
Combination of Histone Deacetylase Inhibitor Panobinostat (LBH589) with β-Catenin Inhibitor Tegavivint (BC2059) Exerts Significant Anti-Myeloma Activity Both In Vitro and In Vivo

 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Primary Samples Ex Vivo Treatment
2.2. Myeloma Cell Lines
2.3. Confocal Microscopy
2.4. XF Mito Stress Analysis
2.5. In Vivo Studies
3. Results
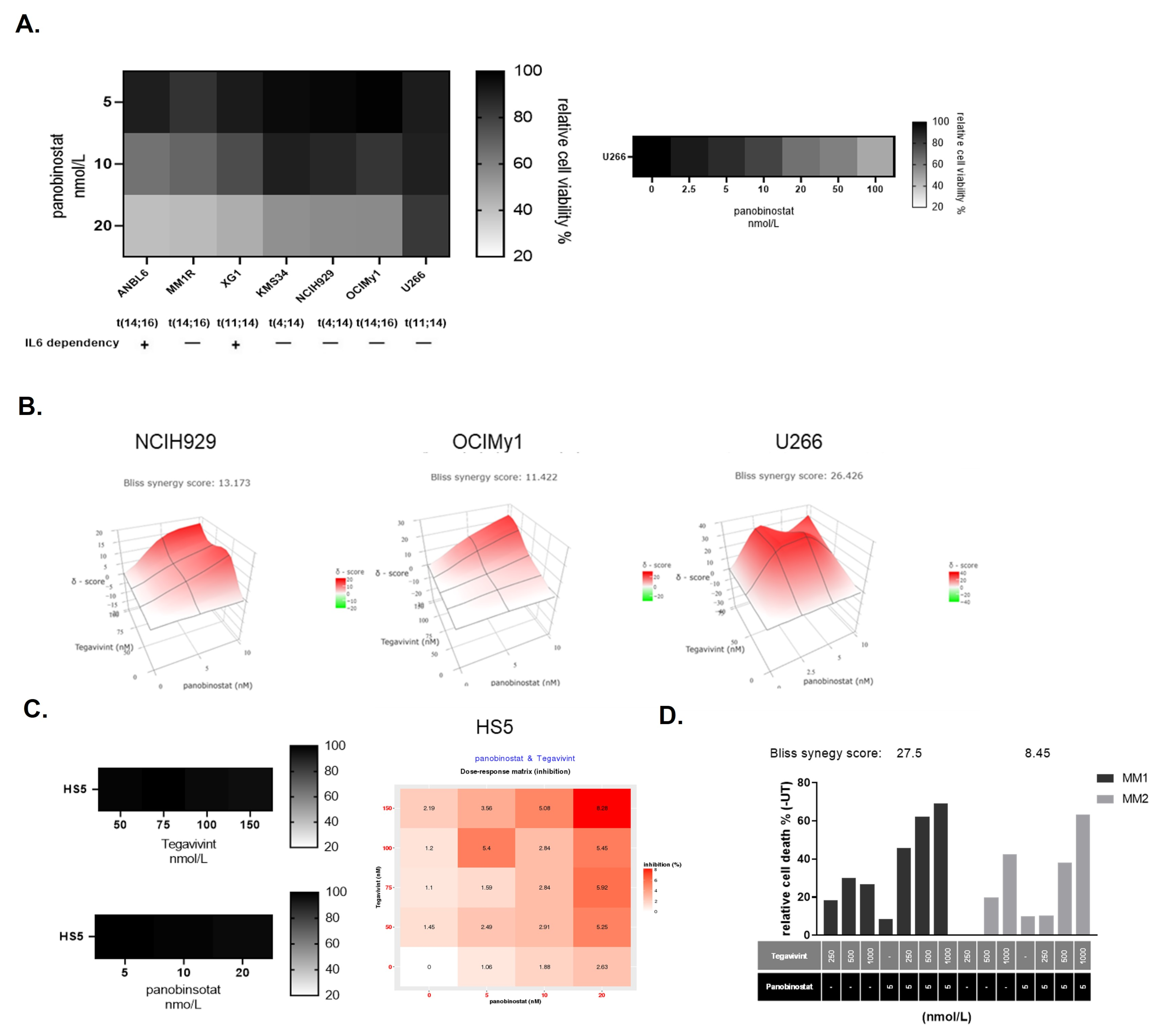
3.1. Low Dose Combinations of Tegavivint and Panobinostat Exert Synergistic Anti-MM Effects In Vitro and Ex Vivo
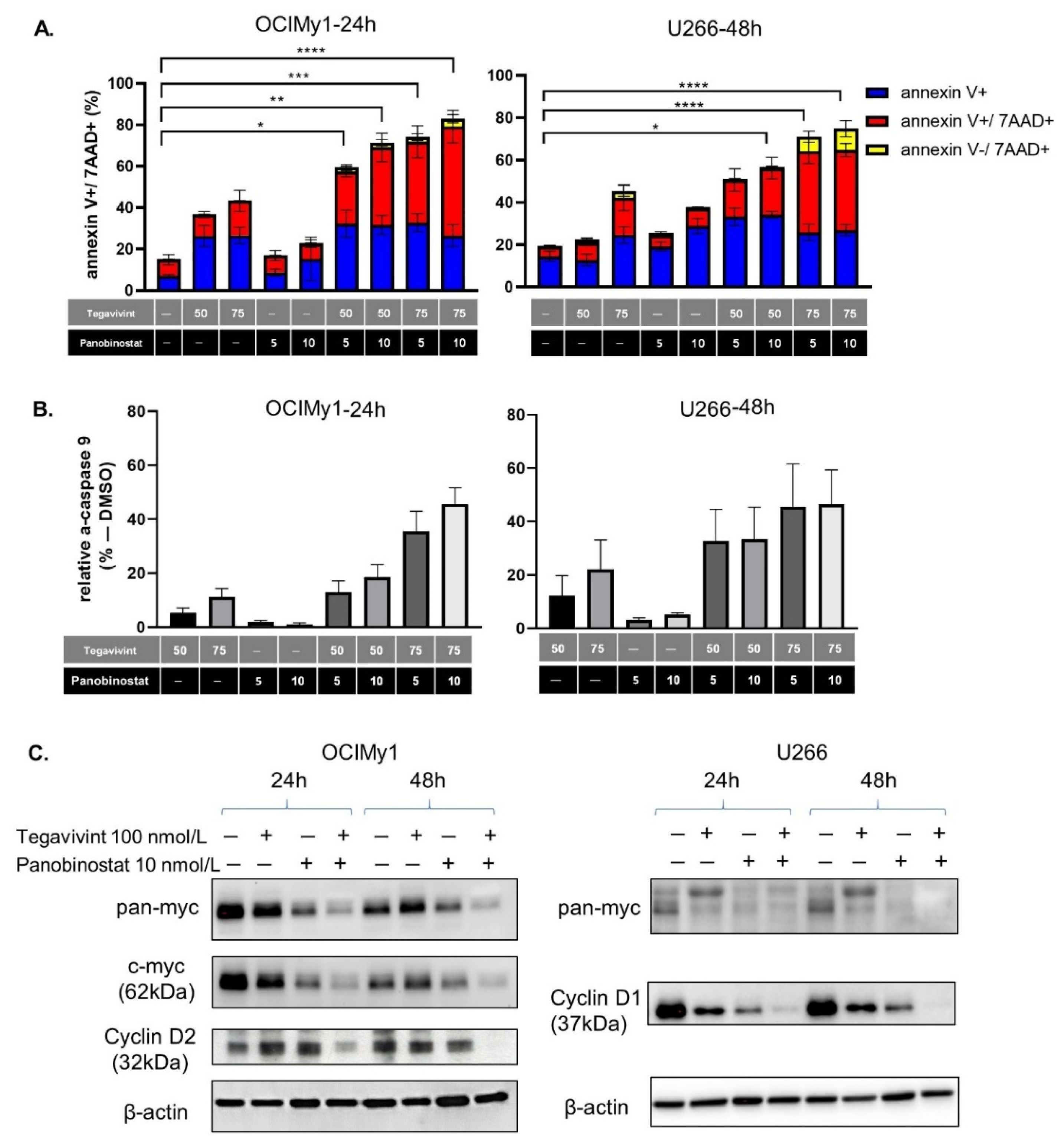
3.2. The Combination of Tegavivint and Panobinostat Induces Apoptosis and Downregulates Wnt Pathway Down-Stream Targets
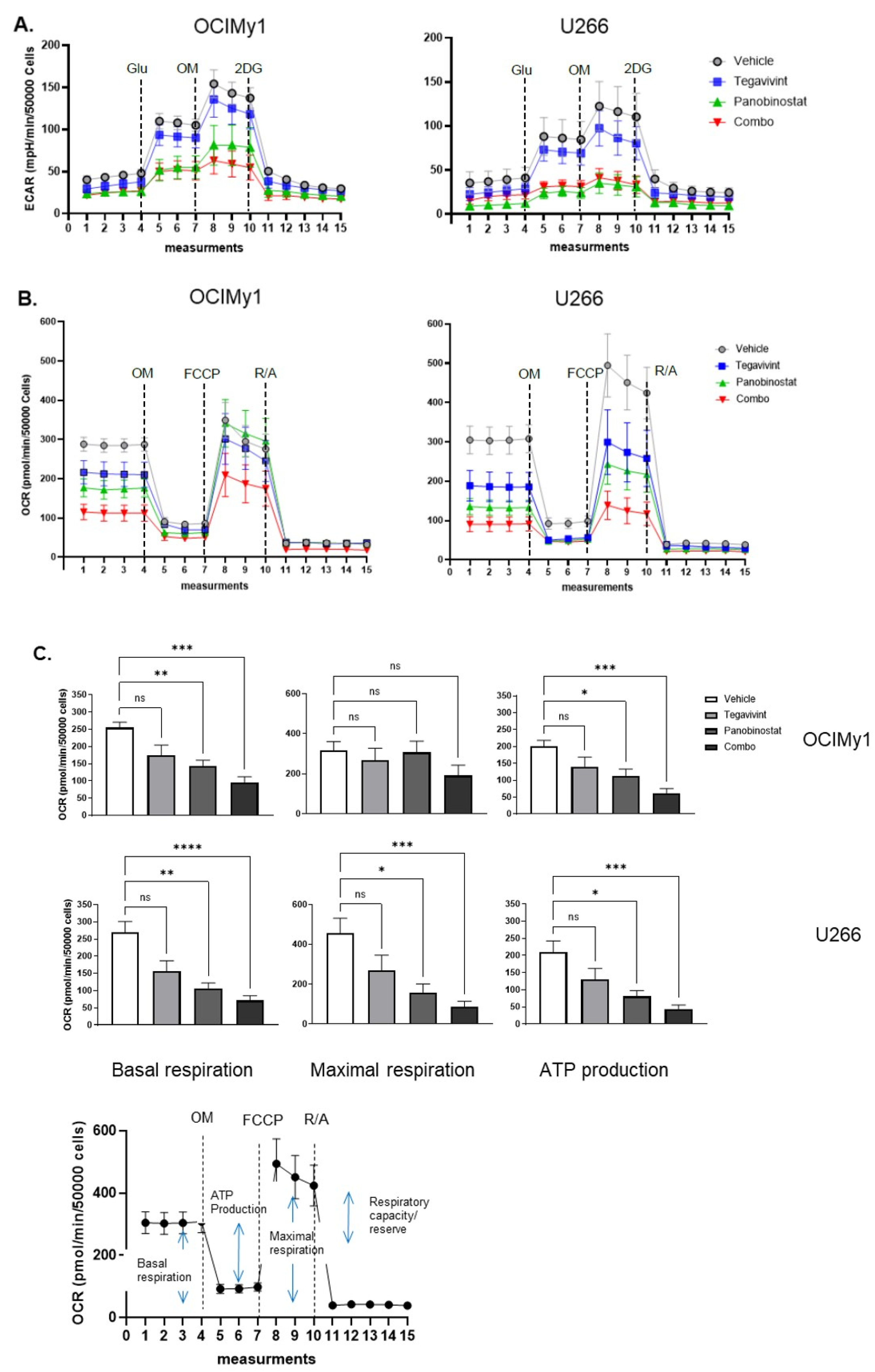
3.3. Tegavivint in Combination with Panobinostat Targets the Mitochondria Fitness of MM Cells
3.4. Tegavivint Overcomes Acquired Panobinostat Resistance in Secondary Bortezomib Resistant MM Cells
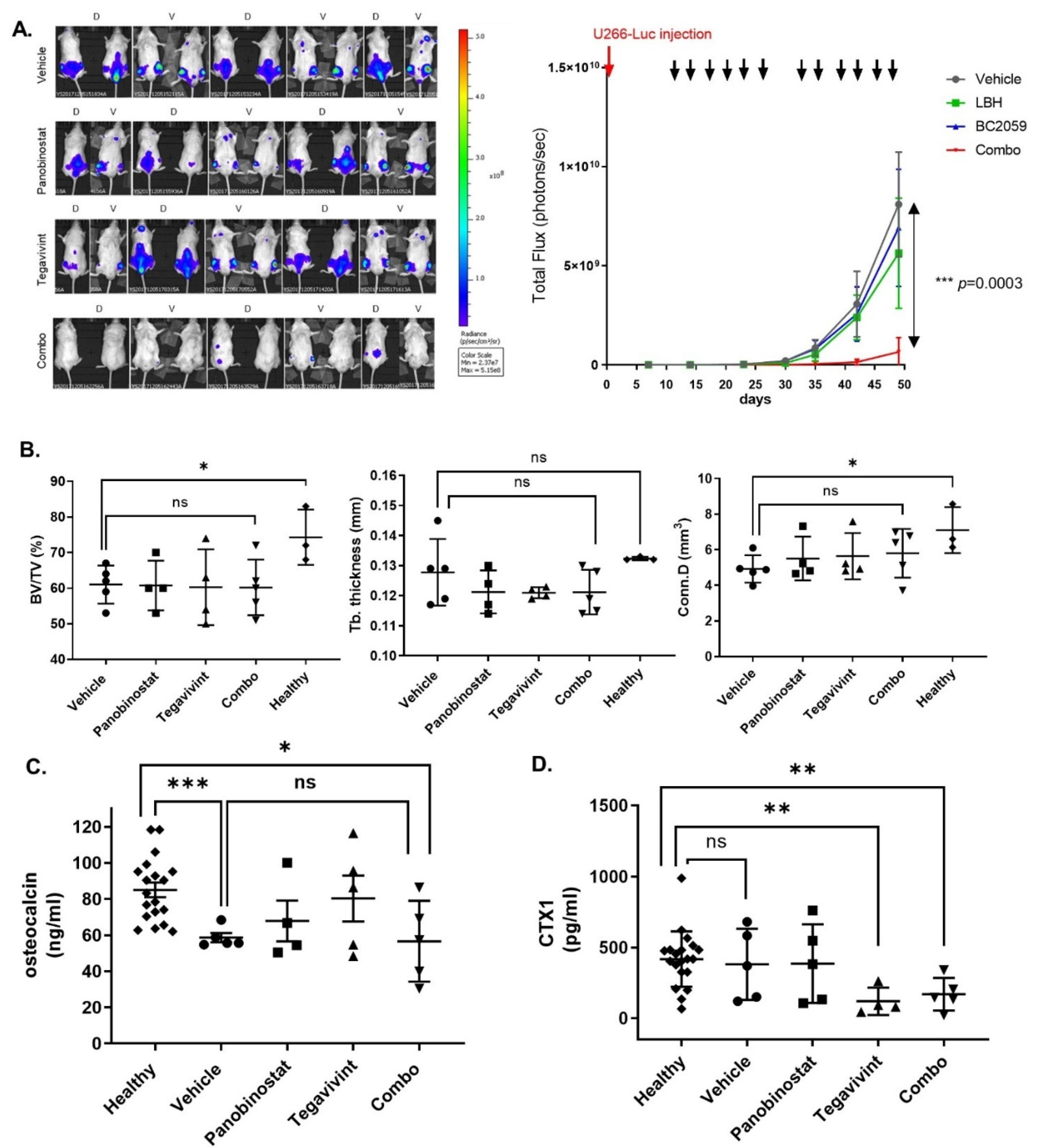
3.5. Tegavivint in Combination with Panobinostat Exerts Potent Anti-MM Effects In Vivo
3.6. Tegavivint in Combination with Panobinostat Is Well Tolerated In Vivo
3.7. Tegavivint in Combination with Panobinostat Does Not Negatively Impact Bone Metabolism In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Robak, P.; Drozdz, I.; Szemraj, J.; Robak, T. Drug resistance in multiple myeloma. Cancer Treat Rev. 2018, 70, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Therneau, T.M.; Gertz, M.A.; Lacy, M.Q.; Dispenzieri, A.; Rajkumar, S.V.; Fonseca, R.; Witzig, T.E.; Lust, J.A.; Larson, D.R.; et al. Clinical course of patients with relapsed multiple myeloma. Mayo Clin. Proc. 2004, 79, 867–874. [Google Scholar] [CrossRef]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Günther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: A multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014, 15, 1195–1206. [Google Scholar] [CrossRef]
