
Past, Current, and Future Strategies to Target ERG Fusion-Positive Prostate Cancer

Abstract
Simple Summary
Abstract
1. Introduction
2. ERG Functions in Development and Normal Physiology
3. ERG Interactors and Post-Translational Modifications
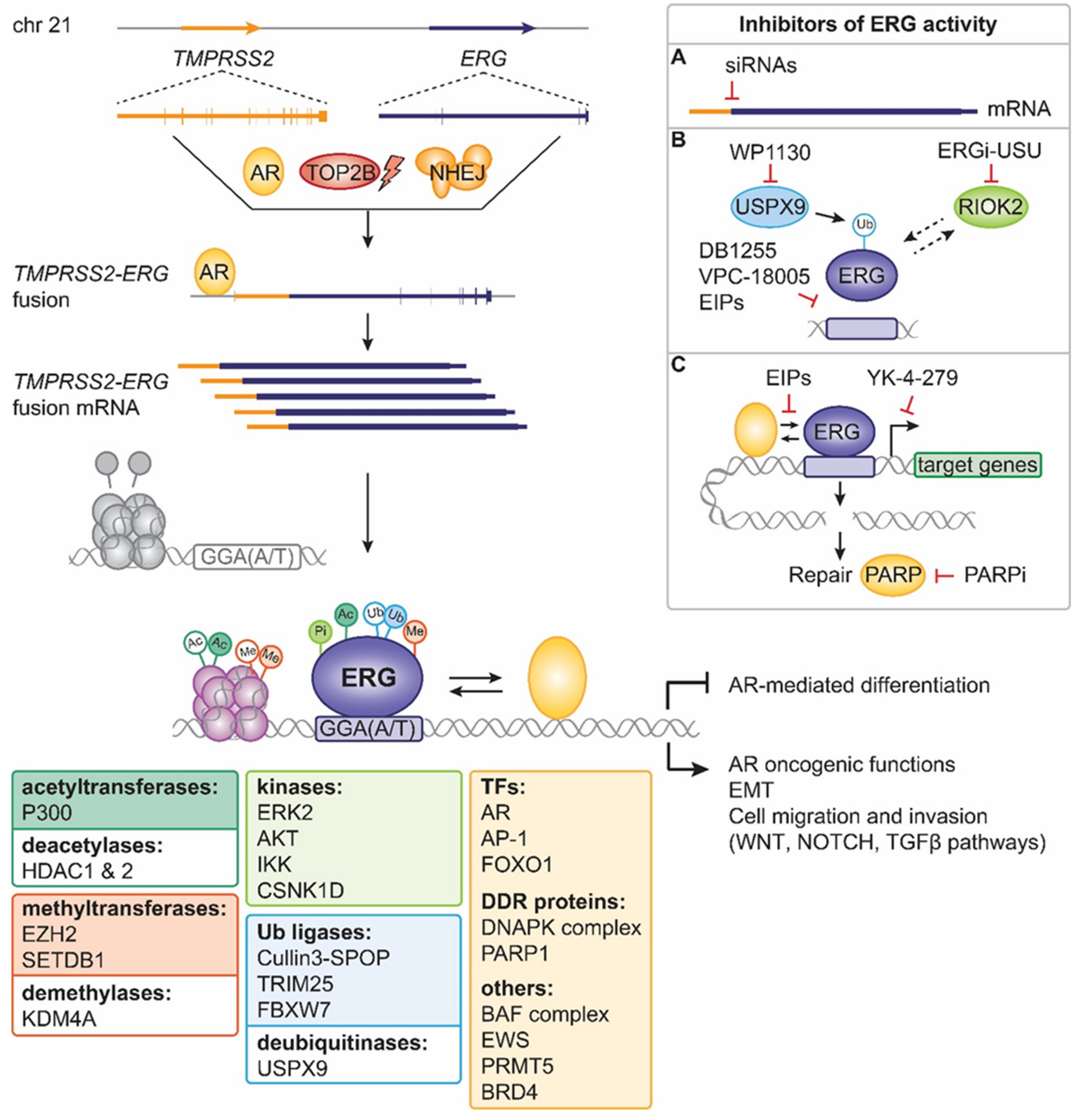
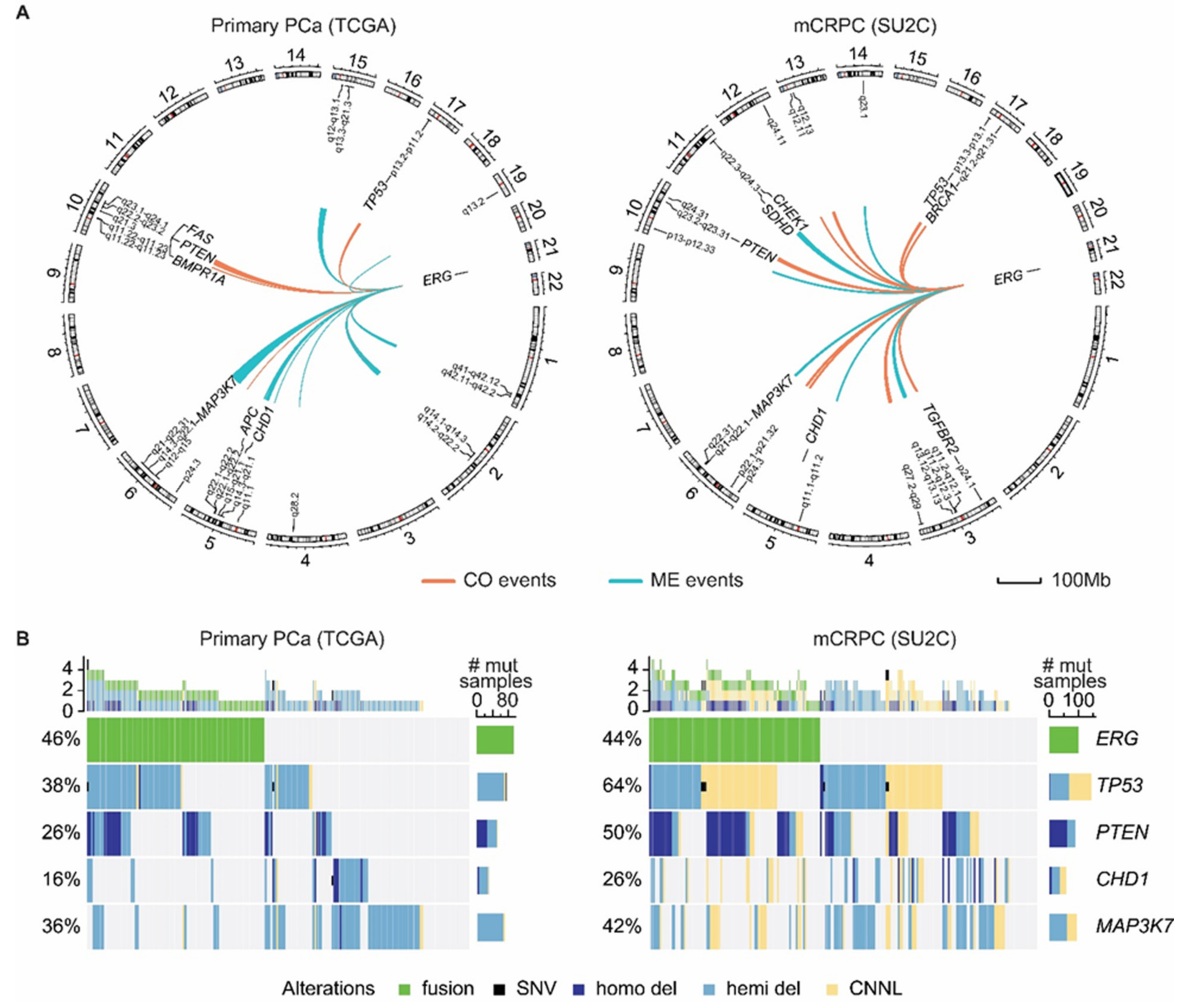
4. ERG Fusions in Prostate Cancer
5. Past Approaches to Target ERG Oncogenic Activity in Prostate Cancer
6. Present Approaches to Exploit ERG in Prostate Cancer
7. Future Strategies to Target ERG-Positive Prostate Cancer
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rao, V.; Papas, T.; Reddy, E. erg, a human ets-related gene on chromosome 21: Alternative splicing, polyadenylation, and translation. Science 1987, 237, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Laudet, V.; Hänni, C.; Stéhelin, D.; Duterque-Coquillaud, M. Molecular phylogeny of the ETS gene family. Oncogene 1999, 18, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Karim, F.D.; Urness, L.D.; Thummel, C.S.; Klemsz, M.J.; McKercher, S.R.; Celada, A.; van Beveren, C.; Maki, R.A.; Gunther, C.V.; Nye, J.A.; et al. The ETS-domain: A new DNA-binding motif that recognizes a purine rich core DNA sequence. Genes Dev. 1990, 4, 1451–1453. [Google Scholar] [CrossRef]
- Clark, J.P.; Cooper, C.S. ETS gene fusions and prostate cancer. Nat. Rev. Urol. 2009, 6, 429–439. [Google Scholar] [CrossRef]
- Findlay, V.J.; LaRue, A.C.; Turner, D.P.; Watson, P.M.; Watson, D.K. Understanding the Role of ETS-Mediated Gene Regulation in Complex Biological Processes. Adv. Cancer Res. 2013, 119, 1–61. [Google Scholar] [CrossRef]
- Sizemore, G.M.; Pitarresi, J.R.; Balakrishnan, S.; Ostrowski, M.C. The ETS family of oncogenic transcription factors in solid tumours. Nat. Rev. Cancer 2017, 17, 337–351. [Google Scholar] [CrossRef]
- Bartel, F.O.; Higuchi, T.; Spyropoulos, D.D. Mouse models in the study of the Ets family of transcription factors. Oncogene 2001, 19, 6443–6454. [Google Scholar] [CrossRef]
- Richardson, L.; Venkataraman, S.; Stevenson, P.; Yang, Y.; Burton, N.; Rao, J.; Fisher, M.; Baldock, R.A.; Davidson, D.R.; Christiansen, J.H. EMAGE mouse embryo spatial gene expression database: 2010 update. Nucleic Acids Res. 2010, 38, D703–D709. [Google Scholar] [CrossRef]
- Hollenhorst, P.C.; Jones, D.A.; Graves, B.J. Expression profiles frame the promoter specificity dilemma of the ETS family of transcription factors. Nucleic Acids Res. 2004, 32, 5693–5702. [Google Scholar] [CrossRef]
- Wei, G.-H.; Badis, G.; Berger, M.F.; Kivioja, T.; Palin, K.; Enge, M.; Bonke, M.; Jolma, A.; Varjosalo, M.; Gehrke, A.R.; et al. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J. 2010, 29, 2147–2160. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 162, 454. [Google Scholar] [CrossRef] [PubMed]
- Baltzinger, M.; Mager-Heckel, A.-M.; Remy, P. Xl erg: Expression Pattern and Overexpression During Development Plead for a Role in Endothelial Cell Differentiation. Dev. Dyn. 1999, 216, 420–433. [Google Scholar] [CrossRef]
- Vlaeminck-Guillem, V.; Carrere, S.; Dewitte, F.; Stehelin, D.; Desbiens, X.; Duterque-Coquillaud, M. The Ets family member Erg gene is expressed in mesodermal tissues and neural crests at fundamental steps during mouse embryogenesis. Mech. Dev. 2000, 91, 331–335. [Google Scholar] [CrossRef]
- Hewett, P.W.; Nishi, K.; Daft, E.L.; Clifford Murray, J. Selective expression of erg isoforms in human endothelial cells. Int. J. Biochem. Cell Biol. 2001, 33, 347–355. [Google Scholar] [CrossRef]
- Ellett, F.; Kile, B.T.; Lieschke, G.J. The role of the ETS factor erg in zebrafish vasculogenesis. Mech. Dev. 2009, 126, 220–229. [Google Scholar] [CrossRef]
- Yuan, L.; Nikolova-Krstevski, V.; Zhan, Y.; Kondo, M.; Bhasin, M.; Varghese, L.; Yano, K.; Carman, C.V.; Aird, W.C.; Oettgen, P. Antiinflammatory Effects of the ETS Factor ERG in Endothelial Cells Are Mediated Through Transcriptional Repression of the Interleukin-8 Gene. Circ. Res. 2009, 104, 1049–1057. [Google Scholar] [CrossRef]
- Vijayaraj, P.; Le Bras, A.; Mitchell, N.; Kondo, M.; Juliao, S.; Wasserman, M.; Beeler, D.; Spokes, K.; Aird, W.C.; Baldwin, H.S.; et al. Erg is a crucial regulator of endocardial-mesenchymal transformation during cardiac valve morphogenesis. Development 2012, 139, 3973–3985. [Google Scholar] [CrossRef]
- Birdsey, G.M.; Shah, A.V.; Dufton, N.; Reynolds, L.E.; Almagro, L.O.; Yang, Y.; Aspalter, I.M.; Khan, S.T.; Mason, J.C.; Dejana, E.; et al. The endothelial transcription factor erg promotes vascular stability and growth through Wnt/β-catenin signaling. Dev. Cell 2015, 32, 82–96. [Google Scholar] [CrossRef]
- Nikolova-Krstevski, V.; Yuan, L.; Le Bras, A.; Vijayaraj, P.; Kondo, M.; Gebauer, I.; Bhasin, M.; Carman, C.V.; Oettgen, P. ERG is required for the differentiation of embryonic stem cells along the endothelial lineage. BMC Dev. Biol. 2009, 9, 72. [Google Scholar] [CrossRef] [PubMed]
- Birdsey, G.M.; Dryden, N.H.; Amsellem, V.; Gebhardt, F.; Sahnan, K.; Haskard, D.O.; Dejana, E.; Mason, J.C.; Randi, A.M. Transcription factor erg regulates angiogenesis and endothelial apoptosis through VE-cadherin. Blood 2008, 111, 3498–3506. [Google Scholar] [CrossRef]
- Birdsey, G.M.; Dryden, N.H.; Shah, A.V.; Hannah, R.; Hall, M.D.; Haskard, D.O.; Parsons, M.; Mason, J.C.; Zvelebil, M.; Gottgens, B.; et al. The transcription factor Erg regulates expression of histone deacetylase 6 and multiple pathways involved in endothelial cell migration and angiogenesis. Blood 2012, 119, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Le Bras, A.; Sacharidou, A.; Itagaki, K.; Zhan, Y.; Kondo, M.; Carman, C.V.; Davis, G.E.; Aird, W.C.; Oettgen, P. ETS-related Gene (ERG) Controls Endothelial Cell Permeability via Transcriptional Regulation of the Claudin 5 (CLDN5) Gene. J. Biol. Chem. 2012, 287, 6582–6591. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.V.; Birdsey, G.M.; Peghaire, C.; Pitulescu, M.E.; Dufton, N.P.; Yang, Y.; Weinberg, I.; Osuna Almagro, L.; Payne, L.; Mason, J.C.; et al. The endothelial transcription factor ERG mediates Angiopoietin-1-dependent control of Notch signalling and vascular stability. Nat. Commun. 2017, 8, 16002. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, F.; Ludbrook, V.J.; Cox, J.; von Carlowitz, I.; Brown, S.; Randi, A.M. Combined genomic and antisense analysis reveals that the transcription factor Erg is implicated in endothelial cell differentiation. Blood 2001, 98, 3332–3339. [Google Scholar] [CrossRef]
- Shah, A.V.; Birdsey, G.M.; Randi, A.M. Regulation of endothelial homeostasis, vascular development and angiogenesis by the transcription factor ERG. Vascul. Pharmacol. 2016, 86, 3–13. [Google Scholar] [CrossRef]
