proteasomal degradation | N/A |
| Clazakizumab | IL-6 | Monoclonal antibody against IL6 ligand | N/A |
| Tocilizumab | IL-6R | Monoclonal antibody against IL6 receptor | Rheumatoid arthritis, systemic juvenile idiopathic arthritis, cytokine release syndrome, COVID-19 |
| Siltuximab | IL-6 | Monoclonal antibody against IL6 ligand | Idiopathic multicentric Castleman disease |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
To, S.Q.; Dmello, R.S.; Richards, A.K.; Ernst, M.; Chand, A.L. STAT3 Signaling in Breast Cancer: Multicellular Actions and Therapeutic Potential. Cancers 2022, 14, 429. https://doi.org/10.3390/cancers14020429
To SQ, Dmello RS, Richards AK, Ernst M, Chand AL. STAT3 Signaling in Breast Cancer: Multicellular Actions and Therapeutic Potential. Cancers. 2022; 14(2):429. https://doi.org/10.3390/cancers14020429
Chicago/Turabian StyleTo, Sarah Q., Rhynelle S. Dmello, Anna K. Richards, Matthias Ernst, and Ashwini L. Chand. 2022. "STAT3 Signaling in Breast Cancer: Multicellular Actions and Therapeutic Potential" Cancers 14, no. 2: 429. https://doi.org/10.3390/cancers14020429
APA StyleTo, S. Q., Dmello, R. S., Richards, A. K., Ernst, M., & Chand, A. L. (2022). STAT3 Signaling in Breast Cancer: Multicellular Actions and Therapeutic Potential. Cancers, 14(2), 429. https://doi.org/10.3390/cancers14020429