- Laubach, J.P.; Schjesvold, F.; Mariz, M.; Dimopoulos, M.A.; Lech-Maranda, E.; Spicka, I.; Hungria, V.T.M.; Shelekhova, T.; Abdo, A.; Jacobasch, L.; et al. Efficacy and safety of oral panobinostat plus subcutaneous bortezomib and oral dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma (PANORAMA 3): An open-label, randomised, phase 2 study. Lancet Oncol. 2021, 22, 142–154. [Google Scholar] [CrossRef]
- Eleutherakis-Papaiakovou, E.; Kanellias, N.; Kastritis, E.; Gavriatopoulou, M.; Terpos, E.; Dimopoulos, M.A. Efficacy of panobinostat for the treatment of multiple myeloma. J. Oncol. 2020, 2020, 7131802. [Google Scholar] [CrossRef] [PubMed]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Spaan, I.; Raymakers, R.A.; van de Stolpe, A.; Peperzak, V. Wnt signaling in multiple myeloma: A central player in disease with therapeutic potential. J. Hematol. Oncol. 2018, 11, 67. [Google Scholar] [CrossRef]
- Savvidou, I.; Khong, T.; Cuddihy, A.; McLean, C.; Horrigan, S.; Spencer, A. β-Catenin Inhibitor BC2059 is efficacious as monotherapy or in combination with proteasome inhibitor bortezomib in multiple myeloma. Mol. Cancer Ther. 2017, 16, 1765–1778. [Google Scholar] [CrossRef] [Green Version]
- Nomura, M.; Rainusso, N.; Lee, Y.C.; Dawson, B.; Coarfa, C.; Han, R.; Larson, J.L.; Shuck, R.; Kurenbekova, L.; Yustein, J.T. Tegavivint and the β-Catenin/ALDH Axis in Chemotherapy-Resistant and Metastatic Osteosarcoma. J. Natl. Cancer Inst. 2019, 111, 1216–1227. [Google Scholar] [CrossRef]