- Dryden, N.H.; Sperone, A.; Martin-Almedina, S.; Hannah, R.L.; Birdsey, G.M.; Khan, S.T.; Layhadi, J.A.; Mason, J.C.; Haskard, D.O.; Göttgens, B.; et al. The Transcription Factor Erg Controls Endothelial Cell Quiescence by Repressing Activity of Nuclear Factor (NF)-κB p65. J. Biol. Chem. 2012, 287, 12331–12342. [Google Scholar] [CrossRef]
- Loughran, S.J.; Kruse, E.A.; Hacking, D.F.; de Graaf, C.A.; Hyland, C.D.; Willson, T.A.; Henley, K.J.; Ellis, S.; Voss, A.K.; Metcalf, D.; et al. The transcription factor Erg is essential for definitive hematopoiesis and the function of adult hematopoietic stem cells. Nat. Immunol. 2008, 9, 810–819. [Google Scholar] [CrossRef]
- Taoudi, S.; Bee, T.; Hilton, A.; Knezevic, K.; Scott, J.; Willson, T.A.; Collin, C.; Thomas, T.; Voss, A.K.; Kile, B.T.; et al. ERG dependence distinguishes developmental control of hematopoietic stem cell maintenance from hematopoietic specification. Genes Dev. 2011, 25, 251–262. [Google Scholar] [CrossRef]
- Anderson, M.K.; Hernandez-Hoyos, G.; Diamond, R.A.; Rothenberg, E.V. Precise developmental regulation of Ets family transcription factors during specification and commitment to the T cell lineage. Development 1999, 126, 3131–3148. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, S.; Taguchi, O.; Seto, M. Promotion and maintenance of leukemia by ERG. Blood 2011, 117, 3858–3868. [Google Scholar] [CrossRef] [PubMed]
- Thoms, J.A.I.; Birger, Y.; Foster, S.; Knezevic, K.; Kirschenbaum, Y.; Chandrakanthan, V.; Jonquieres, G.; Spensberger, D.; Wong, J.W.; Oram, S.H.; et al. ERG promotes T-acute lymphoblastic leukemia and is transcriptionally regulated in leukemic cells by a stem cell enhancer. Blood 2011, 117, 7079–7089. [Google Scholar] [CrossRef]
- Shimizu, K.; Ichikawa, H.; Tojo, A.; Kaneko, Y.; Maseki, N.; Hayashi, Y.; Ohira, M.; Asano, S.; Ohki, M. An ets-related gene, ERG, is rearranged in human myeloid leukemia with t(16;21) chromosomal translocation. Proc. Natl. Acad. Sci. USA 1993, 90, 10280. [Google Scholar] [CrossRef]
- Ichikawa, H.; Shimizu, K.; Hayashi, Y.; Ohki, M. An RNA-binding Protein Gene, TLS/FUS, Is Fused to ERG in Human Myeloid Leukemia with t(16;21) Chromosomal Translocation. Cancer Res. 1994, 54, 2865–2868. [Google Scholar] [PubMed]
- Cox, M.K.; Appelboom, B.L.; Ban, G.I.; Serra, R. Erg cooperates with TGF-β to control mesenchymal differentiation. Exp. Cell Res. 2014, 328, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M.; Higuchi, Y.; Koyama, E.; Enomoto-Iwamoto, M.; Kurisu, K.; Yeh, H.; Abrams, W.R.; Rosenbloom, J.; Pacifici, M. Transcription Factor Erg Variants and Functional Diversification of Chondrocytes during Limb Long Bone Development. J. Cell Biol. 2000, 150, 27–40. [Google Scholar] [CrossRef]
- Currie, S.L.; Lau, D.K.W.; Doane, J.J.; Whitby, F.G.; Okon, M.; McIntosh, L.P.; Graves, B.J. Structured and disordered regions cooperatively mediate DNA-binding autoinhibition of ETS factors ETV1, ETV4 and ETV5. Nucleic Acids Res. 2017, 45, 2223–2241. [Google Scholar] [CrossRef]
- Regan, M.C.; Horanyi, P.S.; Pryor, E.E.; Sarver, J.L.; Cafiso, D.S.; Bushweller, J.H. Structural and dynamic studies of the transcription factor ERG reveal DNA binding is allosterically autoinhibited. Proc. Natl. Acad. Sci. USA 2013, 110, 13374–13379. [Google Scholar] [CrossRef]
- Carrère, S.; Verger, A.; Flourens, A.; Stehelin, D.; Duterque-Coquillaud, M. Erg proteins, transcription factors of the Ets family, form homo, heterodimers and ternary complexes via two distinct domains. Oncogene 1998, 16, 3261–3268. [Google Scholar] [CrossRef]
- Mackereth, C.D.; Schärpf, M.; Gentile, L.N.; MacIntosh, S.E.; Slupsky, C.M.; McIntosh, L.P. Diversity in Structure and Function of the Ets Family PNT Domains. J. Mol. Biol. 2004, 342, 1249–1264. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, N.; Kedage, V.; Hollenhorst, P.C. Comparison of MAPK specificity across the ETS transcription factor family identifies a high-affinity ERK interaction required for ERG function in prostate cells. Cell Commun. Signal. 2015, 13, 12. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Thoms, J.A.I.; Tursky, M.L.; Knezevic, K.; Beck, D.; Chandrakanthan, V.; Suryani, S.; Olivier, J.; Boulton, A.; Glaros, E.N.; et al. MAPK/ERK2 phosphorylates ERG at serine 283 in leukemic cells and promotes stem cell signatures and cell proliferation. Leukemia 2016, 30, 1552–1561. [Google Scholar] [CrossRef] [PubMed]
- Kedage, V.; Strittmatter, B.G.; Dausinas, P.B.; Hollenhorst, P.C. Phosphorylation of the oncogenic transcription factor ERG in prostate cells dissociates polycomb repressive complex 2, allowing target gene activation. J. Biol. Chem. 2017, 292, 17225–17235. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, B.G.; Jerde, T.J.; Hollenhorst, P.C. Ras/ERK and PI3K/AKT signaling differentially regulate oncogenic ERG mediated transcription in prostate cells. PLoS Genet. 2021, 17, e1009708. [Google Scholar] [CrossRef] [PubMed]
- Singareddy, R.; Semaan, L.; Conley-LaComb, M.K.; St. John, J.; Powell, K.; Iyer, M.; Smith, D.; Heilbrun, L.K.; Shi, D.; Sakr, W.; et al. Transcriptional Regulation of CXCR4 in Prostate Cancer: Significance of TMPRSS2-ERG Fusions. Mol. Cancer Res. 2013, 11, 1349–1361. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Ren, S.; Dalangood, S.; Murphy, S.J.; Chang, C.; Pang, X.; Cui, Y.; Wang, L.; Pan, Y.; Zhang, X.; et al. Truncated ERG Oncoproteins from TMPRSS2-ERG Fusions Are Resistant to SPOP-Mediated Proteasome Degradation. Mol. Cell 2015, 59, 904–916. [Google Scholar] [CrossRef]
- Gan, W.; Beck, A.H.; Asara, J.M.; Pandolfi, P.P.; Li, Z.; Inuzuka, H.; Lunardi, A.; Zhang, J.; Dai, X.; Sun, Y.; et al. SPOP Promotes Ubiquitination and Degradation of the ERG Oncoprotein to Suppress Prostate Cancer Progression. Mol. Cell 2015, 59, 917–930. [Google Scholar] [CrossRef]
- Wang, S.; Kollipara, R.K.; Humphries, C.G.; Ma, S.-H.; Hutchinson, R.; Li, R.; Siddiqui, J.; Tomlins, S.A.; Raj, G.V.; Kittler, R.; et al. The ubiquitin ligase TRIM25 targets ERG for degradation in prostate cancer. Oncotarget 2016, 7, 64921–64931. [Google Scholar] [CrossRef]
- Hong, Z.; Zhang, W.; Ding, D.; Huang, Z.; Yan, Y.; Cao, W.; Pan, Y.; Hou, X.; Weroha, S.J.; Karnes, R.J.; et al. DNA Damage Promotes TMPRSS2-ERG Oncoprotein Destruction and Prostate Cancer Suppression via Signaling Converged by GSK3β and WEE1. Mol. Cell 2020, 79, 1008.e4–1023.e4. [Google Scholar] [CrossRef]
- Wang, S.; Kollipara, R.K.; Srivastava, N.; Li, R.; Ravindranathan, P.; Hernandez, E.; Freeman, E.; Humphries, C.G.; Kapur, P.; Lotan, Y.; et al. Ablation of the oncogenic transcription factor ERG by deubiquitinase inhibition in prostate cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 4251–4256. [Google Scholar] [CrossRef] [PubMed]
