
Urokinase Receptor uPAR Downregulation in Neuroblastoma Leads to Dormancy, Chemoresistance and Metastasis

 , , , , ,
, , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Analysis of Neuro2a Cell Sensitivity to Cisplatin and Doxorubicin
2.3. Analysis of Neuro2a Cell Adhesion
2.4. Flow Cytometry
2.5. RNA Isolation, Reverse Transcription and qPCR
2.6. Protein Extraction, Electrophoresis and Western Blot
2.7. Caspase-3 Activity Measurement
2.8. Animal Studies
2.9. Analysis of Lung Metastasis
2.10. Mining Transcriptome Data for Neuroblastoma
2.11. Data and Statistical Analysis
3. Results
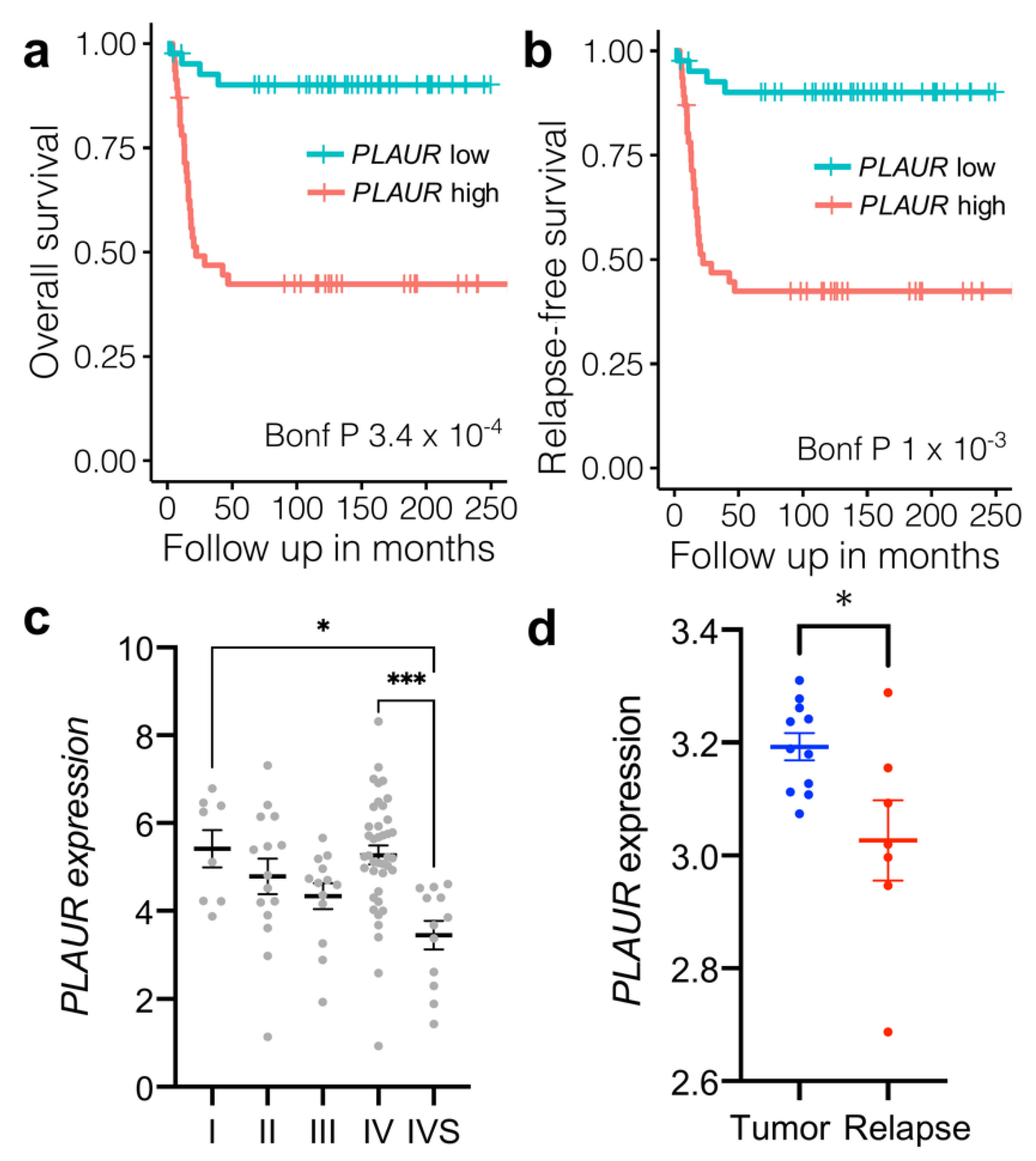
3.1. High Initial PLAUR Expression in Neuroblastoma Predicts Poor Survival; a Decrease in PLAUR Expression Is Associated with Tumor Relapse
3.2. uPAR Downregulation Is Associated with Chemoresistance in Neuroblastoma Neuro2a Cells
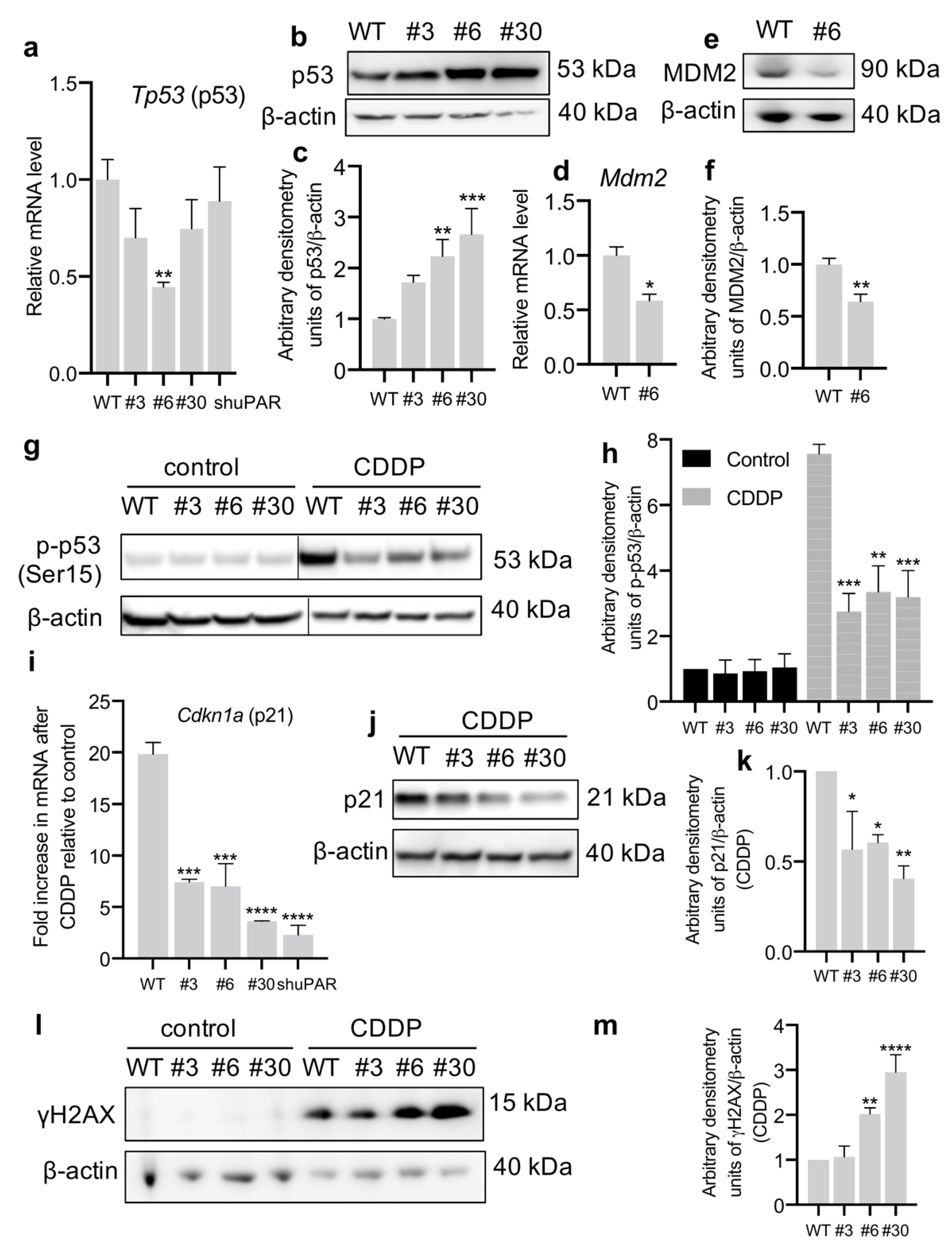
3.3. uPAR-Deficient Neuro2a Cells Exhibit Dormant Phenotype and Decreased p53 Activation upon CDDP Treatment
3.4. uPAR-Deficient Neuroblastoma Cells Have an Increased Metastatic Potential
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Wyganowska-Świątkowska, M.; Tarnowski, M.; Murtagh, D.; Skrzypczak-Jankun, E.; Jankun, J. Proteolysis Is the Most Fundamental Property of Malignancy and Its Inhibition May Be Used Therapeutically (Review). Int. J. Mol. Med. 2019, 43, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, N.; Mihalcioiu, C.; Rabbani, S.A. Multifaceted Role of the Urokinase-Type Plasminogen Activator (UPA) and Its Receptor (UPAR): Diagnostic, Prognostic, and Therapeutic Applications. Front. Oncol. 2018, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Blasi, F.; Carmeliet, P. UPAR: A Versatile Signalling Orchestrator. Nat. Rev. Mol. Cell Biol. 2002, 3, 932–943. [Google Scholar] [CrossRef]
- Eden, G.; Archinti, M.; Furlan, F.; Murphy, R.; Degryse, B. The Urokinase Receptor Interactome. Curr. Pharm. Des. 2011, 17, 1874–1889. [Google Scholar] [CrossRef]
- Huebschman, M.L.; Lane, N.L.; Liu, H.; Sarode, V.R.; Devlin, J.L.; Frenkel, E.P. Molecular Heterogeneity in Adjacent Cells in Triple-Negative Breast Cancer. Breast Cancer 2015, 7, 231–237. [Google Scholar] [CrossRef]
