
Characterisation of the Stromal Microenvironment in Lobular Breast Cancer

 ,
,  , , , , , and
, , , , , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Tissue Processing for LCM and Gene Expression Analysis
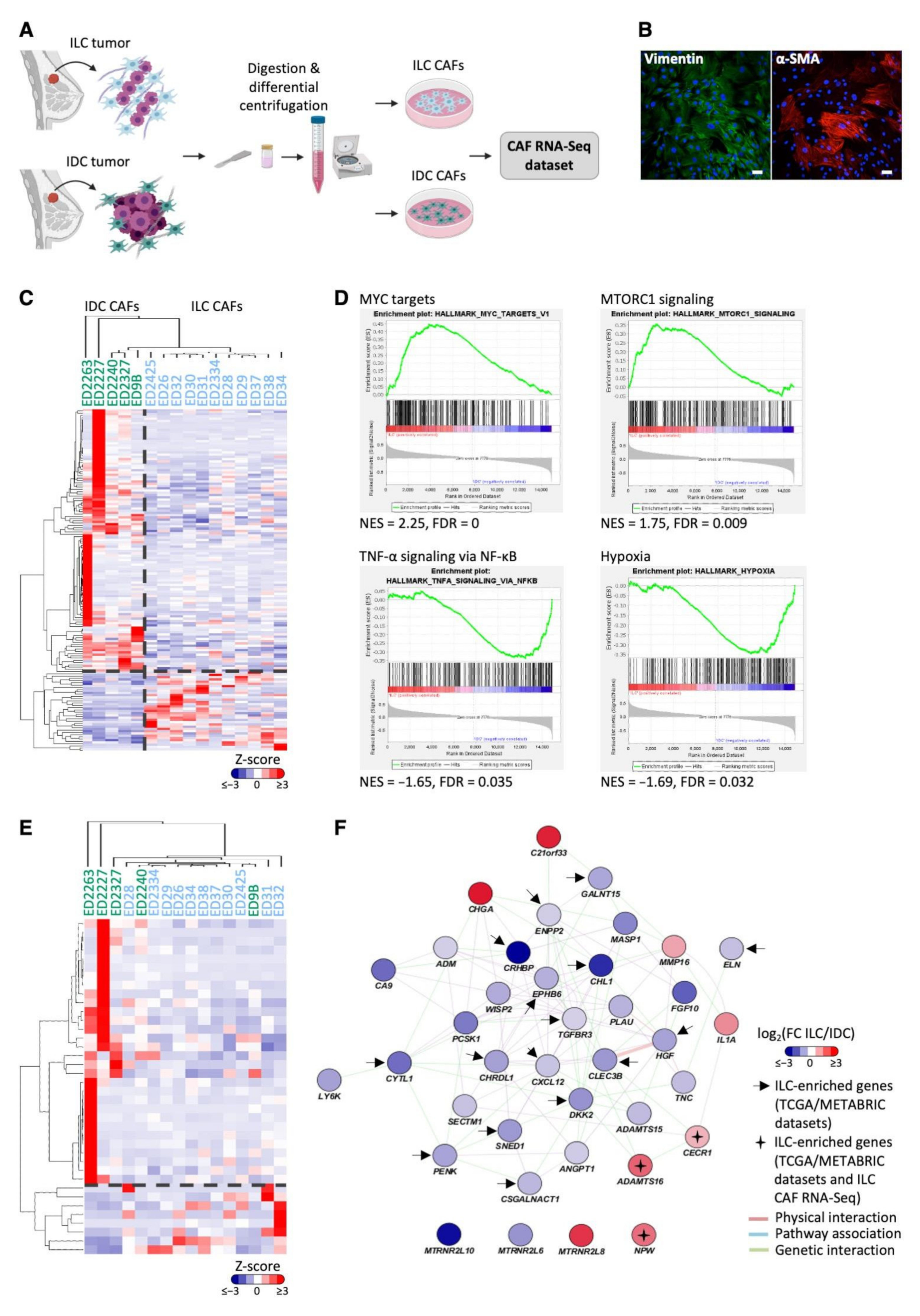
2.2. Primary CAF RNA-Seq Dataset Generation
2.3. Gene Set Enrichment Analysis
2.4. Statistical Analysis
3. Results
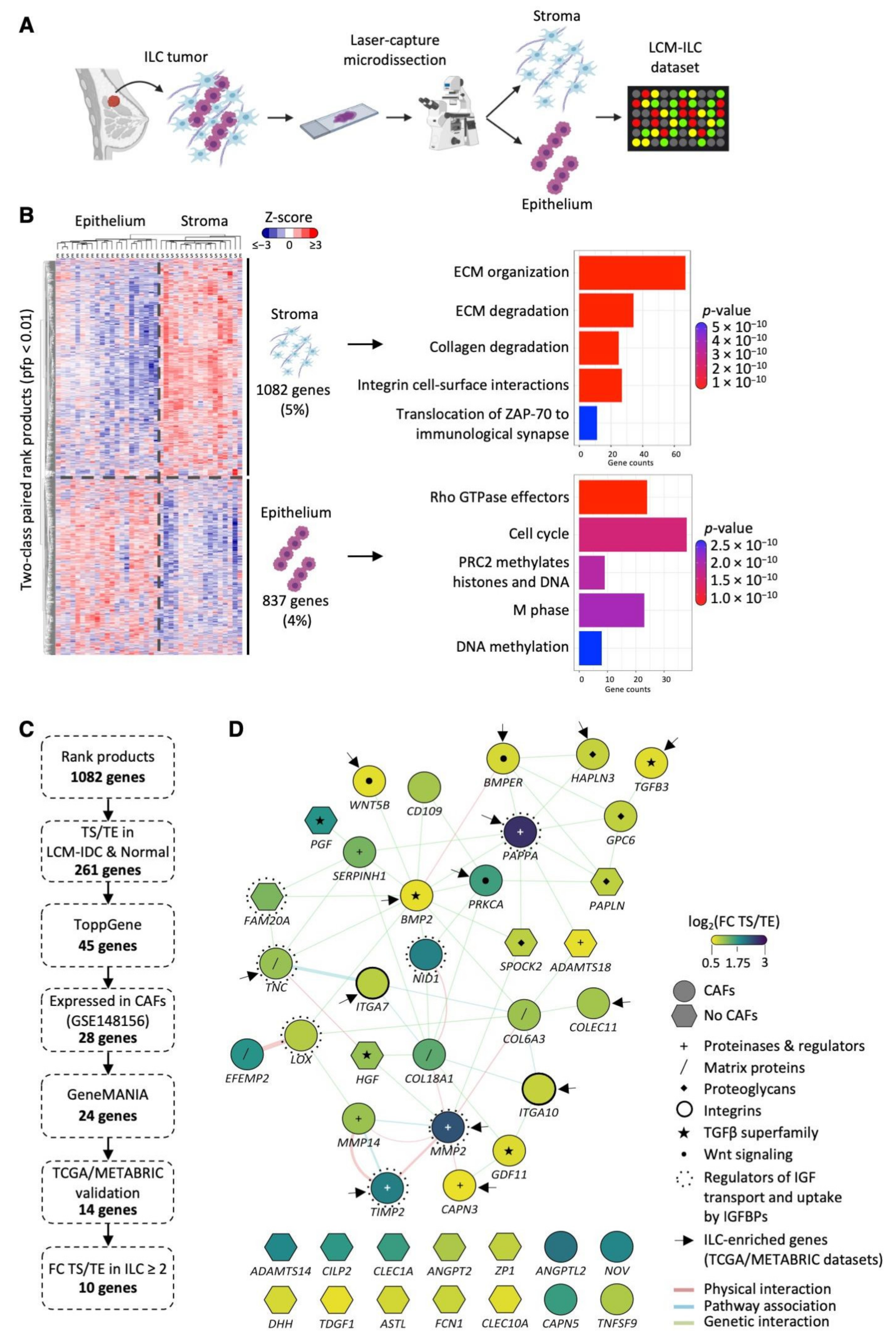
3.1. Generation of an LCM-ILC Dataset
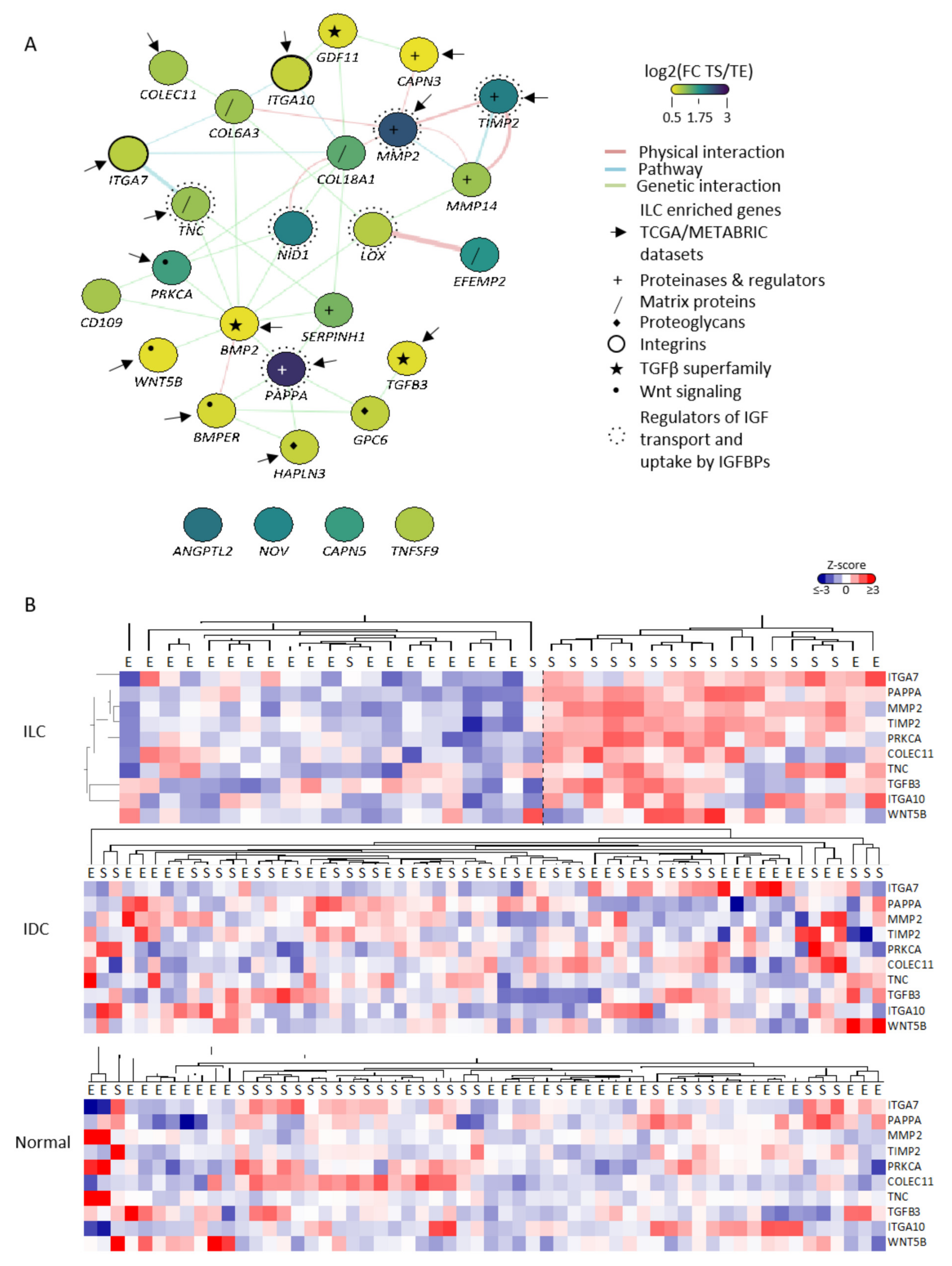
3.2. Identification of TS-ILC Enriched Genes
3.3. ILC-Specific Gene Expression in Primary Cancer-Associated Fibroblasts
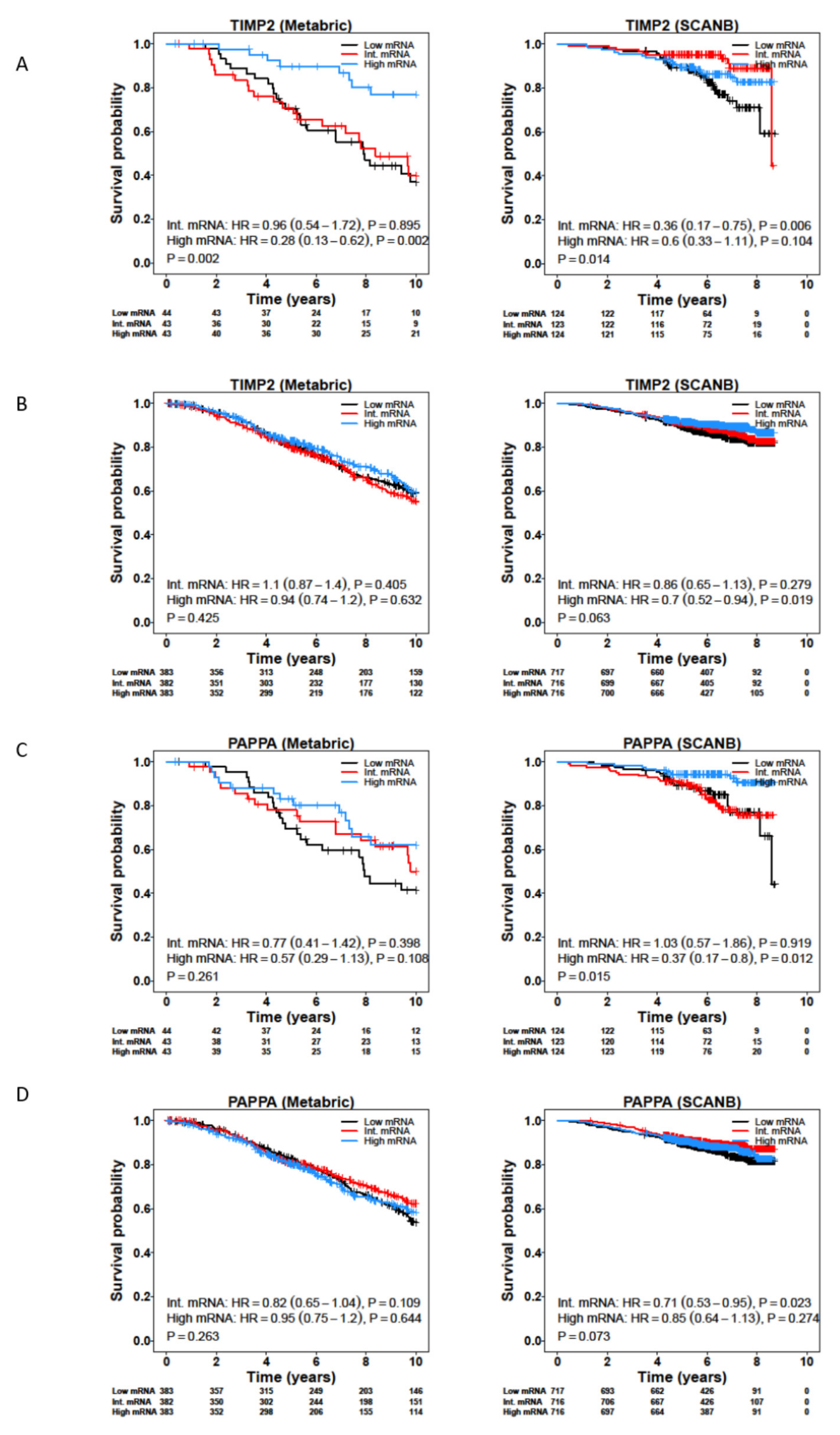
3.4. Survival Analysis of CAF-Associated Genes
3.5. PAPPA Is Predominantly Expressed in the Stroma of ILC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iorfida, M.; Maiorano, E.; Orvieto, E.; Maisonneuve, P.; Bottiglieri, L.; Rotmensz, N.; Montagna, E.; Dellapasqua, S.; Veronesi, P.; Galimberti, V.; et al. Invasive lobular breast cancer: Subtypes and outcome. Breast Cancer Res. Treat. 2012, 133, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Kelly, E.D.; Abraham, J.; Kruse, M. Invasive lobular breast cancer: A review of pathogenesis, diagnosis, management, and future directions of early stage disease. Semin. Oncol. 2019, 46, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Ferlicot, S.; Vincent-Salomon, A.; Médioni, J.; Genin, P.; Rosty, C.; Sigal-Zafrani, B.; Fréneaux, P.; Jouve, M.; Thiery, J.-P.; Sastre-Garau, X. Wide metastatic spreading in infiltrating lobular carcinoma of the breast. Eur. J. Cancer 2004, 40, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Pestalozzi, B.C.; Zahrieh, D.; Mallon, E.; Gusterson, B.A.; Price, K.N.; Gelber, R.D.; Holmberg, S.B.; Lindtner, J.; Snyder, R.; Thürlimann, B.; et al. Distinct Clinical and Prognostic Features of Infiltrating Lobular Carcinoma of the Breast: Combined Results of 15 International Breast Cancer Study Group Clinical Trials. J. Clin. Oncol. 2008, 26, 3006–3014. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Desmedt, C.; Zoppoli, G.; Gundem, G.; Pruneri, G.; Larsimont, D.; Fornili, M.; Fumagalli, D.; Brown, D.; Rothé, F.; Vincent, D.; et al. Genomic Characterization of Primary Invasive Lobular Breast Cancer. J. Clin. Oncol. 2016, 34, 1872–1881. [Google Scholar] [CrossRef] [PubMed]
