
Over-Reduced State of Mitochondria as a Trigger of “β-Oxidation Shuttle” in Cancer Cells


 ,
, 
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Over-Reduced State of Mitochondria and Activation of Fatty Acid Oxidation in Cancer
1.1. mFAO Is Activated in Many Types of Cancer Cells
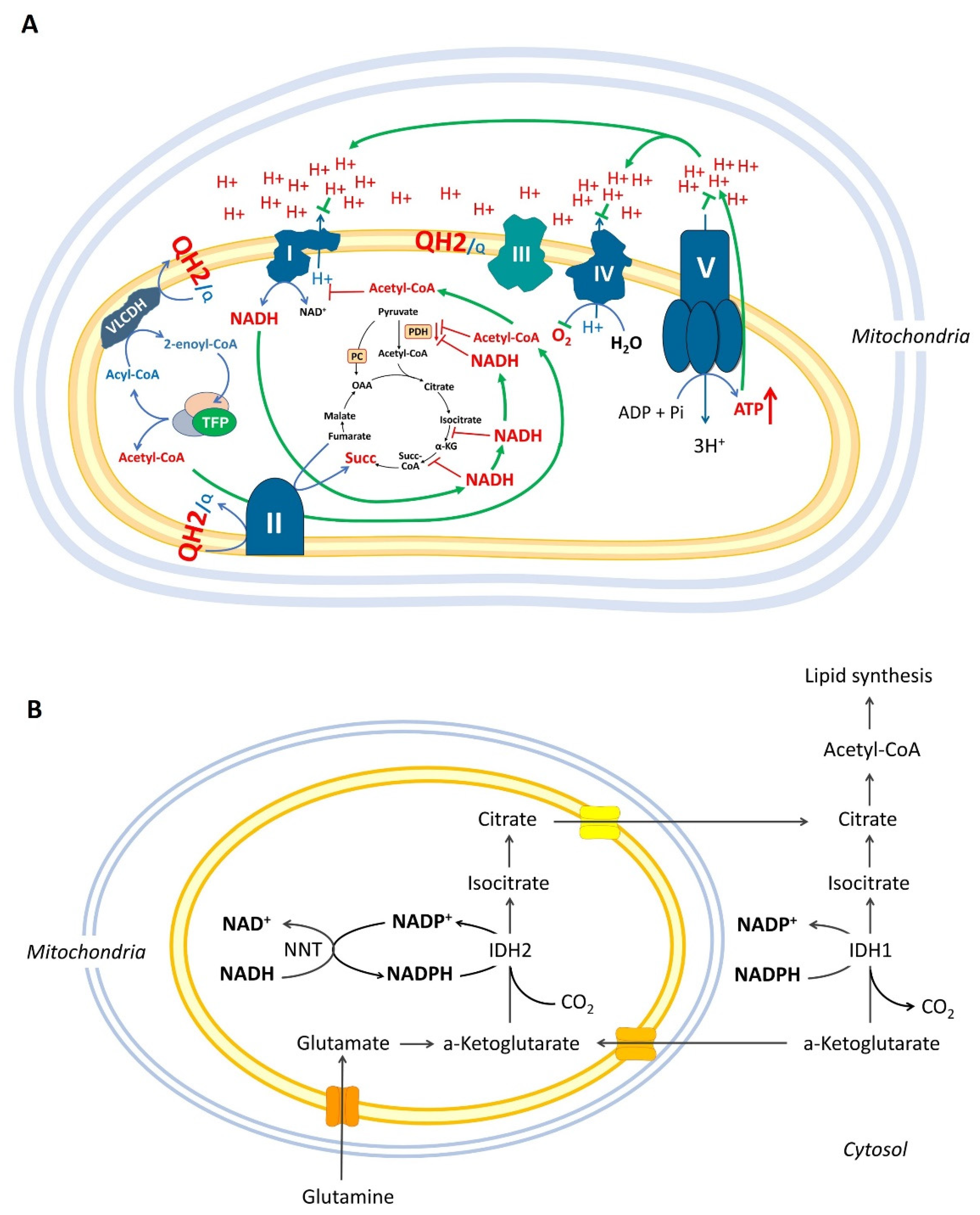
1.2. Mitochondrial β-Oxidation May Inhibit Pyruvate Combustion and Complex I via Acetylation of Mitochondrial Proteins
1.3. Over-Reduced State of Mitochondria Can Inhibit the Krebs Cycle but Not the “β-Oxidation”
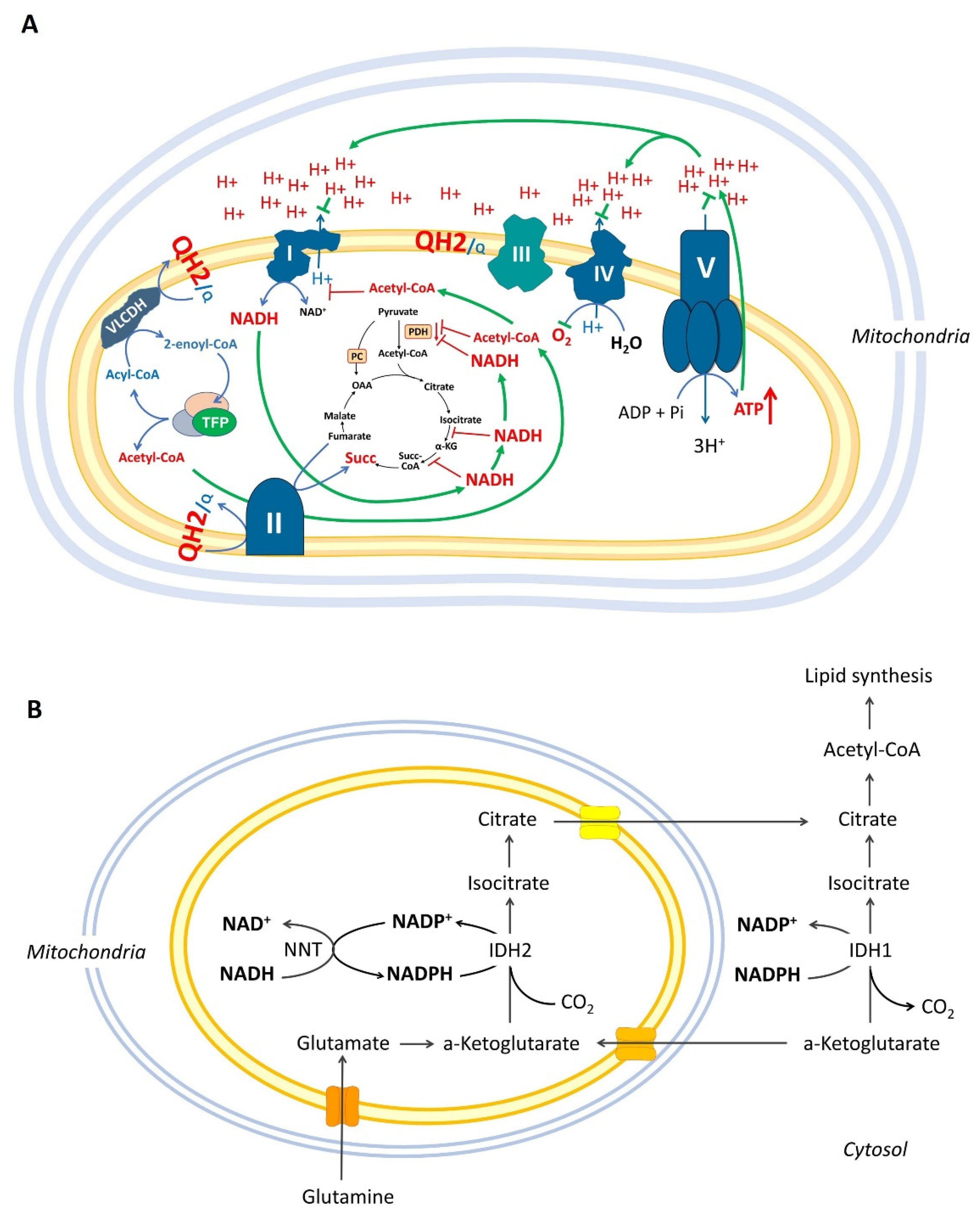
1.4. Reductive Carboxylation, Over-Reduced State of Mitochondrial Matrix, and Krebs Cycle Impairments
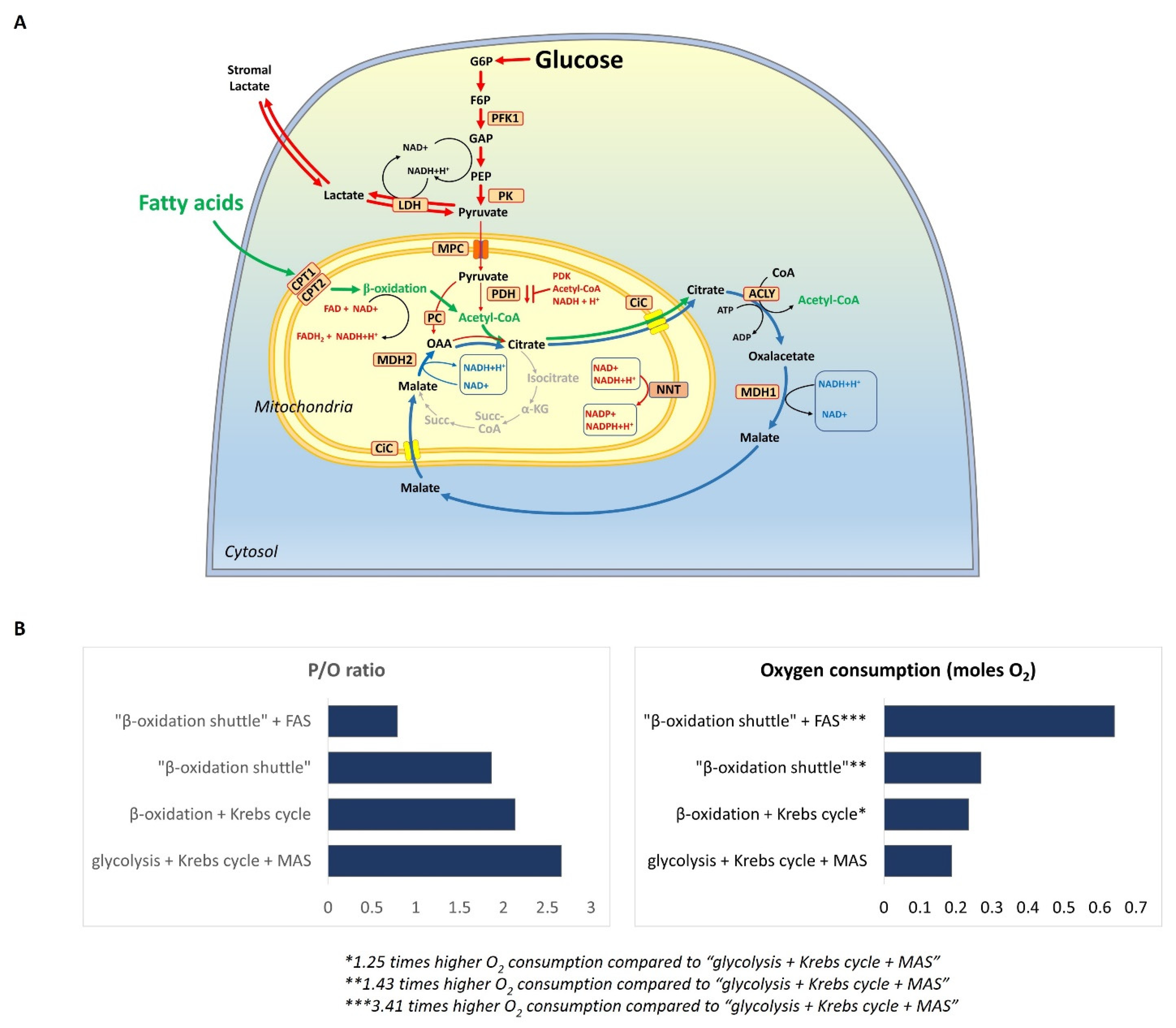
2. Definition of the “β-Oxidation Shuttle” and Its Components: Energy Efficiency
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACAD | acyl-CoA dehydrogenase |
| ACC1 | acetyl-CoA carboxylase |
| ACLY | ATP-dependent citrate lyase |
| ALDH | aldehyde dehydrogenase |
| AMPK | adenosine monophosphate-activated protein kinase |
| CIC | citrate/isocitrate carrier |
| CPT | carnitine palmitoyl transferase |
| CrAT | carnitine acetyltransferase |
| EH | enoyl-CoA hydratase |
| ETC | electron transport chain |
| mFAO | mitochondrial fatty acid oxidation |
| FAS | fatty acid synthesis |
| FH | fumarate hydratase |
| 3HAD | 3-hydroxyacyl-CoA dehydrogenase |
| HIF-1α | hypoxia inducible factor one alpha |
| IDH | isocitrate dehydrogenase |
| KAT | 3-ketothiolases |
| α-KG | alpha-ketoglutarate |
| α-KGDH | alpha-ketoglutarate dehydrogenase |
| LDH | lactate dehydrogenase |
| MCD | malonyl-CoA decarboxylase |
| MDH | malate dehydrogenase |
| NNT | nicotinamide nucleotide transhydrogenase |
| OXPHOS | oxidative phosphorylation |
| PDH | pyruvate dehydrogenase |
| PDK | pyruvate dehydrogenase kinase |
| PPP | pentose phosphate pathway |
| Q10 and Q10H2 | coenzyme Q10 (oxidized and reduced forms) |
| RET | reverse electron transport |
| ROS | reactive oxygen species |
| SDH | succinate dehydrogenase |
| SIRT | sirtuin |
| TFP | mitochondrial trifunctional fatty acid oxidation enzyme |
References
- Chen, K.; Lu, P.; Beeraka, N.M.; Sukocheva, O.A.; Madhunapantula, S.V.; Liu, J.; Sinelnikov, M.Y.; Nikolenko, V.N.; Bulygin, K.V.; Mikhaleva, L.M.; et al. Mitochondrial mutations and mitoepigenetics: Focus on regulation of oxidative stress-induced responses in breast cancers. Semin. Cancer Biol. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.-Y.; Fang, X. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. 2018, 435, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, S.R.; Butler, L.M.; Hoy, A.J. The diversity and breadth of cancer cell fatty acid metabolism. Cancer Metab. 2021, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Patel, S.; Affleck, V.S.; Wilson, I.; Turnbull, D.; Joshi, A.R.; Maxwell, R.; Stoll, E.A. Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro-Oncology 2016, 19, 43–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, C.-Y.; Ann, D.K. When fats commit crimes: Fatty acid metabolism, cancer stemness and therapeutic resistance. Cancer Commun. 2018, 38, 47. [Google Scholar] [CrossRef] [Green Version]