- Zoma, M.; Curti, L.; Shinde, D.; Albino, D.; Mitra, A.; Sgrignani, J.; Mapelli, S.N.; Sandrini, G.; Civenni, G.; Merulla, J.; et al. EZH2-induced lysine K362 methylation enhances TMPRSS2-ERG oncogenic activity in prostate cancer. Nat. Commun. 2021, 12, 4147. [Google Scholar] [CrossRef] [PubMed]
- Hollenhorst, P.C.; Ferris, M.W.; Hull, M.A.; Chae, H.; Kim, S.; Graves, B.J. Oncogenic ETS proteins mimic activated RAS/MAPK signaling in prostate cells. Genes Dev. 2011, 25, 2147–2157. [Google Scholar] [CrossRef]
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136. [Google Scholar] [CrossRef] [PubMed]
- Madison, B.J.; Clark, K.A.; Bhachech, N.; Hollenhorst, P.C.; Graves, B.J.; Currie, S.L. Electrostatic repulsion causes anticooperative DNA binding between tumor suppressor ETS transcription factors and JUN–FOS at composite DNA sites. J. Biol. Chem. 2018, 293, 18624–18635. [Google Scholar] [CrossRef] [PubMed]
- Verger, A.; Buisine, E.; Carrère, S.; Wintjens, R.; Flourens, A.; Coll, J.; Stéhelin, D.; Duterque-Coquillaud, M. Identification of amino acid residues in the ETS transcription factor Erg that mediate Erg-Jun/Fos-DNA ternary complex formation. J. Biol. Chem. 2001, 276, 17181–17189. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Blee, A.M.; Wang, D.; An, J.; Pan, Y.; Yan, Y.; Ma, T.; He, Y.; Dugdale, J.; Hou, X.; et al. Loss of FOXO1 Cooperates with TMPRSS2–ERG Overexpression to Promote Prostate Tumorigenesis and Cell Invasion. Cancer Res. 2017, 77, 6524–6537. [Google Scholar] [CrossRef] [PubMed]
- Kedage, V.; Selvaraj, N.; Nicholas, T.R.; Budka, J.A.; Plotnik, J.P.; Jerde, T.J.; Hollenhorst, P.C. An Interaction with Ewing’s Sarcoma Breakpoint Protein EWS Defines a Specific Oncogenic Mechanism of ETS Factors Rearranged in Prostate Cancer. Cell Rep. 2016, 17, 1289–1301. [Google Scholar] [CrossRef]
- Sandoval, G.J.; Williamson, K.E.; Pop, M.; Hartman, E.; Kadoch, C.; St. Pierre, R.; Pan, J.; Takeda, D.Y.; Garraway, L.A.; Hahn, W.C.; et al. Binding of TMPRSS2-ERG to BAF Chromatin Remodeling Complexes Mediates Prostate Oncogenesis. Mol. Cell 2018, 71, 554.e7–566.e7. [Google Scholar] [CrossRef]
- Arvand, A.; Denny, C.T. Biology of EWS/ETS fusions in Ewing’s family tumors. Oncogene 2001, 20, 5747–5754. [Google Scholar] [CrossRef]
- Peterlin, B.M.; Price, D.H. Controlling the Elongation Phase of Transcription with P-TEFb. Mol. Cell 2006, 23, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Roe, J.S.; Mercan, F.; Rivera, K.; Pappin, D.J.; Vakoc, C.R. BET Bromodomain Inhibition Suppresses the Function of Hematopoietic Transcription Factors in Acute Myeloid Leukemia. Mol. Cell 2015, 58, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- Blee, A.M.; Liu, S.; Wang, L.; Huang, H.; Blee, A.M.; Liu, S.; Wang, L.; Huang, H. BET bromodomain-mediated interaction between ERG and BRD4 promotes prostate cancer cell invasion. Oncotarget 2016, 7, 38319–38332. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-D.; Shin, S.; Janknecht, R. ETS transcription factor ERG cooperates with histone demethylase KDM4A. Oncol. Rep. 2016, 35, 3679–3688. [Google Scholar] [CrossRef]
- Brenner, J.C.; Ateeq, B.; Li, Y.; Yocum, A.K.; Cao, Q.; Asangani, I.A.; Patel, S.; Wang, X.; Liang, H.; Yu, J.; et al. Mechanistic Rationale for Inhibition of Poly(ADP-Ribose) Polymerase in ETS Gene Fusion-Positive Prostate Cancer. Cancer Cell 2011, 19, 664–678. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Yu, J.; Mani, R.S.; Cao, Q.; Brenner, C.J.; Cao, X.; Wang, X.; Wu, L.; Li, J.; Hu, M.; et al. An Integrated Network of Androgen Receptor, Polycomb, and TMPRSS2-ERG Gene Fusions in Prostate Cancer Progression. Cancer Cell 2010, 17, 443–454. [Google Scholar] [CrossRef]
- Chng, K.R.; Chang, C.W.; Tan, S.K.; Yang, C.; Hong, S.Z.; Sng, N.Y.W.; Cheung, E. A transcriptional repressor co-regulatory network governing androgen response in prostate cancers. EMBO J. 2012, 31, 2810–2823. [Google Scholar] [CrossRef]
- Mounir, Z.; Korn, J.M.; Westerling, T.; Lin, F.; Kirby, C.A.; Schirle, M.; McAllister, G.; Hoffman, G.; Ramadan, N.; Hartung, A.; et al. ERG signaling in prostate cancer is driven through PRMT5-dependent methylation of the androgen receptor. eLife 2016, 5, 1–19. [Google Scholar] [CrossRef]
- Yang, L.; Xia, L.; Wu, D.Y.; Wang, H.; Chansky, H.A.; Schubach, W.H.; Hickstein, D.D.; Zhang, Y. Molecular cloning of ESET, a novel histone H3-specific methyltransferase that interacts with ERG transcription factor. Oncogene 2002, 21, 148–152. [Google Scholar] [CrossRef]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef]
- Yeap, L.-S.; Hayashi, K.; Surani, M.A. ERG-associated protein with SET domain (ESET)-Oct4 interaction regulates pluripotency and represses the trophectoderm lineage. Epigenet. Chromatin 2009, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- The GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent Fusion of TMPRSS2 and ETS Transcription Factor Genes in Prostate Cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.; Sabelnykova, V.Y.; Yamaguchi, T.N.; Heisler, L.E.; Livingstone, J.; Huang, V.; Shiah, Y.-J.; Yousif, F.; Lin, X.; Masella, A.P.; et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017, 541, 359–364. [Google Scholar] [CrossRef]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef]
- Mani, R.S.; Iyer, M.K.; Cao, Q.; Brenner, J.C.; Wang, L.; Ghosh, A.; Cao, X.; Lonigro, R.J.; Tomlins, S.A.; Varambally, S.; et al. TMPRSS2-ERG-mediated feed-forward regulation of wild-type ERG in human prostate cancers. Cancer Res. 2011, 71, 5387–5392. [Google Scholar] [CrossRef]
- Clark, J.; Merson, S.; Jhavar, S.; Flohr, P.; Edwards, S.; Foster, C.S.; Eeles, R.; Martin, F.L.; Phillips, D.H.; Crundwell, M.; et al. Diversity of TMPRSS2-ERG fusion transcripts in the human prostate. Oncogene 2006, 26, 2667–2673. [Google Scholar] [CrossRef]
- Svensson, M.A.; Perner, S.; Ohlson, A.L.; Day, J.R.; Groskopf, J.; Kirsten, R.; Sollie, T.; Helenius, G.; Andersson, S.O.; Demichelis, F.; et al. A comparative study of ERG status assessment on DNA, mRNA, and protein levels using unique samples from a swedish biopsy cohort. Appl. Immunohistochem. Mol. Morphol. 2014, 22, 136–141. [Google Scholar] [CrossRef]
- Tu, J.J.; Rohan, S.; Kao, J.; Kitabayashi, N.; Mathew, S.; Chen, Y.-T. Gene fusions between TMPRSS2 and ETS family genes in prostate cancer: Frequency and transcript variant analysis by RT-PCR and FISH on paraffin-embedded tissues. Mod. Pathol. 2007, 20, 921–928. [Google Scholar] [CrossRef]
- Lin, C.; Yang, L.; Tanasa, B.; Hutt, K.; Ju, B.g.; Ohgi, K.; Zhang, J.; Rose, D.W.; Fu, X.D.; Glass, C.K.; et al. Nuclear Receptor-Induced Chromosomal Proximity and DNA Breaks Underlie Specific Translocations in Cancer. Cell 2009, 139, 1069–1083. [Google Scholar] [CrossRef] [PubMed]
- Mani, R.S.; Amin, M.A.; Li, X.; Kalyana-Sundaram, S.; Veeneman, B.A.; Wang, L.; Ghosh, A.; Aslam, A.; Ramanand, S.G.; Rabquer, B.J.; et al. Inflammation-Induced Oxidative Stress Mediates Gene Fusion Formation in Prostate Cancer. Cell Rep. 2016, 17, 2620–2631. [Google Scholar] [CrossRef]