- Noh, H.; Hong, S.; Huang, S. Role of Urokinase Receptor in Tumor Progression and Development. Theranostics 2013, 3, 487–495. [Google Scholar] [CrossRef]
- Montuori, N.; Pesapane, A.; Rossi, F.W.; Giudice, V.; De Paulis, A.; Selleri, C.; Ragno, P. Urokinase Type Plasminogen Activator Receptor (UPAR) as a New Therapeutic Target in Cancer. Transl. Med. UniSa 2016, 15, 15–21. [Google Scholar]
- Rysenkova, K.D.; Semina, E.V.; Karagyaur, M.N.; Shmakova, A.A.; Dyikanov, D.T.; Vasiluev, P.A.; Rubtsov, Y.P.; Rubina, K.A.; Tkachuk, V.A. CRISPR/Cas9 Nickase Mediated Targeting of Urokinase Receptor Gene Inhibits Neuroblastoma Cell Proliferation. Oncotarget 2018, 9, 29414–29430. [Google Scholar] [CrossRef]
- Wang, K.; Xing, Z.-H.; Jiang, Q.-W.; Yang, Y.; Huang, J.-R.; Yuan, M.-L.; Wei, M.-N.; Li, Y.; Wang, S.-T.; Liu, K.; et al. Targeting UPAR by CRISPR/Cas9 System Attenuates Cancer Malignancy and Multidrug Resistance. Front. Oncol. 2019, 9, 80. [Google Scholar] [CrossRef]
- Raghu, H.; Lakka, S.S.; Gondi, C.S.; Mohanam, S.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Suppression of UPA and UPAR Attenuates Angiogenin Mediated Angiogenesis in Endothelial and Glioblastoma Cell Lines. PLoS ONE 2010, 5, e12458. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, A.G.; Holloway, R.W.; Miller, V.A.; Waisman, D.M. Plasmin and Plasminogen System in the Tumor Microenvironment: Implications for Cancer Diagnosis, Prognosis, and Therapy. Cancers 2021, 13, 1838. [Google Scholar] [CrossRef] [PubMed]
- Semina, E.V.; Rubina, K.A.; Shmakova, A.A.; Rysenkova, K.D.; Klimovich, P.S.; Aleksanrushkina, N.A.; Sysoeva, V.Y.; Karagyaur, M.N.; Tkachuk, V.A. Downregulation of UPAR Promotes Urokinase Translocation into the Nucleus and Epithelial to Mesenchymal Transition in Neuroblastoma. J. Cell Physiol. 2020, 235, jcp.29555. [Google Scholar] [CrossRef]
- Mitra, A.; Mishra, L.; Li, S. EMT, CTCs and CSCs in Tumor Relapse and Drug-Resistance. Oncotarget 2015, 6, 10697–10711. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Chen, E.I. Cancer Stem Cells, Tumor Dormancy, and Metastasis. Front. Endocrinol. 2012, 3, 125. [Google Scholar] [CrossRef]