- Corbet, C.; Pinto, A.; Martherus, R.; De Jesus, J.P.S.; Polet, F.; Feron, O. Acidosis Drives the Reprogramming of Fatty Acid Metabolism in Cancer Cells through Changes in Mitochondrial and Histone Acetylation. Cell Metab. 2016, 24, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Gimple, R.C.; Kidwell, R.; Kim, L.J.Y.; Sun, T.; Gromovsky, A.D.; Wu, Q.; Wolf, M.; Lv, D.; Bhargava, S.; Jiang, L.; et al. Glioma Stem Cell–Specific Superenhancer Promotes Polyunsaturated Fatty-Acid Synthesis to Support EGFR Signaling. Cancer Discov. 2019, 9, 1248–1267. [Google Scholar] [CrossRef] [PubMed]
- Duman, C.; Yaqubi, K.; Hoffmann, A.; Acikgöz, A.A.; Korshunov, A.; Bendszus, M.; Herold-Mende, C.; Liu, H.-K.; Alfonso, J. Acyl-CoA-Binding Protein Drives Glioblastoma Tumorigenesis by Sustaining Fatty Acid Oxidation. Cell Metab. 2019, 30, 274–289.e5. [Google Scholar] [CrossRef]
- Sperry, J.; Condro, M.C.; Guo, L.; Braas, D.; Vanderveer-Harris, N.; Kim, K.K.; Pope, W.B.; Divakaruni, A.S.; Lai, A.; Christofk, H.; et al. Glioblastoma Utilizes Fatty Acids and Ketone Bodies for Growth Allowing Progression during Ketogenic Diet Therapy. iScience 2020, 23, 101453. [Google Scholar] [CrossRef]
- Strickland, M.; Stoll, E.A. Metabolic Reprogramming in Glioma. Front. Cell Dev. Biol. 2017, 5, 43. [Google Scholar] [CrossRef] [Green Version]
- Lengyel, E.; Makowski, L.; DiGiovanni, J.; Kolonin, M.G. Cancer as a Matter of Fat: The Crosstalk between Adipose Tissue and Tumors. Trends Cancer 2018, 4, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [Green Version]
- Grunt, T.W.; Lemberger, L.; Colomer, R.; López−Rodríguez, M.L.; Wagner, R. The Pharmacological or Genetic Blockade of Endogenous De Novo Fatty Acid Synthesis Does Not Increase the Uptake of Exogenous Lipids in Ovarian Cancer Cells. Front. Oncol. 2021, 11, 610885. [Google Scholar] [CrossRef] [PubMed]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaugg, K.; Yao, Y.; Reilly, P.T.; Kannan, K.; Kiarash, R.; Mason, J.; Huang, P.; Sawyer, S.K.; Fuerth, B.; Faubert, B.; et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011, 25, 1041–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2009, 120, 142–156. [Google Scholar] [CrossRef] [Green Version]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Corbet, C.; Draoui, N.; Polet, F.; Pinto, A.; Drozak, X.; Riant, O.; Feron, O. The SIRT1/HIF2α Axis Drives Reductive Glutamine Metabolism under Chronic Acidosis and Alters Tumor Response to Therapy. Cancer Res. 2014, 74, 5507–5519. [Google Scholar] [CrossRef] [Green Version]
- LaMonte, G.; Tang, X.; Chen, J.L.-Y.; Wu, J.; Ding, C.-K.C.; Keenan, M.M.; Sangokoya, C.; Kung, H.-N.; Ilkayeva, O.; Boros, L.G.; et al. Acidosis induces reprogramming of cellular metabolism to mitigate oxidative stress. Cancer Metab. 2013, 1, 23. [Google Scholar] [CrossRef] [Green Version]
- Schlaepfer, I.R.; Rider, L.; Rodrigues, L.U.; Gijón, M.A.; Pac, C.T.; Romero, L.; Cimic, A.; Sirintrapun, S.J.; Glodé, L.M.; Eckel, R.H.; et al. Lipid Catabolism via CPT1 as a Therapeutic Target for Prostate Cancer. Mol. Cancer Ther. 2014, 13, 2361–2371. [Google Scholar] [CrossRef] [Green Version]
- Luis, C.; Duarte, F.; Faria, I.; Jarak, I.; Oliveira, P.F.; Alves, M.G.; Soares, R.; Fernandes, R. Warburg Effect Inversion: Adiposity shifts central primary metabolism in MCF-7 breast cancer cells. Life Sci. 2019, 223, 38–46. [Google Scholar] [CrossRef]
