
SOX9 Triggers Different Epithelial to Mesenchymal Transition States to Promote Pancreatic Cancer Progression

 , ,
, ,  , , ,
, , ,  , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Experimental Procedures
2.1. Human Samples
2.2. Cell Culture
2.3. Gene Silencing and Overexpression
2.4. mRNA Expression Analysis
2.5. Western Blot and Immunofluorescence
2.6. In Vitro Assays for Migration and Invasion
2.7. F-Actin Staining
2.8. Colony Formation Assay
2.9. Soft Agar Foci
2.10. In Vivo Carcinogenesis Assays
2.11. Immunohistochemistry
2.12. Expression Microarrays
2.13. Data Evaluation
3. Results
3.1. SOX9 Overexpression Confers Metastatic Potential and Promotes EMT in K-Ras Transformed Cells
3.2. SOX9 Overexpression Promotes EMT and Metastatic Traits in Pancreatic Cancer Cells
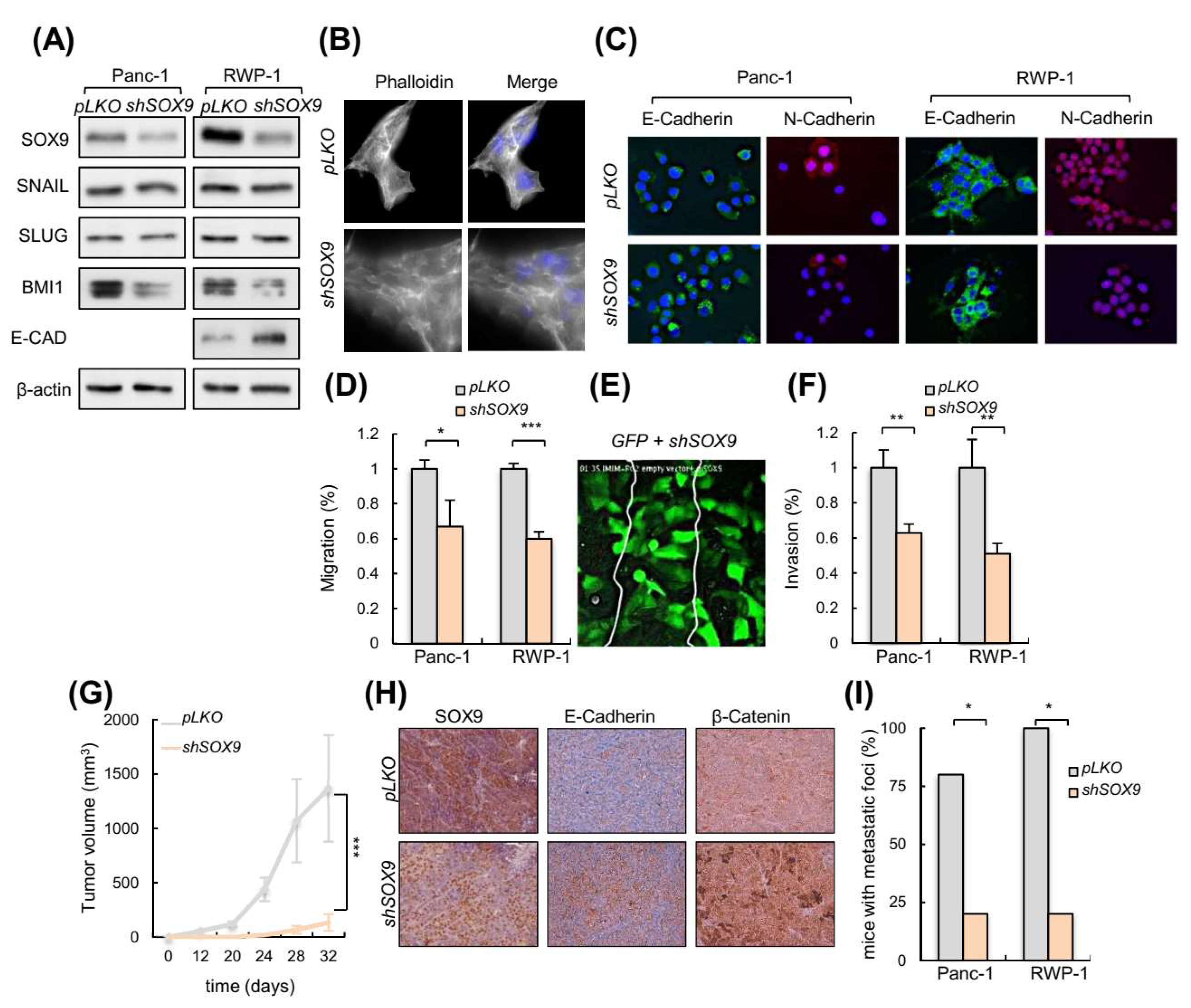
3.3. Silencing of SOX9 Drives MET and Decreases Metastatic Potential
3.4. SOX9 Is Necessary for Pro-Metastatic Cancer Self-Renewal
3.5. Transcriptomics Reveal Multiple Processes Differentially Expressed in SOX9-Silenced Cells
3.6. BMI1 Stem Cell Factor Mediates SOX9 Activities
3.7. High Levels of SOX9 Confer Chemoresistance
3.8. High Levels of SOX9 Are Associated with Reduced Survival and Correlate with EMT Markers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ocana, O.H.; Corcoles, R.; Fabra, A.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Fazilaty, H.; Rago, L.; Kass Youssef, K.; Ocana, O.H.; Garcia-Asencio, F.; Arcas, A.; Galceran, J.; Nieto, M.A. A gene regulatory network to control EMT programs in development and disease. Nat. Commun. 2019, 10, 5115. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Ng Eaton, E.; Weinberg, R.A. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 2015, 525, 256–260. [Google Scholar] [CrossRef] [PubMed]
- van Staalduinen, J.; Baker, D.; Ten Dijke, P.; van Dam, H. Epithelial-mesenchymal-transition-inducing transcription factors: New targets for tackling chemoresistance in cancer? Oncogene 2018, 37, 6195–6211. [Google Scholar] [CrossRef] [PubMed]
- Beck, B.; Lapouge, G.; Rorive, S.; Drogat, B.; Desaedelaere, K.; Delafaille, S.; Dubois, C.; Salmon, I.; Willekens, K.; Marine, J.C.; et al. Different levels of Twist1 regulate skin tumor initiation, stemness, and progression. Cell Stem Cell 2015, 16, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Kamachi, Y.; Kondoh, H. Sox proteins: Regulators of cell fate specification and differentiation. Development 2013, 140, 4129–4144. [Google Scholar] [CrossRef] [PubMed]
- Seymour, P.A.; Freude, K.K.; Dubois, C.L.; Shih, H.P.; Patel, N.A.; Sander, M. A dosage-dependent requirement for Sox9 in pancreatic endocrine cell formation. Dev. Biol. 2008, 323, 19–30. [Google Scholar] [CrossRef]