- Fiskus, W.; Sharma, S.; Saha, S.; Shah, B.; Devaraj, S.G.; Sun, B.; Horrigan, S.; Leveque, C.; Zu, Y.; Iyer, S.; et al. Pre-clinical efficacy of combined therapy with novel β-catenin antagonist BC2059 and histone deacetylase inhibitor against AML cells. Leukemia 2015, 29, 1267–1278. [Google Scholar] [CrossRef] [PubMed]
- Saenz, D.T.; Fiskus, W.; Manshouri, T.; Mill, C.P.; Qian, Y.; Raina, K.; Rajapakshe, K.; Coarfa, C.; Soldi, R.; Bose, P.; et al. Targeting nuclear β-catenin as therapy for post-myeloproliferative neoplasm secondary AML. Leukemia 2019, 33, 1373–1386. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wng, C.Y. TBL1-TBLR1 and beta–catenin recruit each other to Wnt target-gene promoter for transcription activation and oncogenesis. Nat. Cell Biol. 2008, 10, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Soldi, R.; Halder, T.G.; Sampson, S.; Vankayalapati, H.; Weston, A.; Thode, T.; Bhalla, K.N.; Ng, S.; Rodriguez Del Villar, R.; Drenner, K.; et al. The Small Molecule BC-2059 inhibits Wingless/integrated (Wnt)-dependent gene transcription in cancer through disruption of the Transducin β-Like 1- β-Catenin protein complex. J. Pharmacol. Exp. Ther. 2021, 378, 77–86. [Google Scholar] [CrossRef]
- Mithraprabhu, S.; Khong, T.; Spencer, A. Overcoming inherent resistance to histone deacetylase inhibitors in multiple myeloma cells by targeting pathways integral to the actin cytoskeleton. Cell Death Dis. 2014, 5, e1134. [Google Scholar] [CrossRef] [Green Version]
- Ianevski, A.; Giri, K.A.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef]