- German, N.J.; Yoon, H.; Yusuf, R.Z.; Murphy, J.P.; Finley, L.W.; Laurent, G.; Haas, W.; Satterstrom, F.; Guarnerio, J.; Zaganjor, E.; et al. PHD3 Loss in Cancer Enables Metabolic Reliance on Fatty Acid Oxidation via Deactivation of ACC2. Mol. Cell 2016, 63, 1006–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, G.-H.; Takeda, K. Role and regulation of prolyl hydroxylase domain proteins. Cell Death Differ. 2008, 15, 635–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.H.; Kim, N.H.; Yun, J.S.; Cho, E.S.; Cha, Y.H.; Cho, S.B.; Lee, S.-H.; Cha, S.Y.; Kim, S.-Y.; Choi, J.; et al. Snail augments fatty acid oxidation by suppression of mitochondrial ACC2 during cancer progression. Life Sci. Alliance 2020, 3, e202000683. [Google Scholar] [CrossRef]
- Amoedo, N.D.; Sarlak, S.; Obre, E.; Esteves, P.; Bégueret, H.; Kieffer, Y.; Rousseau, B.; Dupis, A.; Izotte, J.; Bellance, N.; et al. Targeting the mitochondrial trifunctional protein restrains tumor growth in oxidative lung carcinomas. J. Clin. Investig. 2021, 131, e133081. [Google Scholar] [CrossRef] [PubMed]
- Camarda, R.; Zhou, A.Y.; Kohnz, R.A.; Balakrishnan, S.; Mahieu, C.; Anderton, B.; Eyob, H.; Kajimura, S.; Tward, A.; Krings, G.; et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat. Med. 2016, 22, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Daniels, G.; Lee, P.; Monaco, M.E. Lipid metabolism in prostate cancer. Am. J. Clin. Exp. Urol. 2014, 2, 111–120. [Google Scholar]
- Tcheng, M.; Roma, A.; Ahmed, N.; Smith, R.W.; Jayanth, P.; Minden, M.D.; Schimmer, A.D.; Hess, D.A.; Hope, K.; Rea, K.A.; et al. Very long chain fatty acid metabolism is required in acute myeloid leukemia. Blood 2021, 137, 3518–3532. [Google Scholar] [CrossRef]
- Kageyama, T.; Nagashio, R.; Ryuge, S.; Matsumoto, T.; Iyoda, A.; Satoh, Y.; Masuda, N.; Jiang, S.-X.; Saegusa, M.; Sato, Y. HADHA is a potential predictor of response to platinum-based chemotherapy for lung cancer. Asian Pac. J. Cancer Prev. 2011, 12, 3457–3463. [Google Scholar]
- De Oliveira, M.P.; Liesa, M. The Role of Mitochondrial Fat Oxidation in Cancer Cell Proliferation and Survival. Cells 2020, 9, 2600. [Google Scholar] [CrossRef]
- Yamamoto, K.; Abe, S.; Honda, A.; Hashimoto, J.; Aizawa, Y.; Ishibashi, S.; Takemura, T.; Hanagata, N.; Yamamoto, M.; Miura, O.; et al. Fatty acid beta oxidation enzyme HADHA is a novel potential therapeutic target in malignant lymphoma. Lab. Investig. 2019, 100, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Mozolewska, P.; Duzowska, K.; Pakiet, A.; Mika, A.; Śledziński, T. Inhibitors of Fatty Acid Synthesis and Oxidation as Potential Anticancer Agents in Colorectal Cancer Treatment. Anticancer Res. 2020, 40, 4843–4856. [Google Scholar] [CrossRef] [PubMed]
- Samudio, I.; Konopleva, M. Targeting leukemia’s “fatty tooth”. Blood 2015, 126, 1874–1875. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, M.; Jonscher, K.; Friedman, J.E. Reduced mitochondrial function in obesity-associated fatty liver: SIRT3 takes on the fat. Aging 2011, 3, 175–178. [Google Scholar] [CrossRef] [Green Version]