- Michaut, M.; Chin, S.F.; Majewski, I.; Severson, T.M.; Bismeijer, T.; De Koning, L.; Peeters, J.K.; Schouten, P.C.; Rueda, O.M.; Bosma, A.J.; et al. Integration of genomic, transcriptomic and proteomic data identifies two biologically distinct subtypes of invasive lobular breast cancer. Sci. Rep. 2016, 6, 18517. [Google Scholar] [CrossRef]
- Shah, S.P.; Morin, R.D.; Khattra, J.; Prentice, L.; Pugh, T.; Burleigh, A.; Delaney, A.; Gelmon, K.; Guliany, R.; Senz, J.; et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 2009, 461, 809–813. [Google Scholar] [CrossRef]
- Mccart Reed, A.E.; Foong, S.; Kutasovic, J.R.; Nones, K.; Waddell, N.; Lakhani, S.R.; Simpson, P.T. The Genomic Landscape of Lobular Breast Cancer. Cancers 2021, 13, 1950. [Google Scholar] [CrossRef]
- Desmedt, C.; Salgado, R.; Fornili, M.; Pruneri, G.; Van den Eynden, G.; Zoppoli, G.; Rothé, F.; Buisseret, L.; Garaud, S.; Willard-Gallo, K.; et al. Immune Infiltration in Invasive Lobular Breast Cancer. J. Natl. Cancer Inst. 2018, 110, 768–776. [Google Scholar] [CrossRef]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Finak, G.; Bertos, N.; Pepin, F.; Sadekova, S.; Souleimanova, M.; Zhao, H.; Chen, H.; Omeroglu, G.; Meterissian, S.; Omeroglu, A.; et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat. Med. 2008, 14, 518–527. [Google Scholar] [CrossRef]
- Finak, G.; Sadekova, S.; Pepin, F.; Hallett, M.; Meterissian, S.; Halwani, F.; Khetani, K.; Souleimanova, M.; Zabolotny, B.; Omeroglu, A.; et al. Gene expression signatures of morphologically normal breast tissue identify basal-like tumors. Breast Cancer Res. 2006, 8, R58. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Wang, Y.E.; Kutnetsov, L.; Partensky, A.; Farid, J.; Quackenbush, J. {WebMeV}: A Cloud Platform for Analyzing and Visualizing Cancer Genomic Data. Cancer Res. 2017, 77, e11–e14. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Vallon-Christersson, J.; Häkkinen, J.; Hegardt, C.; Saal, L.H.; Larsson, C.; Ehinger, A.; Lindman, H.; Olofsson, H.; Sjöblom, T.; Wärnberg, F.; et al. Cross comparison and prognostic assessment of breast cancer multigene signatures in a large population-based contemporary clinical series. Sci. Rep. 2019, 9, 12184. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.