- González-Calle, V.; Keane, N.; Braggio, E.; Fonseca, R. Precision medicine in myeloma: Challenges in defining an actionable approach. Clin. Lymphoma Myeloma Leuk. 2017, 17, 621–630. [Google Scholar] [CrossRef]
- Tateishi, K.; Iafrate, A.J.; Ho, Q.; Curry, W.T.; Batchelor, T.T.; Flaherty, K.T.; Onozato, M.L.; Lelic, N.; Sundaram, S.; Cahill, D.P.; et al. Myc-Driven glycolysis is a therapeutic target in glioblastoma. Clin. Cancer Res. 2016, 22, 4452–4465. [Google Scholar] [CrossRef] [Green Version]
- Dejure, F.R.; Eilers, M. MYC and tumour metabolism: Chicken and egg. EMBO J. 2017, 36, 3409–3420. [Google Scholar] [CrossRef]
- Wardell, S.E.; Ilkayeva, O.R.; Wieman, H.L.; Frigo, D.E.; Rathmell, J.C.; Newgard, C.B.; McDonnell, D.P. Glucose metabolism as a target of histone deacetylase inhibitors. Mol. Endocrinol. 2009, 23, 388–401. [Google Scholar] [CrossRef] [Green Version]
- Vergara, D.; Stanca, E.; Guerra, F.; Priore, P.; Gaballo, A.; Franck, J.; Simeone, P.; Trerotola, M.; De Domenico, S.; Fournier, I.; et al. β-Catenin knockdown affects mitochondrial biogenesis and lipid metabolism in breast cancer cells. Front Physiol. 2017, 8, 544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.C.; Ng, A.; Kim, B.H.; Bianco, A.; Xavier, R.J.; Elledge, S.J. Wnt signaling regulates mitochondrial physiology and insulin sensitivity. Genes Dev. 2010, 24, 1507–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Shen, L.; Li, X.; Liu, J. Efficacy and toxicity of histone deacetylase inhibitors in relapsed/refractory multiple myeloma: Systematic review and meta-analysis of clinical trials. Exp Ther Med. 2019, 18, 1057–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Andel, H.; Kocemba, K.A.; Spaargaren, M.; Pals, S.T. Aberrant Wnt signaling in multiple myeloma: Molecular mechanisms and targeting options. Leukemia 2019, 33, 1063–1075. [Google Scholar] [CrossRef]