- Lantier, L.; Williams, A.S.; Williams, I.M.; Yang, K.K.; Bracy, D.P.; Goelzer, M.; James, F.D.; Gius, D.; Wasserman, D.H. SIRT3 Is Crucial for Maintaining Skeletal Muscle Insulin Action and Protects Against Severe Insulin Resistance in High-Fat–Fed Mice. Diabetes 2015, 64, 3081–3092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koves, T.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O.; Bain, J.R.; Stevens, R.; Dyck, J.R.; Newgard, C.B.; et al. Mitochondrial Overload and Incomplete Fatty Acid Oxidation Contribute to Skeletal Muscle Insulin Resistance. Cell Metab. 2008, 7, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Ramsay, R.R. The carnitine acyltransferases: Modulators of acyl-CoA-dependent reactions. Biochem. Soc. Trans. 2000, 28, 182–186. [Google Scholar] [CrossRef]
- Davies, M.N.; Kjalarsdottir, L.; Thompson, J.W.; Dubois, L.G.; Stevens, R.D.; Ilkayeva, O.R.; Brosnan, M.J.; Rolph, T.P.; Grimsrud, P.A.; Muoio, D.M. The Acetyl Group Buffering Action of Carnitine Acetyltransferase Offsets Macronutrient-Induced Lysine Acetylation of Mitochondrial Proteins. Cell Rep. 2015, 14, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Randle, P.J. Regulatory interactions between lipids and carbohydrates: The glucose fatty acid cycle after 35 years. Diabetes Metab. Rev. 1998, 14, 263–283. [Google Scholar] [CrossRef]
- Eales, K.L.; Hollinshead, K.E.R.; Tennant, D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016, 5, e190. [Google Scholar] [CrossRef] [Green Version]
- Sahlin, K.; Katz, A. The content of NADH in rat skeletal muscle at rest and after cyanide poisoning. Biochem. J. 1986, 239, 245–248. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Hu, Q.; Cheng, F.; Su, N.; Wang, A.; Zou, Y.; Hu, H.; Chen, X.; Zhou, H.-M.; Huang, X.; et al. SoNar, a Highly Responsive NAD+/NADH Sensor, Allows High-Throughput Metabolic Screening of Anti-tumor Agents. Cell Metab. 2015, 21, 777–789. [Google Scholar] [CrossRef] [Green Version]
- Rajagopalan, K.N.; Egnatchik, R.A.; Calvaruso, M.A.; Wasti, A.T.; Padanad, M.S.; Boroughs, L.K.; Ko, B.; Hensley, C.T.; Acar, M.; Hu, Z.; et al. Metabolic plasticity maintains proliferation in pyruvate dehydrogenase deficient cells. Cancer Metab. 2015, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Vatrinet, R.; Leone, G.; De Luise, M.; Girolimetti, G.; Vidone, M.; Gasparre, G.; Porcelli, A.M. The α-ketoglutarate dehydrogenase complex in cancer metabolic plasticity. Cancer Metab. 2017, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Wanders, R.J.; Ruiter, J.P.; IJlst, L.; Waterham, H.R.; Houten, S.M. The enzymology of mitochondrial fatty acid beta-oxidation and its application to follow-up analysis of positive neonatal screening results. J. Inherit. Metab. Dis. 2010, 33, 479–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaton, S.; Middleton, B.; Bartlett, K. Control of mitochondrial β-oxidation: Sensitivity of the trifunctional protein to [NAD+]/[NADH] and [acetyl-CoA]/[CoA]. Biochim. Biophys. Acta 1998, 1429, 230–238. [Google Scholar] [CrossRef]
- Tretter, L.; Adam-Vizi, V. Inhibition of Krebs Cycle Enzymes by Hydrogen Peroxide: A Key Role of α-Ketoglutarate Dehydrogenase in Limiting NADH Production under Oxidative Stress. J. Neurosci. 2000, 20, 8972–8979. [Google Scholar] [CrossRef] [PubMed]
- Tretter, L. Generation of Reactive Oxygen Species in the Reaction Catalyzed by—Ketoglutarate Dehydrogenase. J. Neurosci. 2004, 24, 7771–7778. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Hu, L.; Ma, T.; Yang, J.; Ding, J. Insights into the inhibitory mechanisms of NADH on the αγ heterodimer of human NAD-dependent isocitrate dehydrogenase. Sci. Rep. 2018, 8, 3146. [Google Scholar] [CrossRef]