- Furuyama, K.; Kawaguchi, Y.; Akiyama, H.; Horiguchi, M.; Kodama, S.; Kuhara, T.; Hosokawa, S.; Elbahrawy, A.; Soeda, T.; Koizumi, M.; et al. Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat. Genet. 2011, 43, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Chaboissier, M.C.; Behringer, R.R.; Rowitch, D.H.; Schedl, A.; Epstein, J.A.; de Crombrugghe, B. Essential role of Sox9 in the pathway that controls formation of cardiac valves and septa. Proc. Natl. Acad. Sci. USA 2004, 101, 6502–6507. [Google Scholar] [CrossRef]
- Cheung, M.; Chaboissier, M.C.; Mynett, A.; Hirst, E.; Schedl, A.; Briscoe, J. The transcriptional control of trunk neural crest induction, survival, and delamination. Dev. Cell 2005, 8, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Panda, M.; Tripathi, S.K.; Biswal, B.K. SOX9: An emerging driving factor from cancer progression to drug resistance. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188517. [Google Scholar] [CrossRef]
- Carrasco-Garcia, E.; Lopez, L.; Aldaz, P.; Arevalo, S.; Aldaregia, J.; Egana, L.; Bujanda, L.; Cheung, M.; Sampron, N.; Garcia, I.; et al. SOX9-regulated cell plasticity in colorectal metastasis is attenuated by rapamycin. Sci. Rep. 2016, 6, 32350. [Google Scholar] [CrossRef]
- Huang, J.Q.; Wei, F.K.; Xu, X.L.; Ye, S.X.; Song, J.W.; Ding, P.K.; Zhu, J.; Li, H.F.; Luo, X.P.; Gong, H.; et al. SOX9 drives the epithelial-mesenchymal transition in non-small-cell lung cancer through the Wnt/beta-catenin pathway. J. Transl. Med. 2019, 17, 143. [Google Scholar] [CrossRef] [PubMed]
- Luanpitpong, S.; Li, J.; Manke, A.; Brundage, K.; Ellis, E.; McLaughlin, S.L.; Angsutararux, P.; Chanthra, N.; Voronkova, M.; Chen, Y.C.; et al. SLUG is required for SOX9 stabilization and functions to promote cancer stem cells and metastasis in human lung carcinoma. Oncogene 2016, 35, 2824–2833. [Google Scholar] [CrossRef]
- Guo, W.; Keckesova, Z.; Donaher, J.L.; Shibue, T.; Tischler, V.; Reinhardt, F.; Itzkovitz, S.; Noske, A.; Zurrer-Hardi, U.; Bell, G.; et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 2012, 148, 1015–1028. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Ghaneh, P.; Kleeff, J.; Halloran, C.M.; Raraty, M.; Jackson, R.; Melling, J.; Jones, O.; Palmer, D.H.; Cox, T.F.; Smith, C.J.; et al. The Impact of Positive Resection Margins on Survival and Recurrence Following Resection and Adjuvant Chemotherapy for Pancreatic Ductal Adenocarcinoma. Ann. Surg. 2019, 269, 520–529. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Prevot, P.P.; Simion, A.; Grimont, A.; Colletti, M.; Khalaileh, A.; Van den Steen, G.; Sempoux, C.; Xu, X.; Roelants, V.; Hald, J.; et al. Role of the ductal transcription factors HNF6 and Sox9 in pancreatic acinar-to-ductal metaplasia. Gut 2012, 61, 1723–1732. [Google Scholar] [CrossRef]