- Li Santi, A.; Napolitano, F.; Montuori, N.; Ragno, P. The Urokinase Receptor: A Multifunctional Receptor in Cancer Cell Biology. Therapeutic Implications. Int. J. Mol. Sci. 2021, 22, 4111. [Google Scholar] [CrossRef]
- Mazar, A.P.; Ahn, R.W.; O’Halloran, T.V. Development of Novel Therapeutics Targeting the Urokinase Plasminogen Activator Receptor (UPAR) and Their Translation Toward the Clinic. Curr. Pharm. Des. 2011, 17, 1970–1978. [Google Scholar] [CrossRef]
- Di Mauro, C.; Pesapane, A.; Formisano, L.; Rosa, R.; D’Amato, V.; Ciciola, P.; Servetto, A.; Marciano, R.; Orsini, R.C.; Monteleone, F.; et al. Urokinase-Type Plasminogen Activator Receptor (UPAR) Expression Enhances Invasion and Metastasis in RAS Mutated Tumors. Sci. Rep. 2017, 7, 9388. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Dyikanov, D.; Vasiluev, P.; Rysenkova, K.; Aleksandrushkina, N.; Tyurin-Kuzmin, P.; Kulebyakin, K.; Rubtsov, Y.; Shmakova, A.; Evseeva, M.; Balatskiy, A.; et al. Optimization of CRISPR/Cas9 Technology to Knock-out Genes of Interest in Aneuploid Cell Lines. Tissue Eng. Part C Methods 2019, 25, 168–175. [Google Scholar] [CrossRef]
- Rysenkova, K.D.; Semina, E.V.; Karagyaur, M.N.; Shmakova, A.A.; Dyikanov, D.T.; Rubina, K.A.; Tkachuk, V.A. Use of CRISPR /Cas9 Genome Editing Technology to Suppress Urokinase Receptor Gene Expression in Neuroblastoma Cells. Technol. Living Syst. 2018, 15, 10–19. [Google Scholar]
- Semina, E.; Rubina, K.; Sysoeva, V.; Rysenkova, K.; Klimovich, P.; Plekhanova, O.; Tkachuk, V. Urokinase and Urokinase Receptor Participate in Regulation of Neuronal Migration, Axon Growth and Branching. Eur. J. Cell Biol. 2016, 95, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Klimovich, P.S.; Semina, E.V.; Karagyaur, M.N.; Rysenkova, K.D.; Sysoeva, V.Y.; Mironov, N.A.; Sagaradze, G.D.; Az’muko, A.A.; Popov, V.S.; Rubina, K.A.; et al. Urokinase Receptor Regulates Nerve Regeneration through Its Interaction with A5β1-Integrin. Biomed. Pharmacother. 2020, 125, 110008. [Google Scholar] [CrossRef] [PubMed]
- van Meerloo, J.; Kaspers, G.J.L.; Cloos, J. Cell Sensitivity Assays: The MTT Assay. Methods Mol. Biol. 2011, 731, 237–245. [Google Scholar] [CrossRef] [PubMed]
- RIPA Lysis Buffer. Cold Spring Harb. Protoc. 2017, 2017, pdb.rec101428. [CrossRef]