- Randle, P.J.; Kerbey, A.L.; Espinal, J. Mechanisms decreasing glucose oxidation in diabetes and starvation: Role of lipid fuels and hormones. Diabetes/Metabolism Rev. 1988, 4, 623–638. [Google Scholar] [CrossRef] [PubMed]
- Guarás, A.; Perales-Clemente, E.; Calvo, E.; Acin-Perez, R.; Loureiro, M.; Pujol, C.; Martínez-Carrascoso, I.; Nuñez, E.; Garcia-Marques, F.; Rodríguez-Hernández, Á.; et al. The CoQH2/CoQ Ratio Serves as a Sensor of Respiratory Chain Efficiency. Cell Rep. 2016, 15, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozza, E.D.; Dando, I.; Pacchiana, R.; Liboi, E.; Scupoli, M.T.; Donadelli, M.; Palmieri, M. Regulation of succinate dehydrogenase and role of succinate in cancer. Semin. Cell Dev. Biol. 2020, 98, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-Y.; Huang, T.-W.; Hsieh, Y.-T.; Wang, Y.-F.; Yen, C.-C.; Lee, G.-L.; Yeh, C.-C.; Peng, Y.-J.; Kuo, Y.-Y.; Wen, H.-T.; et al. Cancer-Derived Succinate Promotes Macrophage Polarization and Cancer Metastasis via Succinate Receptor. Mol. Cell 2020, 77, 213–227.e5. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Zhao, T.; Xu, C.; Shi, W.; Geng, B.; Shen, J.; Zhang, C.; Pan, J.; Yang, J.; Hu, S.; et al. Oncometabolite succinate promotes angiogenesis by upregulating VEGF expression through GPR91-mediated STAT3 and ERK activation. Oncotarget 2017, 8, 13174–13185. [Google Scholar] [CrossRef]
- Li, J.; Yang, Y.-L.; Li, L.-Z.; Zhang, L.; Liu, Q.; Liu, K.; Li, P.; Liu, B.; Qi, L.-W. Succinate accumulation impairs cardiac pyruvate dehydrogenase activity through GRP91-dependent and independent signaling pathways: Therapeutic effects of ginsenoside Rb1. Biochim. Biophys. Acta 2017, 1863, 2835–2847. [Google Scholar] [CrossRef]
- Ryan, D.G.; Murphy, M.P.; Frezza, C.; Prag, H.A.; Chouchani, E.T.; O’Neill, L.A.; Mills, E.L. Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat. Metab. 2019, 1, 16–33. [Google Scholar] [CrossRef]
- Serena, C.; Ceperuelo-Mallafré, V.; Keiran, N.; Queipo-Ortuño, M.I.; Bernal, R.; Gomez-Huelgas, R.; Urpi-Sarda, M.; Sabater, M.; Pérez-Brocal, V.; Andrés-Lacueva, C.; et al. Elevated circulating levels of succinate in human obesity are linked to specific gut microbiota. ISME J. 2018, 12, 1642–1657. [Google Scholar] [CrossRef] [Green Version]
- Boden, G. Obesity and Free Fatty Acids. Endocrinol. Metab. Clin. N. Am. 2008, 37, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Gameiro, P.A.; Laviolette, L.A.; Kelleher, J.K.; Iliopoulos, O.; Stephanopoulos, G. Cofactor Balance by Nicotinamide Nucleotide Transhydrogenase (NNT) Coordinates Reductive Carboxylation and Glucose Catabolism in the Tricarboxylic Acid (TCA) Cycle. J. Biol. Chem. 2013, 288, 12967–12977. [Google Scholar] [CrossRef] [Green Version]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.-H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, A.R.; Hu, Z.; Shi, X.; Jiang, L.; Boroughs, L.K.; Kovacs, Z.; Boriack, R.; Rakheja, D.; Sullivan, L.B.; Linehan, W.M.; et al. Oxidation of Alpha-Ketoglutarate Is Required for Reductive Carboxylation in Cancer Cells with Mitochondrial Defects. Cell Rep. 2014, 7, 1679–1690. [Google Scholar] [CrossRef] [Green Version]