- Kopp, J.L.; von Figura, G.; Mayes, E.; Liu, F.F.; Dubois, C.L.; Morris, J.P.t.; Pan, F.C.; Akiyama, H.; Wright, C.V.; Jensen, K.; et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 22, 737–750. [Google Scholar] [CrossRef]
- Chen, N.M.; Singh, G.; Koenig, A.; Liou, G.Y.; Storz, P.; Zhang, J.S.; Regul, L.; Nagarajan, S.; Kuhnemuth, B.; Johnsen, S.A.; et al. NFATc1 Links EGFR Signaling to Induction of Sox9 Transcription and Acinar-Ductal Transdifferentiation in the Pancreas. Gastroenterology 2015, 148, 1024–1034.e1029. [Google Scholar] [CrossRef]
- Sun, L.; Mathews, L.A.; Cabarcas, S.M.; Zhang, X.; Yang, A.; Zhang, Y.; Young, M.R.; Klarmann, K.D.; Keller, J.R.; Farrar, W.L. Epigenetic regulation of SOX9 by the NF-kappaB signaling pathway in pancreatic cancer stem cells. Stem Cells 2013, 31, 1454–1466. [Google Scholar] [CrossRef]
- Camaj, P.; Jackel, C.; Krebs, S.; De Toni, E.N.; Blum, H.; Jauch, K.W.; Nelson, P.J.; Bruns, C.J. Hypoxia-independent gene expression mediated by SOX9 promotes aggressive pancreatic tumor biology. Mol. Cancer Res. 2014, 12, 421–432. [Google Scholar] [CrossRef][Green Version]
- Li, J.; Chen, X.; Zhu, L.; Lao, Z.; Zhou, T.; Zang, L.; Ge, W.; Jiang, M.; Xu, J.; Cao, Y.; et al. SOX9 is a critical regulator of TSPAN8-mediated metastasis in pancreatic cancer. Oncogene 2021, 40, 4884–4893. [Google Scholar] [CrossRef]
- Higashihara, T.; Yoshitomi, H.; Nakata, Y.; Kagawa, S.; Takano, S.; Shimizu, H.; Kato, A.; Furukawa, K.; Ohtsuka, M.; Miyazaki, M. Sex Determining Region Y Box 9 Induces Chemoresistance in Pancreatic Cancer Cells by Induction of Putative Cancer Stem Cell Characteristics and Its High Expression Predicts Poor Prognosis. Pancreas 2017, 46, 1296–1304. [Google Scholar] [CrossRef]
- Tanaka, T.; Kuroki, T.; Adachi, T.; Ono, S.; Hirabaru, M.; Soyama, A.; Kitasato, A.; Takatsuki, M.; Hayashi, T.; Eguchi, S. Evaluation of SOX9 expression in pancreatic ductal adenocarcinoma and intraductal papillary mucinous neoplasm. Pancreas 2013, 42, 488–493. [Google Scholar] [CrossRef]
- Matheu, A.; Collado, M.; Wise, C.; Manterola, L.; Cekaite, L.; Tye, A.J.; Canamero, M.; Bujanda, L.; Schedl, A.; Cheah, K.S.; et al. Oncogenicity of the developmental transcription factor Sox9. Cancer Res. 2012, 72, 1301–1315. [Google Scholar] [CrossRef]
- Santos, J.C.; Carrasco-Garcia, E.; Garcia-Puga, M.; Aldaz, P.; Montes, M.; Fernandez-Reyes, M.; de Oliveira, C.C.; Lawrie, C.H.; Arauzo-Bravo, M.J.; Ribeiro, M.L.; et al. SOX9 Elevation Acts with Canonical WNT Signaling to Drive Gastric Cancer Progression. Cancer Res. 2016, 76, 6735–6746. [Google Scholar] [CrossRef]