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med. 2013, 19, 179–192. [Google Scholar] [CrossRef]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Dimopoulos, M.A. Myeloma bone disease: From biology findings to treatment approaches. Blood 2019, 133, 1534–1539. [Google Scholar] [CrossRef] [Green Version]
- Mithraprabhu, S.; Sirdesai, S.; Chen, M.; Khong, T.; Spencer, A. Circulating tumour DNA analysis for tumour genome characterisation and monitoring disease burden in extramedullary multiple myeloma. Int. J. Mol. Sci. 2018, 19, 1858. [Google Scholar] [CrossRef] [Green Version]
- Bjorklund, C.C.; Ma, W.; Wang, Z.Q.; Davis, R.E.; Kuhn, D.J.; Kornblau, S.M.; Wang, M.; Shah, J.J.; Orlowski, R.Z. Evidence of a role for activation of Wnt/beta-catenin signaling in the resistance of plasma cells to lenalidomide. J. Biol. Chem. 2011, 286, 11009–11020. [Google Scholar] [CrossRef] [Green Version]
- Proto, M.C.; Fiore, D.; Piscopo, C.; Franceschelli, S.; Bizzarro, V.; Laezza, C.; Lauro, G.; Feoli, A.; Tosco, A.; Bifulco, G.; et al. Inhibition of Wnt/β-Catenin pathway and histone acetyltransferase activity by Rimonabant: A therapeutic target for colon cancer. Sci. Rep. 2017, 7, 11678. [Google Scholar] [CrossRef]
- Götze, S.; Coersmeyer, M.; Müller, O.; Sievers, S. Histone deacetylase inhibitors induce attenuation of Wnt signaling and TCF7L2 depletion in colorectal carcinoma cells. Int. J. Oncol. 2014, 45, 1715–1723. [Google Scholar] [CrossRef]
