
Role of CBL Mutations in Cancer and Non-Malignant Phenotype

 , ,
, ,  , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
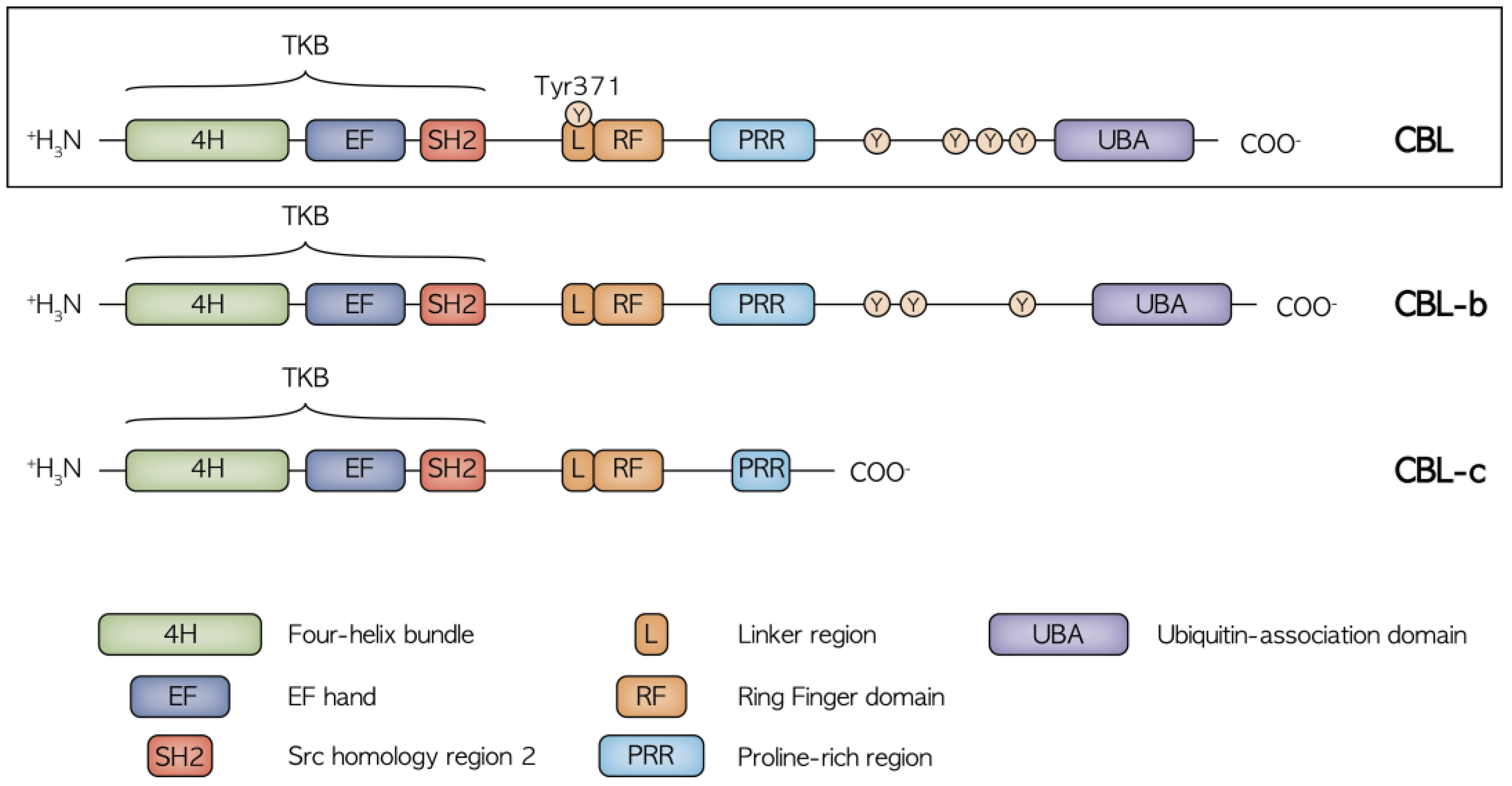
2. Structure and Function of CBL
3. The Role of CBL in Signaling Pathway Modulation
3.1. CBL and JAK2 Signaling
3.2. CBL and EGFR–CBL–CIN85 Axis
3.3. CBL and PI3K/AKT/LYN Interaction
4. CBL in Human Malignancies
4.1. CBL in JMML
4.2. CBL in Hematological Neoplasms
4.3. CBL in Other Malignancies
5. CBL in Non-Malignant Clinical Spectrum
5.1. CBL Syndrome
5.2. Vascular Pathology
5.3. Immunological and Hematological Manifestations
5.4. Coagulative Disorders
5.5. Prenatal Manifestation of CBL Germline Mutations
6. Therapeutic Potential of CBL Targeting
7. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Kratz, C.P.; Jongmans, M.C.; Cavé, H.; Wimmer, K.; Behjati, S.; Guerrini-Rousseau, L.; Milde, T.; Pajtler, K.W.; Golmard, L.; Gauthier-Villars, M.; et al. Predisposition to cancer in children and adolescents. Lancet Child Adolesc. Health 2021, 5, 142–154. [Google Scholar] [CrossRef]
- Huang, K.-L.; Mashl, R.J.; Wu, Y.; Ritter, D.I.; Wang, J.; Oh, C.; Paczkowska, M.; Reynolds, S.; Wyczalkowski, M.A.; Oak, N.; et al. Pathogenic Germline Variants in 10,389 Adult Cancers. Cell 2018, 173, 355–370.e14. [Google Scholar] [CrossRef]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Aman, P.; Zhou, X.; Yergeau, D.A.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef]
- Narumi, S.; Amano, N.; Ishii, T.; Katsumata, N.; Muroya, K.; Adachi, M.; Toyoshima, K.; Tanaka, Y.; Fukuzawa, R.; Miyako, K.; et al. SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat. Genet. 2016, 48, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Bruzzese, A.; Leardini, D.; Masetti, R.; Strocchio, L.; Girardi, K.; Algeri, M.; Del Baldo, G.; Locatelli, F.; Mastronuzzi, A. GATA2 Related Conditions and Predisposition to Pediatric Myelodysplastic Syndromes. Cancers 2020, 12, 2962. [Google Scholar] [CrossRef] [PubMed]
- Tesi, B.; Davidsson, J.; Voss, M.; Rahikkala, E.; Holmes, T.D.; Chiang, S.C.C.; Komulainen-Ebrahim, J.; Gorcenco, S.; Nilsson, A.R.; Ripperger, T.; et al. Gain-of-function SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS, and neurological symptoms. Blood 2017, 129, 2266–2279. [Google Scholar] [CrossRef] [PubMed]