-Y.; Christensen, S.M.; Ghanta, S.; Jeong, J.C.; Bucur, O.; Glass, B.; Montaser-Kouhsari, L.; Knoblauch, N.W.; Bertos, N.; Saleh, S.M.I.; et al. Extensive rewiring of epithelial-stromal co-expression networks in breast cancer. Genome Biol. 2015, 16, 128. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Wang, R.T.; Ahn, S.; Park, C.C.; Smith, D.J. A genome-wide map of human genetic interactions inferred from radiation hybrid genotypes. Genome Res. 2010, 20, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Karlsson, M.J.; Hober, A.; Svensson, A.S.; Scheffel, J.; Kotol, D.; Zhong, W.; Tebani, A.; Strandberg, L.; Edfors, F.; et al. The human secretome. Sci. Signal. 2019, 12, eaaz0274. [Google Scholar] [CrossRef] [PubMed]
- Oxvig, C. The role of {PAPP}-A in the {IGF} system: Location, location, location. J. Cell Commun. Signal. 2015, 9, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Jepsen, M.R.; Kløverpris, S.; Mikkelsen, J.H.; Pedersen, J.H.; Füchtbauer, E.-M.; Laursen, L.S.; Oxvig, C. Stanniocalcin-2 Inhibits Mammalian Growth by Proteolytic Inhibition of the Insulin-like Growth Factor Axis. J. Biol. Chem. 2015, 290, 3430–3439. [Google Scholar] [CrossRef]
- Kløverpris, S.; Mikkelsen, J.H.; Pedersen, J.H.; Jepsen, M.R.; Laursen, L.S.; Petersen, S.V.; Oxvig, C. Stanniocalcin-1 Potently Inhibits the Proteolytic Activity of the Metalloproteinase Pregnancy-associated Plasma Protein-A. J. Biol. Chem. 2015, 290, 21915–21924. [Google Scholar] [CrossRef]
- Loddo, M.; Andryszkiewicz, J.; Rodriguez-Acebes, S.; Stoeber, K.; Jones, A.; Dafou, D.; Apostolidou, S.; Wollenschlaeger, A.; Widschwendter, M.; Sainsbury, R.; et al. Pregnancy-associated plasma protein A regulates mitosis and is epigenetically silenced in breast cancer. J. Pathol. 2014, 233, 344–356. [Google Scholar] [CrossRef]
- Mansfield, A.S.; Visscher, D.W.; Hart, S.N.; Wang, C.; Goetz, M.P.; Oxvig, C.; Conover, C.A. Pregnancy-associated plasma protein-A expression in human breast cancer. Growth Horm. IGF Res. 2014, 24, 264–267. [Google Scholar] [CrossRef]