- Mani, R.-S.; Tomlins, S.A.; Callahan, K.; Ghosh, A.; Nyati, M.K.; Varambally, S.; Palanisamy, N.; Chinnaiyan, A.M. Induced Chromosomal Proximity and Gene Fusions in Prostate Cancer. Science 2009, 326, 1230. [Google Scholar] [CrossRef] [PubMed]
- Haffner, M.C.; Aryee, M.J.; Toubaji, A.; Esopi, D.M.; Albadine, R.; Gurel, B.; Isaacs, W.B.; Bova, G.S.; Liu, W.; Xu, J.; et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat. Genet. 2010, 42, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Baek, G.H.; Ramanand, S.G.; Sharp, A.; Gao, Y.; Yuan, W.; Welti, J.; Rodrigues, D.N.; Dolling, D.; Figueiredo, I.; et al. BRD4 Promotes DNA Repair and Mediates the Formation of TMPRSS2-ERG Gene Rearrangements in Prostate Cancer. Cell Rep. 2018, 22, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Thangapazham, R.; Saenz, F.; Katta, S.; Mohamed, A.A.; Tan, S.H.; Petrovics, G.; Srivastava, S.; Dobi, A. Loss of the NKX3.1 tumorsuppressor promotes the TMPRSS2-ERG fusion gene expression in prostate cancer. BMC Cancer 2014, 14, 16. [Google Scholar] [CrossRef]
- Bowen, C.; Zheng, T.; Gelmann, E.P. NKX3.1 Suppresses TMPRSS2–ERG Gene Rearrangement and Mediates Repair of Androgen Receptor–Induced DNA Damage. Cancer Res. 2015, 75, 2686–2698. [Google Scholar] [CrossRef]
- Clark, J.; Attard, G.; Jhavar, S.; Flohr, P.; Reid, A.; De-Bono, J.; Eeles, R.; Scardino, P.; Cuzick, J.; Fisher, G.; et al. Complex patterns of ETS gene alteration arise during cancer development in the human prostate. Oncogene 2008, 27, 1993–2003. [Google Scholar] [CrossRef]
- Mosquera, J.-M.; Perner, S.; Genega, E.M.; Sanda, M.; Hofer, M.D.; Mertz, K.D.; Paris, P.L.; Simko, J.; Bismar, T.A.; Ayala, G.; et al. Characterization of TMPRSS2-ERG Fusion High-Grade Prostatic Intraepithelial Neoplasia and Potential Clinical Implications. Clin. Cancer Res. 2008, 14, 3380–3385. [Google Scholar] [CrossRef]
- Park, K.; Tomlins, S.A.; Mudaliar, K.M.; Chiu, Y.-L.; Esgueva, R.; Mehra, R.; Suleman, K.; Varambally, S.; Brenner, J.C.; MacDonald, T.; et al. Antibody-Based Detection of ERG Rearrangement-Positive Prostate Cancer. Neoplasia 2010, 12, 590. [Google Scholar] [CrossRef]
- Van Leenders, G.J.; Boormans, J.L.; Vissers, C.J.; Hoogland, A.M.; Bressers, A.A.; Furusato, B.; Trapman, J. Antibody EPR3864 is specific for ERG genomic fusions in prostate cancer: Implications for pathological practice. Mod. Pathol. 2011, 24, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Perner, S.; Mosquera, J.M.; Demichelis, F.; Hofer, M.D.; Paris, P.L.; Simko, J.; Collins, C.; Bismar, T.A.; Chinnaiyan, A.M.; de Marzo, A.M.; et al. TMPRSS2-ERG fusion prostate cancer: An early molecular event associated with invasion. Am. J. Surg. Pathol. 2007, 31, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Cerveira, N.; Ribeiro, F.R.; Peixoto, A.; Costa, V.; Henrique, R.; Jerónimo, C.; Teixeira, M.R. TMPRSS2-ERG Gene Fusion Causing ERG Overexpression Precedes Chromosome Copy Number Changes in Prostate Carcinomas and Paired HGPIN Lesions. Neoplasia 2006, 8, 826. [Google Scholar] [CrossRef] [PubMed]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef]
- Prandi, D.; Baca, S.C.; Romanel, A.; Barbieri, C.E.; Mosquera, J.-M.; Fontugne, J.; Beltran, H.; Sboner, A.; Garraway, L.a.; Rubin, M.a.; et al. Unraveling the clonal hierarchy of somatic genomic aberrations. Genome Biol. 2014, 15, 439. [Google Scholar] [CrossRef]
- Carver, B.S.; Tran, J.; Gopalan, A.; Chen, Z.; Shaikh, S.; Carracedo, A.; Alimonti, A.; Nardella, C.; Varmeh, S.; Scardino, P.T.; et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat. Genet. 2009, 41, 619–624. [Google Scholar] [CrossRef]
- King, J.C.; Xu, J.; Wongvipat, J.; Hieronymus, H.; Carver, B.S.; Leung, D.H.; Taylor, B.S.; Sander, C.; Cardiff, R.D.; Couto, S.S.; et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat. Genet. 2009, 41, 524–526. [Google Scholar] [CrossRef]
- Carver, B.S.; Tran, J.; Chen, Z.; Carracedo-Perez, A.; Alimonti, A.; Nardella, C.; Gopalan, A.; Scardino, P.T.; Cordon-Cardo, C.; Gerald, W.; et al. ETS rearrangements and prostate cancer initiation. Nature 2009, 457, E1. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Laxman, B.; Varambally, S.; Cao, X.; Yu, J.; Helgeson, B.E.; Cao, Q.; Prensner, J.R.; Rubin, M.A.; Shah, R.B.; et al. Role of the TMPRSS2-ERG Gene Fusion in Prostate Cancer. Neoplasia 2008, 10, 177–188. [Google Scholar] [CrossRef]
- Klezovitch, O.; Risk, M.; Coleman, I.; Lucas, J.M.; Null, M.; True, L.D.; Nelson, P.S.; Vasioukhin, V. A causal role for ERG in neoplastic transformation of prostate epithelium. Proc. Natl. Acad. Sci. USA 2008, 105, 2105–2110. [Google Scholar] [CrossRef]
- Baena, E.; Shao, Z.; Linn, D.E.; Glass, K.; Hamblen, M.J.; Fujiwara, Y.; Kim, J.; Nguyen, M.; Zhang, X.; Godinho, F.J.; et al. ETV1 directs androgen metabolism and confers aggressive prostate cancer in targeted mice and patients. Genes Dev. 2013, 27, 683–698. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chi, P.; Rockowitz, S.; Iaquinta, P.J.; Shamu, T.; Shukla, S.; Gao, D.; Sirota, I.; Carver, B.S.; Wongvipat, J.; et al. ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis in response to PTEN loss. Nat. Med. 2013, 19, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Tretiakova, M.S.; Silvis, M.R.; Lucas, J.; Klezovitch, O.; Coleman, I.; Bolouri, H.; Kutyavin, V.I.; Morrissey, C.; True, L.D.; et al. ERG Activates the YAP1 Transcriptional Program and Induces the Development of Age-Related Prostate Tumors. Cancer Cell 2015, 27, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Xin, L.; Goldstein, A.S.; Lawson, D.A.; Teitell, M.A.; Witte, O.N. ETS family transcription factors collaborate with alternative signaling pathways to induce carcinoma from adult murine prostate cells. Proc. Natl. Acad. Sci. USA 2009, 106, 12465–12470. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, J.; Li, C.; Giacomini, C.P.; Salari, K.; Huang, S.; Wang, P.; Ferrari, M.; Hernandez-Boussard, T.; Brooks, J.D.; Pollack, J.R. Genomic profiling reveals alternative genetic pathways of prostate tumorigenesis. Cancer Res. 2007, 67, 8504–8510. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Swennenhuis, J.F.; Olmos, D.; Reid, A.H.M.; Vickers, E.; A’Hern, R.; Levink, R.; Coumans, F.; Moreira, J.; Riisnaes, R.; et al. Characterization of ERG, AR and PTEN Gene Status in Circulating Tumor Cells from Patients with Castration-Resistant Prostate Cancer. Cancer Res. 2009, 69, 2912–2918. [Google Scholar] [CrossRef]
- Nicholas, T.R.; Strittmatter, B.G.; Hollenhorst, P.C. Oncogenic ETS Factors in Prostate Cancer. Adv. Exp. Med. Biol. 2019, 1210, 409–436. [Google Scholar] [CrossRef]