- Filipp, F.V.; Scott, D.A.; Ronai, Z.A.; Osterman, A.L.; Smith, J.W. Reverse TCA cycle flux through isocitrate dehydrogenases 1 and 2 is required for lipogenesis in hypoxic melanoma cells. Pigment Cell Melanoma Res. 2012, 25, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Urra, F.A.; Muñoz, F.; Lovy, A.; Cárdenas, C. The Mitochondrial Complex(I)ty of Cancer. Front. Oncol. 2017, 7, 118. [Google Scholar] [CrossRef]
- Abla, H.; Sollazzo, M.; Gasparre, G.; Iommarini, L.; Porcelli, A.M. The multifaceted contribution of α-ketoglutarate to tumor progression: An opportunity to exploit? Semin. Cell Dev. Biol. 2020, 98, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Chen, J.; Li, H.; Fan, T.; Zheng, X.; Wang, H.; Zhang, N.; Liu, Y.; Luo, X.; Wang, J.; et al. Role of the Mitochondrial Citrate-malate Shuttle in Hras12V-Induced Hepatocarcinogenesis: A Metabolomics-Based Analysis. Metabolites 2020, 10, 193. [Google Scholar] [CrossRef]
- Ronchi, J.A.; Francisco, A.; Passos, L.A.C.; Figueira, T.R.; Castilho, R.F. The Contribution of Nicotinamide Nucleotide Transhydrogenase to Peroxide Detoxification Is Dependent on the Respiratory State and Counterbalanced by Other Sources of NADPH in Liver Mitochondria. J. Biol. Chem. 2016, 291, 20173–20187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rydström, J. Mitochondrial NADPH, transhydrogenase and disease. Biochim. Biophys. Acta 2006, 1757, 721–726. [Google Scholar] [CrossRef] [Green Version]
- Ho, H.-Y.; Lin, Y.-T.; Lin, G.; Wu, P.-R.; Cheng, M.-L. Nicotinamide nucleotide transhydrogenase (NNT) deficiency dysregulates mitochondrial retrograde signaling and impedes proliferation. Redox Biol. 2017, 12, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhuang, Z.; Wu, T.; Lin, J.-C.; Liu, Z.-X.; Zhou, L.-F.; Dai, T.; Lu, L.; Ju, H.-Q. Nicotinamide nucleotide transhydrogenase-mediated redox homeostasis promotes tumor growth and metastasis in gastric cancer. Redox Biol. 2018, 18, 246–255. [Google Scholar] [CrossRef]
- Ward, N.P.; Kang, Y.P.; Falzone, A.; Boyle, T.A.; DeNicola, G.M. Nicotinamide nucleotide transhydrogenase regulates mitochondrial metabolism in NSCLC through maintenance of Fe-S protein function. J. Exp. Med. 2020, 217, e20191689. [Google Scholar] [CrossRef] [Green Version]
- Borst, P. The malate–aspartate shuttle (Borst cycle): How it started and developed into a major metabolic pathway. IUBMB Life 2020, 72, 2241–2259. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Mosaoa, R.; Kasprzyk-Pawelec, A.; Fernandez, H.; Avantaggiati, M. The Mitochondrial Citrate Carrier SLC25A1/CIC and the Fundamental Role of Citrate in Cancer, Inflammation and Beyond. Biomolecules 2021, 11, 141. [Google Scholar] [CrossRef]
- Catalina-Rodriguez, O.; Kolukula, V.K.; Tomita, Y.; Preet, A.; Palmieri, F.; Wellstein, A.; Byers, S.; Giaccia, A.J.; Glasgow, E.; Albanese, C.; et al. The mitochondrial citrate transporter, CIC, is essential for mitochondrial homeostasis. Oncotarget 2012, 3, 1220–1235. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, H.R.; Gadre, S.M.; Tan, M.; Graham, G.T.; Mosaoa, R.; Ongkeko, M.S.; Kim, K.A.; Riggins, R.B.; Parasido, E.; Petrini, I.; et al. The mitochondrial citrate carrier, SLC25A1, drives stemness and therapy resistance in non-small cell lung cancer. Cell Death Differ. 2018, 25, 1239–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolukula, V.K.; Sahu, G.; Wellstein, A.; Rodriguez, O.C.; Preet, A.; Iacobazzi, V.; D’Orazi, G.; Albanese, C.; Palmieri, F.; Avantaggiati, M.L. SLC25A1, or CIC, is a novel transcriptional target of mutant p53 and a negative tumor prognostic marker. Oncotarget 2014, 5, 1212–1225. [Google Scholar] [CrossRef] [Green Version]