- Kumar, S.K.; Harrison, S.J.; Cavo, M.; de la Rubia, J.; Popat, R.; Gasparetto, C.; Hungria, V.; Salwender, H.; Suzuki, K.; Kim, I.; et al. Venetoclax or placebo in combination with bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma (BELLINI): A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2020, 21, 1630–1642. [Google Scholar] [CrossRef]
- Gong, J.N.; Khong, T.; Segal, D.; Yao, Y.; Riffkin, C.D.; Garnier, J.M.; Khaw, S.L.; Lessene, G.; Spencer, A.; Herold, M.J.; et al. Hierarchy for targeting prosurvival BCL2 family proteins in multiple myeloma: Pivotal role of MCL1. Blood 2016, 128, 1834–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luis, T.C.; Ichii, M.; Brugman, M.H.; Kincade, P.; Staal, F.J. Wnt signaling strength regulates normal haematopoiesis and its deregulation is involved in leukemia development. Leukemia 2012, 26, 414–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niida, A.; Hiroko, T.; Kasai, M.; Furukawa, Y.; Nakamura, Y.; Suzuki, Y.; Sugano, S.; Akiyama, T. DKK1, a negative regulator of Wnt signaling, is a target of the β-catenin/TCF pathway. Oncogene 2004, 23, 8520–8526. [Google Scholar] [CrossRef] [Green Version]
- Sottnik, J.L.; Hall, C.L.; Zhang, J.; Keller, E.T. Wnt and Wnt inhibitors in bone metastasis. Bonekey Rep. 2012, 1, 101. [Google Scholar] [CrossRef] [Green Version]
- Nusse, R.; Fuerer, C.; Ching, W.; Harnish, K.; Logan, C.; Zeng, A.; ten Berge, D.; Kalani, Y. Wnt signaling and stem cell control. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Okugawa, H. A new imaging method for confocal microscopy. Proc. SPIE 2008, 6860, 1–7. [Google Scholar] [CrossRef]