- Prokhorova, E.A.; Kopeina, G.S.; Lavrik, I.N.; Zhivotovsky, B. Apoptosis Regulation by Subcellular Relocation of Caspases. Sci. Rep. 2018, 8, 12199. [Google Scholar] [CrossRef]
- Amirkhosravi, A.; Warnes, G.; Biggerstaff, J.; Malik, Z.; May, K.; Francis, J.L. The Effect of Pentoxifylline on Spontaneous and Experimental Metastasis of the Mouse Neuro2a Neuroblastoma. Clin Exp Metastasis. 1997, 15, 453–461. [Google Scholar] [CrossRef]
- Molenaar, J.J.; Koster, J.; Zwijnenburg, D.A.; van Sluis, P.; Valentijn, L.J.; van der Ploeg, I.; Hamdi, M.; van Nes, J.; Westerman, B.A.; van Arkel, J.; et al. Sequencing of Neuroblastoma Identifies Chromothripsis and Defects in Neuritogenesis Genes. Nature 2012, 483, 589–593. [Google Scholar] [CrossRef]
- Schramm, A.; Köster, J.; Assenov, Y.; Althoff, K.; Peifer, M.; Mahlow, E.; Odersky, A.; Beisser, D.; Ernst, C.; Henssen, A.G.; et al. Mutational Dynamics between Primary and Relapse Neuroblastomas. Nat. Genet. 2015, 47, 872–877. [Google Scholar] [CrossRef]
- Gilder, A.S.; Natali, L.; Van Dyk, D.M.; Zalfa, C.; Banki, M.A.; Pizzo, D.P.; Wang, H.; Klemke, R.L.; Mantuano, E.; Gonias, S.L. The Urokinase Receptor Induces a Mesenchymal Gene Expression Signature in Glioblastoma Cells and Promotes Tumor Cell Survival in Neurospheres. Sci. Rep. 2018, 8, 2982. [Google Scholar] [CrossRef]
- Lossi, L.; Castagna, C.; Merighi, A. Caspase-3 Mediated Cell Death in the Normal Development of the Mammalian Cerebellum. Int. J. Mol. Sci. 2018, 19, 3999. [Google Scholar] [CrossRef]
- Bouchard, V.J.; Rouleau, M.; Poirier, G.G. PARP-1, a Determinant of Cell Survival in Response to DNA Damage. Exp. Hematol. 2003, 31, 446–454. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.; Khachfe, H.; Salhab, H.; Fares, Y. Molecular Principles of Metastasis: A Hallmark of Cancer Revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhang, M.; Tang, Y.; Liang, X. Cancer Cell Dormancy: Mechanisms and Implications of Cancer Recurrence and Metastasis. Onco. Targets Ther. 2017, 10, 5219–5228. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-Y.; Nam, J.-S. The Force Awakens: Metastatic Dormant Cancer Cells. Exp. Mol. Med. 2020, 52, 569–581. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A.; Liu, D.; Mignatti, A.; Kovalski, K.; Ossowski, L. Urokinase Receptor and Fibronectin Regulate the ERK MAPK to P38 MAPK Activity Ratios That Determine Carcinoma Cell Proliferation or Dormancy In Vivo. Mol. Biol. Cell 2001, 12, 863–879. [Google Scholar] [CrossRef]