- Damiano, F.; Gnoni, G.V.; Siculella, L. Citrate carrier promoter is target of peroxisome proliferator-activated receptor alpha and gamma in hepatocytes and adipocytes. Int. J. Biochem. Cell Biol. 2012, 44, 659–668. [Google Scholar] [CrossRef]
- Gnoni, G.V.; Priore, P.; Geelen, M.J.H.; Siculella, L. The mitochondrial citrate carrier: Metabolic role and regulation of its activity and expression. IUBMB Life 2009, 61, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Ebadi, M.; Field, C.J.; Lehner, R.; Mazurak, V.C. Chemotherapy diminishes lipid storage capacity of adipose tissue in a preclinical model of colon cancer. Lipids Health Dis. 2017, 16, 1012. [Google Scholar] [CrossRef] [Green Version]
- Khwairakpam, A.D.; Banik, K.; Girisa, S.; Shabnam, B.; Shakibaei, M.; Fan, L.; Arfuso, F.; Monisha, J.; Wang, H.; Mao, X.; et al. The vital role of ATP citrate lyase in chronic diseases. Klin. Wochenschr. 2019, 98, 71–95. [Google Scholar] [CrossRef]
- Icard, P.; Coquerel, A.; Wu, Z.; Gligorov, J.; Fuks, D.; Fournel, L.; Lincet, H.; Simula, L. Understanding the Central Role of Citrate in the Metabolism of Cancer Cells and Tumors: An Update. Int. J. Mol. Sci. 2021, 22, 6587. [Google Scholar] [CrossRef]
- Migita, T.; Okabe, S.; Ikeda, K.; Igarashi, S.; Sugawara, S.; Tomida, A.; Soga, T.; Taguchi, R.; Seimiya, H. Inhibition of ATP citrate lyase induces triglyceride accumulation with altered fatty acid composition in cancer cells. Int. J. Cancer 2013, 135, 37–47. [Google Scholar] [CrossRef]
- Migita, T.; Okabe, S.; Ikeda, K.; Igarashi, S.; Sugawara, S.; Tomida, A.; Taguchi, R.; Soga, T.; Seimiya, H. Inhibition of ATP Citrate Lyase Induces an Anticancer Effect via Reactive Oxygen Species: AMPK as a Predictive Biomarker for Therapeutic Impact. Am. J. Pathol. 2013, 182, 1800–1810. [Google Scholar] [CrossRef]
- Migita, T.; Narita, T.; Nomura, K.; Miyagi, E.; Inazuka, F.; Matsuura, M.; Ushijima, M.; Mashima, T.; Seimiya, H.; Satoh, Y.; et al. ATP Citrate Lyase: Activation and Therapeutic Implications in Non–Small Cell Lung Cancer. Cancer Res. 2008, 68, 8547–8554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sena, L.A.; Denmeade, S.R. Fatty acid synthesis in prostate cancer: Vulnerability or epiphenomenon? Cancer Res. 2021, 81, 4385–4393. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Tao, R.; Gao, X.; Li, T.; Zhou, X.; Guan, K.-L.; Xiong, Y.; Lei, Q.-Y. Acetylation Stabilizes ATP-Citrate Lyase to Promote Lipid Biosynthesis and Tumor Growth. Mol. Cell 2013, 51, 506–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basappa, J.; Citir, M.; Zhang, Q.; Wang, H.Y.; Liu, X.; Melnikov, O.; Yahya, H.; Stein, F.; Muller, R.; Traynor-Kaplan, A.; et al. ACLY is the novel signaling target of PIP2/PIP3 and Lyn in acute myeloid leukemia. Heliyon 2020, 6, e03910. [Google Scholar] [CrossRef]
- Chinopoulos, C. Which way does the citric acid cycle turn during hypoxia? The critical role of α-ketoglutarate dehydrogenase complex. J. Neurosci. Res. 2013, 91, 1030–1043. [Google Scholar] [CrossRef] [Green Version]