- Guerra, C.; Schuhmacher, A.J.; Canamero, M.; Grippo, P.J.; Verdaguer, L.; Perez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Aldaz, P.; Otaegi-Ugartemendia, M.; Saenz-Antonanzas, A.; Garcia-Puga, M.; Moreno-Valladares, M.; Flores, J.M.; Gerovska, D.; Arauzo-Bravo, M.J.; Sampron, N.; Matheu, A.; et al. SOX9 promotes tumor progression through the axis BMI1-p21(CIP). Sci. Rep. 2020, 10, 357. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Hsu, D.S.; Wang, H.W.; Wang, H.J.; Lan, H.Y.; Yang, W.H.; Huang, C.H.; Kao, S.Y.; Tzeng, C.H.; Tai, S.K.; et al. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat. Cell Biol. 2010, 12, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; van Galen, P.; Pedley, N.M.; Lima-Fernandes, E.; Frelin, C.; Davis, T.; Cao, L.; Baiazitov, R.; Du, W.; Sydorenko, N.; et al. Self-renewal as a therapeutic target in human colorectal cancer. Nat. Med. 2014, 20, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Tao, K.; Li, H.; Jin, C.; Song, Z.; Li, J.; Shi, H.; Li, X.; Dang, Z.; Dou, K. Bmi-1 is related to proliferation, survival and poor prognosis in pancreatic cancer. Cancer Sci. 2010, 101, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Proctor, E.; Waghray, M.; Lee, C.J.; Heidt, D.G.; Yalamanchili, M.; Li, C.; Bednar, F.; Simeone, D.M. Bmi1 enhances tumorigenicity and cancer stem cell function in pancreatic adenocarcinoma. PLoS ONE 2013, 8, e55820. [Google Scholar] [CrossRef] [PubMed]
- Karamitopoulou, E.; Pallante, P.; Zlobec, I.; Tornillo, L.; Carafa, V.; Schaffner, T.; Borner, M.; Diamantis, I.; Esposito, F.; Brunner, T.; et al. Loss of the CBX7 protein expression correlates with a more aggressive phenotype in pancreatic cancer. Eur. J. Cancer 2010, 46, 1438–1444. [Google Scholar] [CrossRef]
- Martinez-Garcia, R.; Juan, D.; Rausell, A.; Munoz, M.; Banos, N.; Menendez, C.; Lopez-Casas, P.P.; Rico, D.; Valencia, A.; Hidalgo, M. Transcriptional dissection of pancreatic tumors engrafted in mice. Genome Med. 2014, 6, 27. [Google Scholar] [CrossRef]
- Hong, S.M.; Li, A.; Olino, K.; Wolfgang, C.L.; Herman, J.M.; Schulick, R.D.; Iacobuzio-Donahue, C.; Hruban, R.H.; Goggins, M. Loss of E-cadherin expression and outcome among patients with resectable pancreatic adenocarcinomas. Mod. Pathol. 2011, 24, 1237–1247. [Google Scholar] [CrossRef]
- Handra-Luca, A.; Hong, S.M.; Walter, K.; Wolfgang, C.; Hruban, R.; Goggins, M. Tumour epithelial vimentin expression and outcome of pancreatic ductal adenocarcinomas. Br. J. Cancer 2011, 104, 1296–1302. [Google Scholar] [CrossRef]