- Moleirinho, S.; Chang, N.; Sims, A.H.; Tilston-Lünel, A.M.; Angus, L.; Steele, A.; Boswell, V.; Barnett, S.C.; Ormandy, C.; Faratian, D.; et al. KIBRA exhibits MST-independent functional regulation of the Hippo signaling pathway in mammals. Oncogene 2013, 32, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Derksen, P.W.B.; Liu, X.; Saridin, F.; van der Gulden, H.; Zevenhoven, J.; Evers, B.; van Beijnum, J.R.; Griffioen, A.W.; Vink, J.; Krimpenfort, P.; et al. Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell 2006, 10, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, J.H.; Resch, Z.T.; Kalra, B.; Savjani, G.; Kumar, A.; Conover, C.A.; Oxvig, C. Indirect targeting of IGF receptor signaling in vivo by substrate-selective inhibition of PAPP-A proteolytic activity. Oncotarget 2014, 5, 1014–1025. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Miki, Y.; Miyashita, M.; Hata, S.; Takahashi, Y.; Rai, Y.; Sagara, Y.; Ohi, Y.; Hirakawa, H.; Tamaki, K.; et al. Tumor microenvironment in invasive lobular carcinoma: Possible therapeutic targets. Breast Cancer Res. Treat. 2015, 155, 65–75. [Google Scholar] [CrossRef]
- Teo, K.; Gómez-Cuadrado, L.; Tenhagen, M.; Byron, A.; Rätze, M.; van Amersfoort, M.; Renes, J.; Strengman, E.; Mandoli, A.; Singh, A.A.; et al. E-cadherin loss induces targetable autocrine activation of growth factor signalling in lobular breast cancer. Sci. Rep. 2018, 8, 15454. [Google Scholar] [CrossRef]
- Qian, X.; Karpova, T.; Sheppard, A.M.; McNally, J.; Lowy, D.R. E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. EMBO J. 2004, 23, 1739–1784. [Google Scholar] [CrossRef]
- Nagle, A.M.; Levine, K.M.; Tasdemir, N.; Scott, J.A.; Burlbaugh, K.; Kehm, J.; Katz, T.A.; Boone, D.N.; Jacobsen, B.M.; Atkinson, J.M.; et al. Loss of E-cadherin Enhances IGF1-IGF1R Pathway Activation and Sensitizes Breast Cancers to Anti-IGF1R/InsR Inhibitors. Clin. Cancer Res. 2018, 24, 5165–5177. [Google Scholar] [CrossRef]
- Burke, K.; Tang, P.; Brown, E. Second harmonic generation reveals matrix alterations during breast tumor progression. J. Biomed. Opt. 2012, 18, 31106. [Google Scholar] [CrossRef]
- De Andrade Natal, R.; Paiva, G.R.; Pelegati, V.B.; Marenco, L.; Alvarenga, C.A.; Vargas, R.F.; Derchain, S.F.; Sarian, L.O.; Franchet, C.; Cesar, C.L.; et al. Exploring Collagen Parameters in Pure Special Types of Invasive Breast Cancer. Sci. Rep. 2019, 9, 7715. [Google Scholar] [CrossRef]
- Castellana, B.; Escuin, D.; Peiró, G.; Garcia-Valdecasas, B.; Vázquez, T.; Pons, C.; Pérez-Olabarria, M.; Barnadas, A.; Lerma, E. ASPN and GJB2 are implicated in the mechanisms of invasion of ductal breast carcinomas. J. Cancer 2012, 3, 175–183. [Google Scholar] [CrossRef]