- Fahien, L.A.; Kmiotek, E.H.; MacDonald, M.J.; Fibich, B.; Mandic, M. Regulation of malate dehydrogenase activity by glutamate, citrate, alpha-ketoglutarate, and multienzyme interaction. J. Biol. Chem. 1988, 263, 10687–10697. [Google Scholar] [CrossRef]
- Ma, Y.; Tian, P.; Chen, Z.; Yue, D.; Liu, C.; Li, C.; Chen, C.; Zhang, H.; Liu, H.; Zhang, Z.; et al. Urinary malate dehydrogenase 2 is a new biomarker for early detection of non-small-cell lung cancer. Cancer Sci. 2021, 112, 2349–2360. [Google Scholar] [CrossRef] [PubMed]
- Hanse, E.A.; Ruan, C.; Kachman, M.; Wang, D.; Lowman, X.H.; Kelekar, A. Cytosolic malate dehydrogenase activity helps support glycolysis in actively proliferating cells and cancer. Oncogene 2017, 36, 3915–3924. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.H.; Lee, S.-H.; Hong, D.; Lee, J.-S.; Ahn, H.-S.; Ahn, J.-H.; Seong, T.W.; Lee, C.-H.; Jang, H.; Hong, K.M.; et al. Aldehyde dehydrogenase is used by cancer cells for energy metabolism. Exp. Mol. Med. 2016, 48, e272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, A.; Siddiqui, A.; Vazakidou, M.E.; Napoli, F.; Böttcher, M.; Menchicchi, B.; Raza, U.; Saatci, O.; Krebs, A.M.; Ferrazzi, F.; et al. Polyol Pathway Links Glucose Metabolism to the Aggressiveness of Cancer Cells. Cancer Res. 2018, 78, 1604–1618. [Google Scholar] [CrossRef] [Green Version]
- Krause, N.; Wegner, A. Fructose Metabolism in Cancer. Cells 2020, 9, 2635. [Google Scholar] [CrossRef]
- Nakagawa, T.; Lanaspa, M.A.; Millan, I.S.; Fini, M.; Rivard, C.J.; Sanchez-Lozada, L.G.; Andres-Hernando, A.; Tolan, D.R.; Johnson, R.J. Fructose contributes to the Warburg effect for cancer growth. Cancer Metab. 2020, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef] [Green Version]
- Hinkle, P.C. P/O ratios of mitochondrial oxidative phosphorylation. Biochim. Biophys. Acta 2005, 1706, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Peng, Z.; Hu, Q.; Xu, L.; Zou, X.; Yu, Y.; Huang, D.; Yi, P. Berberine Suppressed Tumor Growth through Regulating Fatty Acid Metabolism and Triggering Cell Apoptosis via Targeting FABPs. Evidence-Based Complement. Altern. Med. 2020, 2020, 6195050. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhelev, Z.; Sumiyoshi, A.; Aoki, I.; Lazarova, D.; Vlaykova, T.; Higashi, T.; Bakalova, R. Over-Reduced State of Mitochondria as a Trigger of “β-Oxidation Shuttle” in Cancer Cells. Cancers 2022, 14, 871. https://doi.org/10.3390/cancers14040871
Zhelev Z, Sumiyoshi A, Aoki I, Lazarova D, Vlaykova T, Higashi T, Bakalova R. Over-Reduced State of Mitochondria as a Trigger of “β-Oxidation Shuttle” in Cancer Cells. Cancers. 2022; 14(4):871. https://doi.org/10.3390/cancers14040871
Chicago/Turabian StyleZhelev, Zhivko, Akira Sumiyoshi, Ichio Aoki, Dessislava Lazarova, Tatyana Vlaykova, Tatsuya Higashi, and Rumiana Bakalova. 2022. "Over-Reduced State of Mitochondria as a Trigger of “β-Oxidation Shuttle” in Cancer Cells" Cancers 14, no. 4: 871. https://doi.org/10.3390/cancers14040871
APA StyleZhelev, Z., Sumiyoshi, A., Aoki, I., Lazarova, D., Vlaykova, T., Higashi, T., & Bakalova, R. (2022). Over-Reduced State of Mitochondria as a Trigger of “β-Oxidation Shuttle” in Cancer Cells. Cancers, 14(4), 871. https://doi.org/10.3390/cancers14040871