- Tuveson, D.A.; Neoptolemos, J.P. Understanding metastasis in pancreatic cancer: A call for new clinical approaches. Cell 2012, 148, 21–23. [Google Scholar] [CrossRef]
- Kaushik, G.; Seshacharyulu, P.; Rauth, S.; Nallasamy, P.; Rachagani, S.; Nimmakayala, R.K.; Vengoji, R.; Mallya, K.; Chirravuri-Venkata, R.; Singh, A.B.; et al. Selective inhibition of stemness through EGFR/FOXA2/SOX9 axis reduces pancreatic cancer metastasis. Oncogene 2021, 40, 848–862. [Google Scholar] [CrossRef]
- Wang, G.; Lunardi, A.; Zhang, J.; Chen, Z.; Ala, U.; Webster, K.A.; Tay, Y.; Gonzalez-Billalabeitia, E.; Egia, A.; Shaffer, D.R.; et al. Zbtb7a suppresses prostate cancer through repression of a Sox9-dependent pathway for cellular senescence bypass and tumor invasion. Nat. Genet. 2013, 45, 739–746. [Google Scholar] [CrossRef]
- Cheng, P.F.; Shakhova, O.; Widmer, D.S.; Eichhoff, O.M.; Zingg, D.; Frommel, S.C.; Belloni, B.; Raaijmakers, M.I.; Goldinger, S.M.; Santoro, R.; et al. Methylation-dependent SOX9 expression mediates invasion in human melanoma cells and is a negative prognostic factor in advanced melanoma. Genome Biol. 2015, 16, 42. [Google Scholar] [CrossRef] [PubMed]
- Larsimont, J.C.; Youssef, K.K.; Sanchez-Danes, A.; Sukumaran, V.; Defrance, M.; Delatte, B.; Liagre, M.; Baatsen, P.; Marine, J.C.; Lippens, S.; et al. Sox9 Controls Self-Renewal of Oncogene Targeted Cells and Links Tumor Initiation and Invasion. Cell Stem Cell 2015, 17, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Laughney, A.M.; Hu, J.; Campbell, N.R.; Bakhoum, S.F.; Setty, M.; Lavallee, V.P.; Xie, Y.; Masilionis, I.; Carr, A.J.; Kottapalli, S.; et al. Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat. Med. 2020, 26, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Shepherd, J.; Zhao, D.; Bollu, L.R.; Tahaney, W.M.; Hill, J.; Zhang, Y.; Mazumdar, A.; Brown, P.H. SOX9 is Essential for Triple-Negative Breast Cancer Cell Survival and Metastasis. Mol. Cancer Res. 2020, 18, 1825–1838. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Huang, H.; Shi, G.; Zhao, L.; Li, T.; Zhang, Z.; Liu, R.; Hu, Y.; Liu, H.; Yu, J.; et al. TGF-beta1-SOX9 axis-inducible COL10A1 promotes invasion and metastasis in gastric cancer via epithelial-to-mesenchymal transition. Cell Death Dis. 2018, 9, 849. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Yasuchika, K.; Ishii, T.; Miyauchi, Y.; Kojima, H.; Yamaoka, R.; Katayama, H.; Yoshitoshi, E.Y.; Ogiso, S.; Kita, S.; et al. SOX9 is a novel cancer stem cell marker surrogated by osteopontin in human hepatocellular carcinoma. Sci. Rep. 2016, 6, 30489. [Google Scholar] [CrossRef] [PubMed]