- Wasmuth, E.V.; Hoover, E.A.; Antar, A.; Klinge, S.; Chen, Y.; Sawyers, C.L. Modulation of androgen receptor DNA binding activity through direct interaction with the ETS transcription factor ERG. Proc. Natl. Acad. Sci. USA 2020, 117, 8584–8592. [Google Scholar] [CrossRef]
- Shah, N.; Kesten, N.; Font-Tello, A.; Chang, M.E.K.; Vadhi, R.; Lim, K.; Flory, M.R.; Cejas, P.; Mohammed, H.; Long, H.W.; et al. ERG-Mediated Coregulator Complex Formation Maintains Androgen Receptor Signaling in Prostate Cancer. Cancer Res. 2020, 80, 4612–4619. [Google Scholar] [CrossRef]
- Powell, K.; Semaan, L.; Conley-LaComb, M.K.; Asangani, I.; Wu, Y.-M.; Ginsburg, K.B.; Williams, J.; Squire, J.A.; Maddipati, K.R.; Cher, M.L.; et al. ERG/AKR1C3/AR Constitutes a Feed-Forward Loop for AR Signaling in Prostate Cancer Cells. Clin. Cancer Res. 2015, 21, 2569–2579. [Google Scholar] [CrossRef]
- Rickman, D.S.; Soong, T.D.; Moss, B.; Mosquera, J.M.; Dlabal, J.; Terry, S.; MacDonald, T.Y.; Tripodi, J.; Bunting, K.; Najfeld, V.; et al. Oncogene-mediated alterations in chromatin conformation. Proc. Natl. Acad. Sci. USA 2012, 109, 9083–9088. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Yuan, Q.; Di, W.; Xia, X.; Liu, Z.; Mao, N.; Li, L.; Li, C.; He, J.; Li, Y.; et al. ERG orchestrates chromatin interactions to drive prostate cell fate reprogramming. J. Clin. Investig. 2020, 130, 5924–5941. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Lee, H.H.; Choi, K.; Moon, Y.J.; Heo, J.E.; Ham, W.S.; Jang, W.S.; Rha, K.H.; Cho, N.H.; Giancotti, F.G.; et al. Prostate epithelial genes define therapy-relevant prostate cancer molecular subtype. Prostate Cancer Prostatic Dis. 2021, 24, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- You, S.; Knudsen, B.S.; Erho, N.; Alshalalfa, M.; Takhar, M.; Ashab, H.A.D.; Davicioni, E.; Karnes, R.J.; Klein, E.A.; Den, R.B.; et al. Integrated Classification of Prostate Cancer Reveals a Novel Luminal Subtype with Poor Outcome. Cancer Res. 2016, 76, 4948–4958. [Google Scholar] [CrossRef]
- Zhao, S.G.; Chang, S.L.; Erho, N.; Yu, M.; Lehrer, J.; Alshalalfa, M.; Speers, C.; Cooperberg, M.R.; Kim, W.; Ryan, C.J.; et al. Associations of Luminal and Basal Subtyping of Prostate Cancer with Prognosis and Response to Androgen Deprivation Therapy. JAMA Oncol. 2017, 3, 1663–1672. [Google Scholar] [CrossRef]
- Cai, C.; Wang, H.; He, H.H.; Chen, S.; He, L.; Ma, F.; Mucci, L.; Wang, Q.; Fiore, C.; Sowalsky, A.G.; et al. ERG induces androgen receptor-mediated regulation of SOX9 in prostate cancer. J. Clin. Investig. 2013, 123, 1109–1122. [Google Scholar] [CrossRef]
- Mounir, Z.; Lin, F.; Lin, V.G.; Korn, J.M.; Yu, Y.; Valdez, R.; Aina, O.H.; Buchwalter, G.; Jaffe, A.B.; Korpal, M.; et al. TMPRSS2:ERG blocks neuroendocrine and luminal cell differentiation to maintain prostate cancer proliferation. Oncogene 2015, 34, 3815–3825. [Google Scholar] [CrossRef]
- Sun, C.; Dobi, A.; Mohamed, A.; Li, H.; Thangapazham, R.L.; Furusato, B.; Shaheduzzaman, S.; Tan, S.-H.; Vaidyanathan, G.; Whitman, E.; et al. TMPRSS2-ERG fusion, a common genomic alteration in prostate cancer activates C-MYC and abrogates prostate epithelial differentiation. Oncogene 2008, 27, 5348–5353. [Google Scholar] [CrossRef]
- Cai, J.; Kandagatla, P.; Singareddy, R.; Kropinski, A.; Sheng, S.; Cher, M.L.; Chinni, S.R. Androgens Induce Functional CXCR4 through ERG Factor Expression in TMPRSS2-ERG Fusion-Positive Prostate Cancer Cells. Transl. Oncol. 2010, 3, 195. [Google Scholar] [CrossRef]
- Wu, L.; Zhao, J.C.; Kim, J.; Jin, H.-J.; Wang, C.-Y.; Yu, J. ERG Is a Critical Regulator of Wnt/LEF1 Signaling in Prostate Cancer. Cancer Res. 2013, 73, 6068–6079. [Google Scholar] [CrossRef]
- Gupta, S.; Iljin, K.; Sara, H.; Mpindi, J.P.; Mirtti, T.; Vainio, P.; Rantala, J.; Alanen, K.; Nees, M.; Kallioniemi, O. FZD4 as a Mediator of ERG Oncogene–Induced WNT Signaling and Epithelial-to-Mesenchymal Transition in Human Prostate Cancer Cells. Cancer Res. 2010, 70, 6735–6745. [Google Scholar] [CrossRef] [PubMed]
- Iljin, K.; Wolf, M.; Edgren, H.; Gupta, S.; Kilpinen, S.; Skotheim, R.I.; Peltola, M.; Smit, F.; Verhaegh, G.; Schalken, J.; et al. TMPRSS2 Fusions with Oncogenic ETS Factors in Prostate Cancer Involve Unbalanced Genomic Rearrangements and Are Associated with HDAC1 and Epigenetic Reprogramming. Cancer Res. 2006, 66, 10242–10246. [Google Scholar] [CrossRef] [PubMed]
- Ratz, L.; Laible, M.; Kacprzyk, L.A.; Wittig-Blaich, S.M.; Tolstov, Y.; Duensing, S.; Altevogt, P.; Klauck, S.M.; Sültmann, H.; Ratz, L.; et al. TMPRSS2:ERG gene fusion variants induce TGF-β signaling and epithelial to mesenchymal transition in human prostate cancer cells. Oncotarget 2017, 8, 25115–25130. [Google Scholar] [CrossRef] [PubMed]
- Leshem, O.; Madar, S.; Kogan-Sakin, I.; Kamer, I.; Goldstein, I.; Brosh, R.; Cohen, Y.; Jacob-Hirsch, J.; Ehrlich, M.; Ben-Sasson, S.; et al. TMPRSS2/ERG Promotes Epithelial to Mesenchymal Transition through the ZEB1/ZEB2 Axis in a Prostate Cancer Model. PLoS ONE 2011, 6, e21650. [Google Scholar] [CrossRef] [PubMed]
- Brase, J.C.; Johannes, M.; Mannsperger, H.; Fälth, M.; Metzger, J.; Kacprzyk, L.A.; Andrasiuk, T.; Gade, S.; Meister, M.; Sirma, H.; et al. TMPRSS2-ERG -specific transcriptional modulation is associated with prostate cancer biomarkers and TGF-β signaling. BMC Cancer 2011, 11, 507. [Google Scholar] [CrossRef]
- Kron, K.J.; Murison, A.; Zhou, S.; Huang, V.; Yamaguchi, T.N.; Shiah, Y.J.; Fraser, M.; van der Kwast, T.; Boutros, P.C.; Bristow, R.G.; et al. TMPRSS2-ERG fusion co-opts master transcription factors and activates NOTCH signaling in primary prostate cancer. Nat. Genet. 2017, 49, 1336–1345. [Google Scholar] [CrossRef]
- Mohamed, A.A.; Tan, S.-H.; Xavier, C.P.; Katta, S.; Huang, W.; Ravindranath, L.; Jamal, M.; Li, H.; Srivastava, M.; Srivatsan, E.S.; et al. Synergistic Activity with NOTCH Inhibition and Androgen Ablation in ERG-Positive Prostate Cancer Cells. Mol. Cancer Res. 2017, 15, 1308–1317. [Google Scholar] [CrossRef]
- Wang, J.; Cai, Y.; Shao, L.; Siddiqui, J.; Palanisamy, N.; Li, R.; Ren, C.; Ayala, G.; Ittmann, M. Activation of NF-κB by TMPRSS2/ERG Fusion Isoforms through Toll-Like Receptor-4. Cancer Res. 2011, 71, 1325–1333. [Google Scholar] [CrossRef]
- Zhang, L.; Shao, L.; Creighton, C.J.; Zhang, Y.; Xin, L.; Ittmann, M.; Wang, J.; Zhang, L.; Shao, L.; Creighton, C.J.; et al. Function of phosphorylation of NF-kB p65 ser536 in prostate cancer oncogenesis. Oncotarget 2015, 6, 6281–6294. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Mehra, R.; Rhodes, D.R.; Cao, X.; Wang, L.; Dhanasekaran, S.M.; Kalyana-Sundaram, S.; Wei, J.T.; Rubin, M.A.; Pienta, K.J.; et al. Integrative molecular concept modeling of prostate cancer progression. Nat. Genet. 2007, 39, 41–51. [Google Scholar] [CrossRef]