- Kordowski, F.; Kolarova, J.; Schafmayer, C.; Buch, S.; Goldmann, T.; Marwitz, S.; Kugler, C.; Scheufele, S.; Gassling, V.; Németh, C.G.; et al. Aberrant DNA methylation of ADAMTS16 in colorectal and other epithelial cancers. BMC Cancer 2018, 18, 796. [Google Scholar] [CrossRef] [PubMed]
- Nyante, S.J.; Wang, T.; Tan, X.; Ozdowski, E.F.; Lawton, T.J. Quantitative expression of MMPs 2, 9, 14, and collagen IV in LCIS and paired normal breast tissue. Sci. Rep. 2019, 9, 13432. [Google Scholar] [CrossRef] [PubMed]
- Daniele, A.; Abbate, I.; Oakley, C.; Casamassima, P.; Savino, E.; Casamassima, A.; Sciortino, G.; Fazio, V.; Gadaleta-Caldarola, G.; Catino, A.; et al. Clinical and prognostic role of matrix metalloproteinase-2, -9 and their inhibitors in breast cancer and liver diseases: A review. Int. J. Biochem. Cell Biol. 2016, 77, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Mustafa, D.; Zheng, P.-P.; Van Der Weiden, M.; Sacchetti, A.; Brandt, M.; Chrifi, I.; Tempel, D.; Leenen, P.J.M.; Duncker, D.J.; et al. Activation of CECR1 in M2-like TAMs promotes paracrine stimulation-mediated glial tumor progression. Neuro-Oncology 2017, 19, 648–659. [Google Scholar] [CrossRef]
- Zhu, C.; Chrifi, I.; Mustafa, D.; Van Der Weiden, M.; Leenen, P.; Duncker, D.J.; Kros, J.M.; Cheng, C. CECR1-mediated cross talk between macrophages and vascular mural cells promotes neovascularization in malignant glioma. Nat. Publ. Gr. 2017, 36, 5356–5368. [Google Scholar] [CrossRef]
- Kutryb-Zajac, B.; Harasim, G.; Jedrzejewska, A.; Krol, O.; Braczko, A.; Jablonska, P.; Mierzejewska, P.; Zielinski, J.; Slominska, E.M.; Smolenski, R.T. Macrophage-derived adenosine deaminase 2 correlates with m2 macrophage phenotype in triple negative breast cancer. Int. J. Mol. Sci. 2021, 22, 3764. [Google Scholar] [CrossRef]
- Kutryb-Zajac, B.; Koszalka, P.; Mierzejewska, P.; Bulinska, A.; Zabielska, M.A.; Brodzik, K.; Skrzypkowska, A.; Zelazek, L.; Pelikant-Malecka, I.; Slominska, E.M.; et al. Adenosine deaminase inhibition suppresses progression of 4T1 murine breast cancer by adenosine receptor-dependent mechanisms. J. Cell. Mol. Med. 2018, 22, 5939–5954. [Google Scholar] [CrossRef]