- Wei, Y.; Eble, J.A.; Wang, Z.; Kreidberg, J.A.; Chapman, H.A. Urokinase Receptors Promote Β1 Integrin Function through Interactions with Integrin A3β1. Mol. Biol. Cell 2001, 12, 2975–2986. [Google Scholar] [CrossRef]
- Zheng, H.-C. The Molecular Mechanisms of Chemoresistance in Cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef]
- Ozaki, T.; Nakagawara, A. Role of P53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef]
- Loughery, J.; Cox, M.; Smith, L.M.; Meek, D.W. Critical Role for P53-Serine 15 Phosphorylation in Stimulating Transactivation at P53-Responsive Promoters. Nucleic Acids Res. 2014, 42, 7666–7680. [Google Scholar] [CrossRef]
- Mah, L.-J.; El-Osta, A.; Karagiannis, T.C. ΓH2AX: A Sensitive Molecular Marker of DNA Damage and Repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Halaby, M.-J.; Hakem, A.; Li, L.; El Ghamrasni, S.; Venkatesan, S.; Hande, P.M.; Sanchez, O.; Hakem, R. Synergistic Interaction of Rnf8 and P53 in the Protection against Genomic Instability and Tumorigenesis. PLoS Genet. 2013, 9, e1003259. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, R.K.; Varshney, A.K.; Yadava, P.K. Diversity and Functional Evolution of the Plasminogen Activator System. Biomed. Pharmacother. 2018, 98, 886–898. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Tang, C.-H.; Kim, Y.; Robillard, L.; Zhang, F.; Kugler, M.C.; Chapman, H.A. Urokinase Receptors Are Required for 5beta1 Integrin-Mediated Signaling in Tumor Cells. J. Biol. Chem. 2006, 282, 3929–3939. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Ghiso, J.A.; Estrada, Y.; Liu, D.; Ossowski, L. ERK(MAPK) Activity as a Determinant of Tumor Growth and Dormancy; Regulation by P38(SAPK). Cancer Res. 2003, 63, 1684–1695. [Google Scholar] [CrossRef]
- Chandrasekar, N.; Mohanam, S.; Gujrati, M.; Olivero, W.C.; Dinh, D.H.; Rao, J.S. Downregulation of UPA Inhibits Migration and PI3k/Akt Signaling in Glioblastoma Cells. Oncogene 2003, 22, 392–400. [Google Scholar] [CrossRef]