- Demichelis, F.; Setlur, S.R.; Beroukhim, R.; Perner, S.; Korbel, J.O.; Lafargue, C.J.; Dorothee, P.; Pina, C.; Hofer, M.D.; Sboner, A.; et al. Distinct Genomic Aberrations Associated with ERG Rearranged Prostate Cancer. Genes Chromosomes Cancer 2009, 48, 366–380. [Google Scholar] [CrossRef] [PubMed]
- Bose, R.; Karthaus, W.R.; Armenia, J.; Abida, W.; Iaquinta, P.J.; Zhang, Z.; Wongvipat, J.; Wasmuth, E.V.; Shah, N.; Sullivan, P.S.; et al. ERF mutations reveal a balance of ETS factors controlling prostate oncogenesis. Nature 2017, 546, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cai, Y.; Yu, W.; Ren, C.; Spencer, D.M.; Ittmann, M. Pleiotropic biological activities of alternatively spliced TMPRSS2/ERG fusion gene transcripts. Cancer Res. 2008, 68, 8516–8524. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Qiao, Y.; Asangani, I.A.; Ateeq, B.; Poliakov, A.; Cieślik, M.; Pitchiaya, S.; Chakravarthi, B.V.S.K.; Cao, X.; Jing, X.; et al. Development of Peptidomimetic Inhibitors of the ERG Gene Fusion Product in Prostate Cancer. Cancer Cell 2017, 31, 532–548.e7. [Google Scholar] [CrossRef]
- Darnell, J.E. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 740–749. [Google Scholar] [CrossRef]
- Henley, M.J.; Koehler, A.N. Advances in targeting ‘undruggable’ transcription factors with small molecules. Nat. Rev. Drug Discov. 2021, 20, 669–688. [Google Scholar] [CrossRef]
- Chatterjee, P.; Choudhary, G.S.; Alswillah, T.; Xiong, X.; Heston, W.D.; Magi-Galluzzi, C.; Zhang, J.; Klein, E.A.; Almasan, A. The TMPRSS2–ERG Gene Fusion Blocks XRCC4-Mediated Nonhomologous End-Joining Repair and Radiosensitizes Prostate Cancer Cells to PARP Inhibition. Mol. Cancer Ther. 2015, 14, 1896–1906. [Google Scholar] [CrossRef]
- Han, S.; Brenner, J.C.; Sabolch, A.; Jackson, W.; Speers, C.; Wilder-Romans, K.; Knudsen, K.E.; Lawrence, T.S.; Chinnaiyan, A.M.; Feng, F.Y. Targeted Radiosensitization of ETS Fusion-Positive Prostate Cancer through PARP1 Inhibition. Neoplasia 2013, 15, 1207. [Google Scholar] [CrossRef]
- Chatterjee, P.; Choudhary, G.S.; Sharma, A.; Singh, K.; Heston, W.D.; Ciezki, J.; Klein, E.A.; Almasan, A. PARP Inhibition Sensitizes to Low Dose-Rate Radiation TMPRSS2-ERG Fusion Gene-Expressing and PTEN-Deficient Prostate Cancer Cells. PLoS ONE 2013, 8, e60408. [Google Scholar] [CrossRef]
- Hussain, M.; Daignault-Newton, S.; Twardowski, P.W.; Albany, C.; Stein, M.N.; Kunju, L.P.; Siddiqui, J.; Wu, Y.-M.; Robinson, D.; Lonigro, R.J.; et al. Targeting Androgen Receptor and DNA Repair in Metastatic Castration-Resistant Prostate Cancer: Results from NCI 9012. J. Clin. Oncol. 2018, 36, 991–999. [Google Scholar] [CrossRef]
- Pra, A.D.; Lalonde, E.; Sykes, J.; Warde, F.; Ishkanian, A.; Meng, A.; Maloff, C.; Srigley, J.; Joshua, A.M.; Petrovics, G.; et al. TMPRSS2-ERG Status Is Not Prognostic Following Prostate Cancer Radiotherapy: Implications for Fusion Status and DSB Repair. Clin. Cancer Res. 2013, 19, 5202–5209. [Google Scholar] [CrossRef]
- Molife, L.R.; Attard, G.; Fong, P.C.; Karavasilis, V.; Reid, A.H.M.; Patterson, S.; Riggs, C.E.; Higano, C.; Stadler, W.M.; McCulloch, W.; et al. Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC). Ann. Oncol. 2010, 21, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Bradley, D.; Rathkopf, D.; Dunn, R.; Stadler, W.M.; Liu, G.; Smith, D.C.; Pili, R.; Zwiebel, J.; Scher, H.; Hussain, M. Vorinostat in advanced prostate cancer patients progressing on prior chemotherapy (National Cancer Institute Trial 6862): Trial results and interleukin-6 analysis: A study by the Department of Defense Prostate Cancer Clinical Trial Consortium and Universi. Cancer 2009, 115, 5541–5549. [Google Scholar] [CrossRef] [PubMed]
- Eigl, B.J.; North, S.; Winquist, E.; Finch, D.; Wood, L.; Sridhar, S.S.; Powers, J.; Good, J.; Sharma, M.; Squire, J.A.; et al. A phase II study of the HDAC inhibitor SB939 in patients with castration resistant prostate cancer: NCIC clinical trials group study IND195. Investig. New Drugs 2015, 33, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Erkizan, H.V.; Kong, Y.; Merchant, M.; Schlottmann, S.; Barber-Rotenberg, J.S.; Yuan, L.; Abaan, O.D.; Chou, T.; Dakshanamurthy, S.; Brown, M.L.; et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat. Med. 2009, 15, 750–756. [Google Scholar] [CrossRef]
- Rahim, S.; Beauchamp, E.M.; Kong, Y.; Brown, M.L.; Toretsky, J.A.; Üren, A. YK-4-279 Inhibits ERG and ETV1 Mediated Prostate Cancer Cell Invasion. PLoS ONE 2011, 6, e19343. [Google Scholar] [CrossRef]
- Winters, B.; Brown, L.; Coleman, I.; Nguyen, H.; Minas, T.Z.; Kollath, L.; Vasioukhin, V.; Nelson, P.; Corey, E.; Üren, A.; et al. Inhibition of ERG Activity in Patient-derived Prostate Cancer Xenografts by YK-4-279. Anticancer Res. 2017, 37, 3385–3396. [Google Scholar] [CrossRef]
- Nhili, R.; Peixoto, P.; Depauw, S.; Flajollet, S.; Dezitter, X.; Munde, M.M.; Ismail, M.A.; Kumar, A.; Farahat, A.A.; Stephens, C.E.; et al. Targeting the DNA-binding activity of the human ERG transcription factor using new heterocyclic dithiophene diamidines. Nucleic Acids Res. 2013, 41, 125–138. [Google Scholar] [CrossRef]
- Butler, M.S.; Roshan-Moniri, M.; Hsing, M.; Lau, D.; Kim, A.; Yen, P.; Mroczek, M.; Nouri, M.; Lien, S.; Axerio-Cilies, P.; et al. Discovery and characterization of small molecules targeting the DNA-binding ETS domain of ERG in prostate cancer. Cancer Res. 2017, 8, 42438–42454. [Google Scholar] [CrossRef]
- Mohamed, A.A.; Xavier, C.P.; Sukumar, G.; Tan, S.-H.; Ravindranath, L.; Seraj, N.; Kumar, V.; Sreenath, T.; McLeod, D.G.; Petrovics, G.; et al. Identification of a Small Molecule That Selectively Inhibits ERG-Positive Cancer Cell Growth. Cancer Res. 2018, 78, 3659–3671. [Google Scholar] [CrossRef]
- Eldhose, B.; Pandrala, M.; Xavier, C.; Mohamed, A.A.; Srivastava, S.; Sunkara, A.D.; Dobi, A.; Malhotra, S.V. New Selective Inhibitors of ERG Positive Prostate Cancer: ERGi-USU-6 Salt Derivatives. ACS Med. Chem. Lett. 2021, 12, 1703–1709. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Tekedereli, I.; Wang, J.; Yuca, E.; Tsang, S.; Sood, A.; Lopez-Berestein, G.; Ozpolat, B.; Ittmann, M. Highly Specific Targeting of the TMPRSS2/ERG Fusion Gene Using Liposomal Nanovectors. Clin. Cancer Res. 2012, 18, 6648–6657. [Google Scholar] [CrossRef]
- Abou-Ouf, H.; Zhao, L.; Bismar, T.A. ERG expression in prostate cancer: Biological relevance and clinical implication. J. Cancer Res. Clin. Oncol. 2016, 142, 1781–1793. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Dalton, J.T.; Narayanan, R.; Barbieri, C.E.; Hancock, M.L.; Bostwick, D.G.; Steiner, M.S.; Rubin, M.A. TMPRSS2:ERG Gene Fusion Predicts Subsequent Detection of Prostate Cancer in Patients with High-Grade Prostatic Intraepithelial Neoplasia. J. Clin. Oncol. 2013, 32, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Demichelis, F.; Fall, K.; Perner, S.; Andrén, O.; Schmidt, F.; Setlur, S.R.; Hoshida, Y.; Mosquera, J.M.; Pawitan, Y.; Lee, C.; et al. TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene 2007, 26, 4596–4599. [Google Scholar] [CrossRef] [PubMed]