- Doube, M.; Kłosowski, M.M.; Arganda-Carreras, I.; Cordelières, F.P.; Dougherty, R.P.; Jackson, J.S.; Schmid, B.; Hutchinson, J.R.; Shefelbine, S.J. BoneJ: Free and extensible bone image analysis in ImageJ. Bone 2010, 47, 1076–1079. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savvidou, I.; Khong, T.; Whish, S.; Carmichael, I.; Sepehrizadeh, T.; Mithraprabhu, S.; Horrigan, S.K.; de Veer, M.; Spencer, A. Combination of Histone Deacetylase Inhibitor Panobinostat (LBH589) with β-Catenin Inhibitor Tegavivint (BC2059) Exerts Significant Anti-Myeloma Activity Both In Vitro and In Vivo. Cancers 2022, 14, 840. https://doi.org/10.3390/cancers14030840
Savvidou I, Khong T, Whish S, Carmichael I, Sepehrizadeh T, Mithraprabhu S, Horrigan SK, de Veer M, Spencer A. Combination of Histone Deacetylase Inhibitor Panobinostat (LBH589) with β-Catenin Inhibitor Tegavivint (BC2059) Exerts Significant Anti-Myeloma Activity Both In Vitro and In Vivo. Cancers. 2022; 14(3):840. https://doi.org/10.3390/cancers14030840
Chicago/Turabian StyleSavvidou, Ioanna, Tiffany Khong, Sophie Whish, Irena Carmichael, Tara Sepehrizadeh, Sridurga Mithraprabhu, Stephen K. Horrigan, Michael de Veer, and Andrew Spencer. 2022. "Combination of Histone Deacetylase Inhibitor Panobinostat (LBH589) with β-Catenin Inhibitor Tegavivint (BC2059) Exerts Significant Anti-Myeloma Activity Both In Vitro and In Vivo" Cancers 14, no. 3: 840. https://doi.org/10.3390/cancers14030840
APA StyleSavvidou, I., Khong, T., Whish, S., Carmichael, I., Sepehrizadeh, T., Mithraprabhu, S., Horrigan, S. K., de Veer, M., & Spencer, A. (2022). Combination of Histone Deacetylase Inhibitor Panobinostat (LBH589) with β-Catenin Inhibitor Tegavivint (BC2059) Exerts Significant Anti-Myeloma Activity Both In Vitro and In Vivo. Cancers, 14(3), 840. https://doi.org/10.3390/cancers14030840