- Subramanian, R.; Gondi, C.S.; Lakka, S.S.; Jutla, A.; Rao, J.S. SiRNA-Mediated Simultaneous Downregulation of UPA and Its Receptor Inhibits Angiogenesis and Invasiveness Triggering Apoptosis in Breast Cancer Cells. Int. J. Oncol. 2006, 28, 831–839. [Google Scholar] [CrossRef][Green Version]
- Kenny, H.A.; Leonhardt, P.; Ladanyi, A.; Yamada, S.D.; Montag, A.; Im, H.K.; Jagadeeswaran, S.; Shaw, D.E.; Mazar, A.P.; Lengyel, E. Targeting the Urokinase Plasminogen Activator Receptor Inhibits Ovarian Cancer Metastasis. Clin. Cancer Res. 2011, 17, 459–471. [Google Scholar] [CrossRef]
- Rysenkova, K.D.; Klimovich, P.S.; Shmakova, A.A.; Karagyaur, M.N.; Ivanova, K.A.; Aleksandrushkina, N.A.; Tkachuk, V.A.; Rubina, K.A.; Semina, E.V. Urokinase Receptor Deficiency Results in EGFR-Mediated Failure to Transmit Signals for Cell Survival and Neurite Formation in Mouse Neuroblastoma Cells. Cell Signal. 2020, 75, 109741. [Google Scholar] [CrossRef]
- Li, P.; Gao, Y.; Ji, Z.; Zhang, X.; Xu, Q.; Li, G.; Guo, Z.; Zheng, B.; Guo, X. Role of Urokinase Plasminogen Activator and Its Receptor in Metastasis and Invasion of Neuroblastoma. J. Pediatric Surg. 2004, 39, 1512–1519. [Google Scholar] [CrossRef]
- Keeratichamroen, S.; Lirdprapamongkol, K.; Svasti, J. Mechanism of ECM-Induced Dormancy and Chemoresistance in A549 Human Lung Carcinoma Cells. Oncol. Rep. 2018, 39, 1765–1774. [Google Scholar] [CrossRef] [PubMed]
- Ghiso, J.A.A.; Kovalski, K.; Ossowski, L. Tumor Dormancy Induced by Downregulation of Urokinase Receptor in Human Carcinoma Involves Integrin and MAPK Signaling. J. Cell Biol. 1999, 147, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Cao, S.; Liu, Z.; Ye, X.; Chen, L.; Meng, S. RNAi-Mediated Downregulation of UPAR Synergizes with Targeting of HER2 through the ERK Pathway in Breast Cancer Cells. Int. J. Cancer 2010, 127, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Ossowski, L.; Aguirre Ghiso, J.; Liu, D.; Estrada, Y.; Rosenbaum, S. The Role of ERK and P38 in Regulation of Tumor Dormancy. Cancer Res. 2004, 64, 1318–1319. [Google Scholar]