- Berg, K.D.; Vainer, B.; Thomsen, F.B.; Røder, M.A.; Gerds, T.A.; Toft, B.G.; Brasso, K.; Iversen, P. ERG Protein Expression in Diagnostic Specimens Is Associated with Increased Risk of Progression During Active Surveillance for Prostate Cancer. Eur. Urol. 2014, 66, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Reig, Ò.; Marín-Aguilera, M.; Carrera, G.; Jiménez, N.; Paré, L.; García-Recio, S.; Gaba, L.; Pereira, M.V.; Fernández, P.; Prat, A.; et al. TMPRSS2-ERG in Blood and Docetaxel Resistance in Metastatic Castration-resistant Prostate Cancer. Eur. Urol. 2016, 70, 709–713. [Google Scholar] [CrossRef]
- Galletti, G.; Matov, A.; Beltran, H.; Fontugne, J.; Miguel Mosquera, J.; Cheung, C.; Macdonald, T.Y.; Sung, M.; O’Toole, S.; Kench, J.G.; et al. ERG induces taxane resistance in castration-resistant prostate cancer. Nat. Commun. 2014, 5, 5548. [Google Scholar] [CrossRef]
- Taylor, R.A.; Fraser, M.; Livingstone, J.; Espiritu, S.M.G.; Thorne, H.; Huang, V.; Lo, W.; Shiah, Y.J.; Yamaguchi, T.N.; Sliwinski, A.; et al. Germline BRCA2 mutations drive prostate cancers with distinct evolutionary trajectories. Nat. Commun. 2017, 8, 13671. [Google Scholar] [CrossRef]
- El Gammal, A.T.; Brüchmann, M.; Zustin, J.; Isbarn, H.; Hellwinkel, O.J.C.; Köllermann, J.; Sauter, G.; Simon, R.; Wilczak, W.; Schwarz, J.; et al. Chromosome 8p Deletions and 8q Gains are Associated with Tumor Progression and Poor Prognosis in Prostate Cancer. Clin. Cancer Res. 2010, 16, 56–64. [Google Scholar] [CrossRef]
- Annala, M.; Struss, W.J.; Warner, E.W.; Beja, K.; Vandekerkhove, G.; Wong, A.; Khalaf, D.; Seppälä, I.L.; So, A.; Lo, G.; et al. Treatment Outcomes and Tumor Loss of Heterozygosity in Germline DNA Repair-deficient Prostate Cancer. Eur. Urol. 2017, 72, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Laxman, B.; Tomlins, S.A.; Mehra, R.; Morris, D.S.; Wang, L.; Helgeson, B.E.; Shah, R.B.; Rubin, M.A.; Wei, J.T.; Chinnaiyan, A.M. Noninvasive Detection of TMPRSS2:ERG Fusion Transcripts in the Urine of Men with Prostate Cancer. Neoplasia 2006, 8, 885–888. [Google Scholar] [CrossRef] [PubMed]
- Hessels, D.; Smit, F.P.; Verhaegh, G.W.; Witjes, J.A.; Cornel, E.B.; Schalken, J.A. Detection of TMPRSS2-ERG Fusion Transcripts and Prostate Cancer Antigen 3 in Urinary Sediments May Improve Diagnosis of Prostate Cancer. Clin. Cancer Res. 2007, 13, 5103–5108. [Google Scholar] [CrossRef] [PubMed]
- Laxman, B.; Morris, D.S.; Yu, J.; Siddiqui, J.; Cao, J.; Mehra, R.; Lonigro, R.J.; Tsodikov, A.; Wei, J.T.; Tomlins, S.A.; et al. A First-Generation Multiplex Biomarker Analysis of Urine for the Early Detection of Prostate Cancer. Cancer Res. 2008, 68, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.; Skog, J.; Nordstrand, A.; Baranov, V.; Mincheva-Nilsson, L.; Breakefield, X.O.; Widmark, A. Prostate cancer-derived urine exosomes: A novel approach to biomarkers for prostate cancer. Br. J. Cancer 2009, 100, 1603–1607. [Google Scholar] [CrossRef] [PubMed]
- Rostad, K.; Hellwinkel, O.J.; Haukaas, S.A.; Halvorsen, O.J.; Oyan, A.M.; Haese, A.; Budaus, L.; Albrecht, H.; Akslen, L.A.; Schlomm, T.; et al. TMPRSS2:ERG fusion transcripts in urine from prostate cancer patients correlate with a less favorable prognosis. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2009, 117, 575–582. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Aubin, S.M.J.; Siddiqui, J.; Lonigro, R.J.; Sefton-Miller, L.; Miick, S.; Williamsen, S.; Hodge, P.; Meinke, J.; Blase, A.; et al. Urine TMPRSS2:ERG Fusion Transcript Stratifies Prostate Cancer Risk in Men with Elevated Serum PSA. Sci. Transl. Med. 2011, 3, 94ra72. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Day, J.R.; Lonigro, R.J.; Hovelson, D.H.; Siddiqui, J.; Kunju, L.P.; Dunn, R.L.; Meyer, S.; Hodge, P.; Groskopf, J.; et al. Urine TMPRSS2:ERG Plus PCA3 for Individualized Prostate Cancer Risk Assessment. Eur. Urol. 2016, 70, 45–53. [Google Scholar] [CrossRef]
- Leyten, G.H.J.M.; Hessels, D.; Jannink, S.A.; Smit, F.P.; De Jong, H.; Cornel, E.B.; de Reijke, T.M.; Vergunst, H.; Kil, P.; Knipscheer, B.C.; et al. Prospective Multicentre Evaluation of PCA3 and TMPRSS2-ERG Gene Fusions as Diagnostic and Prognostic Urinary Biomarkers for Prostate Cancer. Eur. Urol. 2014, 65, 534–542. [Google Scholar] [CrossRef]
- Bernasocchi, T.; El Tekle, G.; Bolis, M.; Mutti, A.; Vallerga, A.; Brandt, L.P.; Spriano, F.; Svinkina, T.; Zoma, M.; Ceserani, V.; et al. Dual functions of SPOP and ERG dictate androgen therapy responses in prostate cancer. Nat. Commun. 2021, 12, 734. [Google Scholar] [CrossRef]
- Guo, Z.-Q.; Zheng, T.; Chen, B.; Luo, C.; Ouyang, S.; Gong, S.; Li, J.; Mao, L.-L.; Lian, F.; Yang, Y.; et al. Small-molecule targeting of E3 ligase adaptor SPOP in kidney cancer. Cancer Cell 2016, 30, 474–484. [Google Scholar] [CrossRef]
- Teply, B.A.; Wang, H.; Luber, B.; Sullivan, R.; Rifkind, I.; Bruns, A.; Spitz, A.; DeCarli, M.; Sinibaldi, V.; Pratz, C.F.; et al. Bipolar androgen therapy in men with metastatic castration-resistant prostate cancer after progression on enzalutamide: An open-label, phase 2, multicohort study. Lancet Oncol. 2018, 19, 76–86. [Google Scholar] [CrossRef]
- Chen, J.; Joshua, A.M.; Denmeade, S.R.; Antonarakis, E.S.; Crumbaker, M. High dose testosterone in men with metastatic castrate-resistant prostate cancer (mCRPC) and homologous recombination deficiency (HRD). J. Clin. Oncol. 2019, 37, TPS5095. [Google Scholar] [CrossRef]
- Chen, Z.H.; Yu, Y.P.; Zuo, Z.H.; Nelson, J.B.; Michalopoulos, G.K.; Monga, S.; Liu, S.; Tseng, G.; Luo, J.H. Targeting genomic rearrangements in tumor cells through Cas9-mediated insertion of a suicide gene. Nat. Biotechnol. 2017, 35, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Selvanathan, S.P.; Moseley, E.; Graham, G.T.; Jessen, K.; Lannutti, B.; Üren, A.; Toretsky, J.A. TK-216: A novel, first-in-class, small molecule inhibitor of EWS-FLI1 in early clinical development, for the treatment of Ewing Sarcoma [abstract]. Cancer Res. 2017, 77, 694. [Google Scholar] [CrossRef]
- Gayvert, K.M.; Dardenne, E.; Cheung, C.; Boland, M.R.; Lorberbaum, T.; Wanjala, J.; Chen, Y.; Rubin, M.A.; Tatonetti, N.P.; Rickman, D.S.; et al. A Computational Drug Repositioning Approach for Targeting Oncogenic Transcription Factors. Cell Rep. 2016, 15, 2348–2356. [Google Scholar] [CrossRef]