- Liao, T.T.; Yang, M.H. Hybrid Epithelial/Mesenchymal State in Cancer Metastasis: Clinical Significance and Regulatory Mechanisms. Cells 2020, 9, 623. [Google Scholar] [CrossRef]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Quint, K.; Tonigold, M.; Di Fazio, P.; Montalbano, R.; Lingelbach, S.; Ruckert, F.; Alinger, B.; Ocker, M.; Neureiter, D. Pancreatic cancer cells surviving gemcitabine treatment express markers of stem cell differentiation and epithelial-mesenchymal transition. Int. J. Oncol. 2012, 41, 2093–2102. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef]
- Shah, A.N.; Summy, J.M.; Zhang, J.; Park, S.I.; Parikh, N.U.; Gallick, G.E. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann. Surg. Oncol. 2007, 14, 3629–3637. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Feldmann, G.; Suarez-Gauthier, A.; Rasheed, Z.; Solomon, A.; Zou, G.M.; Rubio-Viqueira, B.; Garcia-Garcia, E.; Lopez-Rios, F.; Matsui, W.; et al. A direct pancreatic cancer xenograft model as a platform for cancer stem cell therapeutic development. Mol. Cancer Ther. 2009, 8, 310–314. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Holtzinger, A.; Jagan, I.; BeGora, M.; Lohse, I.; Ngai, N.; Nostro, C.; Wang, R.; Muthuswamy, L.B.; Crawford, H.C.; et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat. Med. 2015, 21, 1364–1371. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrasco-Garcia, E.; Lopez, L.; Moncho-Amor, V.; Carazo, F.; Aldaz, P.; Collado, M.; Bell, D.; Gaafar, A.; Karamitopoulou, E.; Tzankov, A.; et al. SOX9 Triggers Different Epithelial to Mesenchymal Transition States to Promote Pancreatic Cancer Progression. Cancers 2022, 14, 916. https://doi.org/10.3390/cancers14040916
Carrasco-Garcia E, Lopez L, Moncho-Amor V, Carazo F, Aldaz P, Collado M, Bell D, Gaafar A, Karamitopoulou E, Tzankov A, et al. SOX9 Triggers Different Epithelial to Mesenchymal Transition States to Promote Pancreatic Cancer Progression. Cancers. 2022; 14(4):916. https://doi.org/10.3390/cancers14040916
Chicago/Turabian StyleCarrasco-Garcia, Estefania, Lidia Lopez, Veronica Moncho-Amor, Fernando Carazo, Paula Aldaz, Manuel Collado, Donald Bell, Ayman Gaafar, Eva Karamitopoulou, Alexandar Tzankov, and et al. 2022. "SOX9 Triggers Different Epithelial to Mesenchymal Transition States to Promote Pancreatic Cancer Progression" Cancers 14, no. 4: 916. https://doi.org/10.3390/cancers14040916
APA StyleCarrasco-Garcia, E., Lopez, L., Moncho-Amor, V., Carazo, F., Aldaz, P., Collado, M., Bell, D., Gaafar, A., Karamitopoulou, E., Tzankov, A., Hidalgo, M., Rubio, Á., Serrano, M., Lawrie, C. H., Lovell-Badge, R., & Matheu, A. (2022). SOX9 Triggers Different Epithelial to Mesenchymal Transition States to Promote Pancreatic Cancer Progression. Cancers, 14(4), 916. https://doi.org/10.3390/cancers14040916