- Heits, N.; Heinze, T.; Bernsmeier, A.; Kerber, J.; Hauser, C.; Becker, T.; Kalthoff, H.; Egberts, J.H.; Braun, F. Influence of mTOR-inhibitors and mycophenolic acid on human cholangiocellular carcinoma and cancer associated fibroblasts. BMC Cancer 2016, 16, 322. [Google Scholar] [CrossRef]
- Zang, C.; Eucker, J.; Habbel, P.; Neumann, C.; Schulz, C.O.; Bangemann, N.; Kissner, L.; Riess, H.; Liu, H. Targeting multiple tyrosine kinase receptors with dovitinib blocks invasion and the interaction between tumor cells and cancer-associated fibroblasts in breast cancer. Cell Cycle 2015, 14, 1291–1299. [Google Scholar] [CrossRef][Green Version]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef]
- Prithviraj, P.; Anaka, M.; Thompson, E.W.; Sharma, R.; Walkiewicz, M.; Tutuka, C.S.A.; Behren, A.; Kannourakis, G.; Jayachandran, A. Aberrant pregnancy-associated plasma protein-A expression in breast cancers prognosticates clinical outcomes. Sci. Rep. 2020, 10, 13779. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, F.; Orsetti, B.; Nègre, V.; Finetti, P.; Rougé, C.; Ahomadegbe, J.C.; Bibeau, F.; Mathieu, M.C.; Treilleux, I.; Jacquemier, J.; et al. Lobular and ductal carcinomas of the breast have distinct genomic and expression profiles. Oncogene 2008, 27, 5359–5372. [Google Scholar] [CrossRef] [PubMed]
- Ekyalongo, R.C.; Yee, D. Revisiting the IGF-1R as a breast cancer target. NPJ Precis. Oncol. 2017, 1, 14. [Google Scholar] [CrossRef] [PubMed]
- Heitzeneder, S.; Sotillo, E.; Shern, J.F.; Sindiri, S.; Xu, P.; Jones, R.; Pollak, M.; Noer, P.R.; Lorette, J.; Fazli, L.; et al. Pregnancy-Associated Plasma Protein-A (PAPP-A) in Ewing Sarcoma: Role in Tumor Growth and Immune Evasion. J. Natl. Cancer Inst. 2019, 111, 970–982. [Google Scholar] [CrossRef]
- Ryan, A.J.; Napoletano, S.; Fitzpatrick, P.A.; Currid, C.A.; O’Sullivan, N.C.; Harmey, J.H. Expression of a protease-resistant insulin-like growth factor-binding protein-4 inhibits tumour growth in a murine model of breast cancer. Br. J. Cancer 2009, 101, 278–286. [Google Scholar] [CrossRef]
- Smith, Y.E.; Toomey, S.; Napoletano, S.; Kirwan, G.; Schadow, C.; Chubb, A.J.; Mikkelsen, J.H.; Oxvig, C.; Harmey, J.H. Recombinant PAPP-A resistant insulin-like growth factor binding protein 4 (dBP4) inhibits angiogenesis and metastasis in a murine model of breast cancer. BMC Cancer 2018, 18, 1016. [Google Scholar] [CrossRef]