- Shao, J.; Yan, Y.; Ding, D.; Wang, D.; He, Y.; Pan, Y.; Yan, W.; Kharbanda, A.; Li, H.; Huang, H. Destruction of DNA-Binding Proteins by Programmable Oligonucleotide PROTAC (O’PROTAC): Effective Targeting of LEF1 and ERG. Adv. Sci. 2021, 8, 2102555. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Alshalalfa, M.; Davicioni, E.; Erho, N.; Yousefi, K.; Zhao, S.; Haddad, Z.; Den, R.B.; Dicker, A.P.; Trock, B.J.; et al. Characterization of 1577 Primary Prostate Cancers Reveals Novel Biological and Clinicopathologic Insights into Molecular Subtypes. Eur. Urol. 2015, 68, 555–567. [Google Scholar] [CrossRef]
- Kluth, M.; Meyer, D.; Krohn, A.; Freudenthaler, F.; Bauer, M.; Salomon, G.; Heinzer, H.; Michl, U.; Steurer, S.; Simon, R.; et al. Heterogeneity and chronology of 6q15 deletion and ERG-fusion in prostate cancer. Oncotarget 2016, 7, 3897–3904. [Google Scholar] [CrossRef][Green Version]
- Minner, S.; Graefen, M.; Sauter, G.; Burkhardt, L.; Kluth, M.; Simon, R.; Schlomm, T.; Krohn, A.; Steuber, T.; Masser, S.; et al. CHD1 Is a 5q21 Tumor Suppressor Required for ERG Rearrangement in Prostate Cancer. Cancer Res. 2013, 73, 2795–2805. [Google Scholar] [CrossRef]
- Metzger, E.; Willmann, D.; McMillan, J.; Forne, I.; Metzger, P.; Gerhardt, S.; Petroll, K.; von Maessenhausen, A.; Urban, S.; Schott, A.K.; et al. Assembly of methylated KDM1A and CHD1 drives androgen receptor–dependent transcription and translocation. Nat. Struct. Mol. Biol. 2016, 23, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Gopalan, A.; Ran, L.; Zheng, D.; Li, D.; Sboner, A.; Chi, P.; Mosquera, J.M.; Cyrta, J.; Shukla, S.; Walczak, E.G.; et al. Aberrant Activation of a Gastrointestinal Transcriptional Circuit in Prostate Cancer Mediates Castration Resistance. Cancer Cell 2017, 32, 792.e7–806.e7. [Google Scholar] [CrossRef]
- Blattner, M.; Liu, D.; Robinson, B.D.; Huang, D.; Poliakov, A.; Gao, D.; Nataraj, S.; Deonarine, L.D.; Augello, M.A.; Sailer, V.; et al. SPOP Mutation Drives Prostate Tumorigenesis In Vivo through Coordinate Regulation of PI3K/mTOR and AR Signaling. Cancer Cell 2017, 31, 436–451. [Google Scholar] [CrossRef] [PubMed]
- Shoag, J.; Liu, D.; Blattner, M.; Sboner, A.; Park, K.; Deonarine, L.; Robinson, B.D.; Mosquera, J.M.; Chen, Y.; Rubin, M.A.; et al. SPOP mutation drives prostate neoplasia without stabilizing oncogenic transcription factor ERG. J. Clin. Investig. 2017, 128, 381–386. [Google Scholar] [CrossRef]
- Fedrizzi, T.; Ciani, Y.; Lorenzin, F.; Cantore, T.; Gasperini, P.; Demichelis, F. Fast mutual exclusivity algorithm nominates potential synthetic lethal gene pairs through brute force matrix product computations. Comput. Struct. Biotechnol. J. 2021, 19, 4394–4403. [Google Scholar] [CrossRef]
- Ciani, Y.; Fedrizzi, T.; Prandi, D.; Lorenzin, F.; Locallo, A.; Gasperini, P.; Franceschini, G.M.; Benelli, M.; Elemento, O.; Fava, L.; et al. Allele-Specific Genomics is an Orthogonal Feature in the Landscape of Primary Tumors Phenotypes. Cell Syst. 2021. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Jinek, M.; East, A.; Cheng, A.; Lin, S.; Ma, E.; Doudna, J. RNA-programmed genome editing in human cells. eLife 2013, 2, e00471. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.Y.; Zhao, Y.-T.; Lamonica, J.M.; Zhou, Z. Locus-specific histone deacetylation using a synthetic CRISPR-Cas9-based HDAC. Nat. Commun. 2017, 8, 15315. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed]
- Burslem, G.M.; Crews, C.M. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181, 102–114. [Google Scholar] [CrossRef]
- Schapira, M.; Calabrese, M.F.; Bullock, A.N.; Crews, C.M. Targeted protein degradation: Expanding the toolbox. Nat. Rev. Drug Discov. 2019, 18, 949–963. [Google Scholar] [CrossRef]
- Salami, J.; Alabi, S.; Willard, R.R.; Vitale, N.J.; Wang, J.; Dong, H.; Jin, M.; McDonnell, D.P.; Crew, A.P.; Neklesa, T.K.; et al. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun. Biol. 2018, 1, 100. [Google Scholar] [CrossRef]
- Han, X.; Wang, C.; Qin, C.; Xiang, W.; Fernandez-Salas, E.; Yang, C.-Y.; Wang, M.; Zhao, L.; Xu, T.; Chinnaswamy, K.; et al. Discovery of ARD-69 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Androgen Receptor (AR) for the Treatment of Prostate Cancer. J. Med. Chem. 2019, 62, 941–964. [Google Scholar] [CrossRef]
- Kregel, S.; Wang, C.; Han, X.; Xiao, L.; Fernandez-Salas, E.; Bawa, P.; McCollum, B.L.; Wilder-Romans, K.; Apel, I.J.; Cao, X.; et al. Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Neoplasia 2020, 22, 111–119. [Google Scholar] [CrossRef]
- Neklesa, T.; Snyder, L.B.; Willard, R.R.; Vitale, N.; Pizzano, J.; Gordon, D.A.; Bookbinder, M.; Macaluso, J.; Dong, H.; Ferraro, C.; et al. ARV-110: An oral androgen receptor PROTAC degrader for prostate cancer. J. Clin. Oncol. 2019, 37, 259. [Google Scholar] [CrossRef]
- Petrylak, D.P.; Gao, X.; Vogelzang, N.J.; Garfield, M.H.; Taylor, I.; Moore, M.D.; Peck, R.A.; III, H.A.B. First-in-human phase I study of ARV-110, an androgen receptor (AR) PROTAC degrader in patients (pts) with metastatic castrate-resistant prostate cancer (mCRPC) following enzalutamide (ENZ) and/or abiraterone (ABI). J. Clin. Oncol. 2020, 38, 3500. [Google Scholar] [CrossRef]
- Kim, G.-Y.; Song, C.W.; Yang, Y.-S.; Lee, N.-R.; Yoo, H.-S.; Son, S.H.; Lee, S.J.; Park, J.S.; Lee, J.K.; Inn, K.-S.; et al. Chemical Degradation of Androgen Receptor (AR) Using Bicalutamide Analog-Thalidomide PROTACs. Molecules 2021, 26, 2525. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, H.; Kaniskan, H.Ü.; Xie, L.; Chen, X.; Jin, J.; Wei, W. TF-PROTACs Enable Targeted Degradation of Transcription Factors. J. Am. Chem. Soc. 2021, 143, 8902–8910. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Therapeutic Strategy | References |
|---|---|
| SPOP inhibition | [170,171] |
| Treatment with supraphysiological androgen levels | [170,172,173] |
| CRISPR-based breakpoint specific insertion of suicide genes (e.g., HSV1-tk) | [174] |
| TK-216 (derivative of the YK-4-279 inhibitor developed for EWS-FLI1) | [145,146,147,175] |
| Dexamethasone (glucocorticoid receptor (GR) agonist with anti-inflammatory activity) | [176] |
| PROTACs (ERG binding motif linked to E3 ligase recruiting element) | [177] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorenzin, F.; Demichelis, F. Past, Current, and Future Strategies to Target ERG Fusion-Positive Prostate Cancer. Cancers 2022, 14, 1118. https://doi.org/10.3390/cancers14051118
Lorenzin F, Demichelis F. Past, Current, and Future Strategies to Target ERG Fusion-Positive Prostate Cancer. Cancers. 2022; 14(5):1118. https://doi.org/10.3390/cancers14051118
Chicago/Turabian StyleLorenzin, Francesca, and Francesca Demichelis. 2022. "Past, Current, and Future Strategies to Target ERG Fusion-Positive Prostate Cancer" Cancers 14, no. 5: 1118. https://doi.org/10.3390/cancers14051118
APA StyleLorenzin, F., & Demichelis, F. (2022). Past, Current, and Future Strategies to Target ERG Fusion-Positive Prostate Cancer. Cancers, 14(5), 1118. https://doi.org/10.3390/cancers14051118