- Gutova, M.; Najbauer, J.; Gevorgyan, A.; Metz, M.Z.; Weng, Y.; Shih, C.-C.; Aboody, K.S. Identification of UPAR-Positive Chemoresistant Cells in Small Cell Lung Cancer. PLoS ONE 2007, 2, e243. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wang, L.; Wang, Y.; Zhuo, Y.; Li, H.; Chen, J.; Chen, W. Overexpression of CD147 Contributes to the Chemoresistance of Head and Neck Squamous Cell Carcinoma Cells. J. Oral Pathol. Med. 2013, 42, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Eastman, B.M.; Jo, M.; Webb, D.L.; Takimoto, S.; Gonias, S.L. A Transformation in the Mechanism by Which the Urokinase Receptor Signals Provides a Selection Advantage for Estrogen Receptor-Expressing Breast Cancer Cells in the Absence of Estrogen. Cell Signal. 2012, 24, 1847–1855. [Google Scholar] [CrossRef]
- Hu, J.; Jo, M.; Cavenee, W.K.; Furnari, F.; VandenBerg, S.R.; Gonias, S.L. Crosstalk between the Urokinase-Type Plasminogen Activator Receptor and EGF Receptor Variant III Supports Survival and Growth of Glioblastoma Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 15984–15989. [Google Scholar] [CrossRef]
- Wykosky, J.; Hu, J.; Gomez, G.G.; Taylor, T.; Villa, G.R.; Pizzo, D.; VandenBerg, S.R.; Thorne, A.H.; Chen, C.C.; Mischel, P.S.; et al. A Urokinase Receptor-Bim Signaling Axis Emerges During EGFR Inhibitor Resistance in Mutant EGFR Glioblastoma. Cancer Res. 2015, 75, 394–404. [Google Scholar] [CrossRef]
- Varadhachary, G.R. Carcinoma of Unknown Primary Origin. Gastrointest Cancer Res. 2007, 1, 229–235. [Google Scholar] [PubMed]
- Madsen, C.D.; Ferraris, G.M.S.; Andolfo, A.; Cunningham, O.; Sidenius, N. UPAR-Induced Cell Adhesion and Migration: Vitronectin Provides the Key. J. Cell Biol. 2007, 177, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Kren, A.; Baeriswyl, V.; Lehembre, F.; Wunderlin, C.; Strittmatter, K.; Antoniadis, H.; Fässler, R.; Cavallaro, U.; Christofori, G. Increased Tumor Cell Dissemination and Cellular Senescence in the Absence of Β1-Integrin Function. EMBO J. 2007, 26, 2832–2842. [Google Scholar] [CrossRef] [PubMed]
- Biagioni, A.; Laurenzana, A.; Chillà, A.; Del Rosso, M.; Andreucci, E.; Poteti, M.; Bani, D.; Guasti, D.; Fibbi, G.; Margheri, F. UPAR Knockout Results in a Deep Glycolytic and OXPHOS Reprogramming in Melanoma and Colon Carcinoma Cell Lines. Cells 2020, 9, 308. [Google Scholar] [CrossRef] [PubMed]