- Espelund, U.; Renehan, A.G.; Cold, S.; Oxvig, C.; Lancashire, L.; Su, Z.; Flyvbjerg, A.; Frystyk, J. Prognostic relevance and performance characteristics of serum IGFBP-2 and PAPP-A in women with breast cancer: A long-term Danish cohort study. Cancer Med. 2018, 7, 2391–2404. [Google Scholar] [CrossRef]
- Creighton, C.J.; Casa, A.; Lazard, Z.W.; Huang, S.; Tsimelzon, A.; Hilsenbeck, S.G.; Osborne, C.K.; Lee, A.V. Insulin-like growth factor-I activates gene transcription programs strongly associated with poor breast cancer prognosis. J. Clin. Oncol. 2008, 26, 4078–4085. [Google Scholar] [CrossRef]
- Mu, L.; Tuck, D.; Katsaros, D.; Lu, L.; Schulz, V.; Perincheri, S.; Menato, G.; Scarampi, L.; Harris, L.; Yu, H. Favorable outcome associated with an IGF-1 ligand signature in breast cancer. Breast Cancer Res. Treat. 2012, 133, 321–331. [Google Scholar] [CrossRef]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Pearce, D.A.; Nirmal, A.J.; Freeman, T.C.; Sims, A.H. Continuous Biomarker Assessment by Exhaustive Survival Analysis. bioRxiv 2018. [Google Scholar] [CrossRef]
- Gyrup, C.; Christiansen, M.; Oxvig, C. Quantification of Proteolytically Active Pregnancy-Associated Plasma Protein-A with an Assay Based on Quenched Fluorescence. Clin. Chem. 2007, 53, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Gyrup, C.; Oxvig, C. Quantitative Analysis of Insulin-like Growth Factor-Modulated Proteolysis of Insulin-like Growth Factor Binding Protein-4 and -5 by Pregnancy-Associated Plasma Protein-A. Biochemistry 2007, 46, 1972–1980. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Log2 fold Change (TS/TE) | METABRIC | TCGA Microarray | TCGA RNA-Seq |
|---|---|---|---|---|
| PAPPA | 2.59 | ILC (*) | ILC (*) | ILC (*) |
| MMP2 | 2.29 | ns | ILC (*) | ILC (***) |
| ANGPTL2 | 2.02 | ILC (*) | ns | ILC (***) |
| TIMP2 | 1.92 | ns | ns | ILC (*) |
| NID1 | 1.89 | ns | ns | ns |
| NOV | 1.85 | ns | ILC (***) | ILC (***) |
| EFEMP2 | 1.71 | IDC (***) | ns | ILC (***) |
| CAPN5 | 1.61 | ILC (*) | ns | ILC (***) |
| PRKCA | 1.57 | ILC (*) | ILC (*) | ILC (***) |
| COL18A1 | 1.41 | IDC (*) | ns | ILC (***) |
| SERPINH1 | 1.25 | IDC (*) | ns | ns |
| TNC | 1.07 | ns | ILC (*) | ILC (*) |
| MMP14 | 1.05 | ns | ns | ns |
| COL6A3 | 1.04 | ns | ns | ns |
| COLEC11 | 1.00 | ILC (*) | ns | ns |