- Stepanenko, A.A.; Vassetzky, Y.S.; Kavsan, V.M. Antagonistic Functional Duality of Cancer Genes. Gene 2013, 529, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Masucci, M.T.; Minopoli, M.; Di Carluccio, G.; Motti, M.L.; Carriero, M.V. Therapeutic Strategies Targeting Urokinase and Its Receptor in Cancer. Cancers 2022, 14, 498. [Google Scholar] [CrossRef]
- Metrangolo, V.; Ploug, M.; Engelholm, L.H. The Urokinase Receptor (UPAR) as a “Trojan Horse” in Targeted Cancer Therapy: Challenges and Opportunities. Cancers 2021, 13, 5376. [Google Scholar] [CrossRef]
- Nalla, A.K.; Gorantla, B.; Gondi, C.S.; Lakka, S.S.; Rao, J.S. Targeting MMP-9, UPAR, and Cathepsin B Inhibits Invasion, Migration and Activates Apoptosis in Prostate Cancer Cells. Cancer Gene Ther. 2010, 17, 599–613. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shmakova, A.A.; Klimovich, P.S.; Rysenkova, K.D.; Popov, V.S.; Gorbunova, A.S.; Karpukhina, A.A.; Karagyaur, M.N.; Rubina, K.A.; Tkachuk, V.A.; Semina, E.V. Urokinase Receptor uPAR Downregulation in Neuroblastoma Leads to Dormancy, Chemoresistance and Metastasis. Cancers 2022, 14, 994. https://doi.org/10.3390/cancers14040994
Shmakova AA, Klimovich PS, Rysenkova KD, Popov VS, Gorbunova AS, Karpukhina AA, Karagyaur MN, Rubina KA, Tkachuk VA, Semina EV. Urokinase Receptor uPAR Downregulation in Neuroblastoma Leads to Dormancy, Chemoresistance and Metastasis. Cancers. 2022; 14(4):994. https://doi.org/10.3390/cancers14040994
Chicago/Turabian StyleShmakova, Anna A., Polina S. Klimovich, Karina D. Rysenkova, Vladimir S. Popov, Anna S. Gorbunova, Anna A. Karpukhina, Maxim N. Karagyaur, Kseniya A. Rubina, Vsevolod A. Tkachuk, and Ekaterina V. Semina. 2022. "Urokinase Receptor uPAR Downregulation in Neuroblastoma Leads to Dormancy, Chemoresistance and Metastasis" Cancers 14, no. 4: 994. https://doi.org/10.3390/cancers14040994
APA StyleShmakova, A. A., Klimovich, P. S., Rysenkova, K. D., Popov, V. S., Gorbunova, A. S., Karpukhina, A. A., Karagyaur, M. N., Rubina, K. A., Tkachuk, V. A., & Semina, E. V. (2022). Urokinase Receptor uPAR Downregulation in Neuroblastoma Leads to Dormancy, Chemoresistance and Metastasis. Cancers, 14(4), 994. https://doi.org/10.3390/cancers14040994