| CD109 | 0.99 | IDC (*) | ns | ns |
| TNFSF9 | 0.94 | IDC (*) | ns | ns |
| LOX | 0.90 | ns | ns | ns |
| ITGA7 | 0.87 | ILC (*) | ILC (***) | ILC (***) |
| GPC6 | 0.83 | IDC (*) | ns | IDC (***) |
| HAPLN3 | 0.81 | ns | ns | ILC (***) |
| ITGA10 | 0.81 | ILC (***) | ILC (***) | ILC (***) |
| BMPER | 0.72 | ns | ns | ILC (***) |
| GDF11 | 0.67 | IDC (***) | ns | ns |
| TGFB3 | 0.65 | ILC (***) | ILC (*) | ILC (***) |
| WNT5B | 0.63 | ns | ns | ILC (*) |
| BMP2 | 0.61 | ns | ILC (*) | ILC (***) |
| CAPN3 | 0.59 | ns | ns | ILC (***) |
| Genes | Log2 Fold Change (ILC/IDC) | p-Value | METABRIC | TCGA Microarray | TCGA RNA-Seq |
|---|---|---|---|---|---|
| CECR1 | 1.36 | 0.042 | ns | ILC (*) | ILC (*) |
| MMP16 | 1.66 | 0.024 | ns | ns | ns |
| IL1A | 2.20 | 0.014 | ns | ns | ns |
| NPW | 2.82 | 0.007 | NA | ILC (*) | ns |
| ADAMTS16 | 3.07 | 0.044 | ns | ILC (*) | ILC (*) |
| GATD3A | 4.49 | 0.027 | ns | ns | ns |
| CHGA | 4.69 | 0.028 | ns | ns | IDC (***) |
| MTRNR2L8 | 4.08 | 0.003 | NA | NA | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Cuadrado, L.; Bullock, E.; Mabruk, Z.; Zhao, H.; Souleimanova, M.; Noer, P.R.; Turnbull, A.K.; Oxvig, C.; Bertos, N.; Byron, A.; et al. Characterisation of the Stromal Microenvironment in Lobular Breast Cancer. Cancers 2022, 14, 904. https://doi.org/10.3390/cancers14040904
Gómez-Cuadrado L, Bullock E, Mabruk Z, Zhao H, Souleimanova M, Noer PR, Turnbull AK, Oxvig C, Bertos N, Byron A, et al. Characterisation of the Stromal Microenvironment in Lobular Breast Cancer. Cancers. 2022; 14(4):904. https://doi.org/10.3390/cancers14040904
Chicago/Turabian StyleGómez-Cuadrado, Laura, Esme Bullock, Zeanap Mabruk, Hong Zhao, Margarita Souleimanova, Pernille Rimmer Noer, Arran K. Turnbull, Claus Oxvig, Nicholas Bertos, Adam Byron, and et al. 2022. "Characterisation of the Stromal Microenvironment in Lobular Breast Cancer" Cancers 14, no. 4: 904. https://doi.org/10.3390/cancers14040904
APA StyleGómez-Cuadrado, L., Bullock, E., Mabruk, Z., Zhao, H., Souleimanova, M., Noer, P. R., Turnbull, A. K., Oxvig, C., Bertos, N., Byron, A., Dixon, J. M., Park, M., Haider, S., Natrajan, R., Sims, A. H., & Brunton, V. G. (2022). Characterisation of the Stromal Microenvironment in Lobular Breast Cancer. Cancers, 14(4), 904. https://doi.org/10.3390/cancers14040904