- Kirwan, M.; Walne, A.J.; Plagnol, V.; Velangi, M.; Ho, A.; Hossain, U.; Vulliamy, T.; Dokal, I. Exome Sequencing Identifies Autosomal-Dominant SRP72 Mutations Associated with Familial Aplasia and Myelodysplasia. Am. J. Hum. Genet. 2012, 90, 888–892. [Google Scholar] [CrossRef]
- Schlegelberger, B.; Heller, P.G. RUNX1 deficiency (familial platelet disorder with predisposition to myeloid leukemia, FPDMM). Semin. Hematol. 2017, 54, 75–80. [Google Scholar] [CrossRef]
- Kales, S.C.; Ryan, P.E.; Nau, M.M.; Lipkowitz, S. Cbl and Human Myeloid Neoplasms: The Cbl Oncogene Comes of Age: Figure 1. Cancer Res. 2010, 70, 4789–4794. [Google Scholar] [CrossRef]
- Levkowitz, G.; Waterman, H.; Zamir, E.; Kam, Z.; Oved, S.; Langdon, W.Y.; Beguinot, L.; Geiger, B.; Yarden, Y. c-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev. 1998, 12, 3663–3674. [Google Scholar] [CrossRef]
- Liyasova, M.S.; Ma, K.; Lipkowitz, S. Molecular Pathways: Cbl Proteins in Tumorigenesis and Antitumor Immunity—Opportunities for Cancer Treatment. Clin. Cancer Res. 2014, 21, 1789–1794. [Google Scholar] [CrossRef]
- Ahmed, S.F.; Buetow, L.; Gabrielsen, M.; Lilla, S.; Sibbet, G.J.; Sumpton, D.; Zanivan, S.; Hedley, A.; Clark, W.; Huang, D.T. E3 ligase-inactivation rewires CBL interactome to elicit oncogenesis by hijacking RTK–CBL–CIN85 axis. Oncogene 2021, 40, 2149–2164. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.H.; Dikic, I. The Cbl interactome and its functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 907–919. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Shih, L.-Y.; Suzuki, T.; Otsu, M.; Nakauchi, H.; Koeffler, H.P.; Sanada, M. Deregulated Intracellular Signaling by Mutated c-CBL in Myeloid Neoplasms. Clin. Cancer Res. 2010, 16, 3825–3831. [Google Scholar] [CrossRef]
- Lv, K.; Jiang, J.; Donaghy, R.; Riling, C.R.; Cheng, Y.; Chandra, V.; Rozenova, K.; An, W.; Mohapatra, B.C.; Goetz, B.T.; et al. CBL family E3 ubiquitin ligases control JAK2 ubiquitination and stability in hematopoietic stem cells and myeloid malignancies. Genes Dev. 2017, 31, 1007–1023. [Google Scholar] [CrossRef]
- Liu, C.-S.; Yang-Yen, H.-F.; Suen, C.-S.; Hwang, M.-J.; Yen, J.J.-Y. Cbl-mediated K63-linked ubiquitination of JAK2 enhances JAK2 phosphorylation and signal transduction. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef]
- Javadi, M.; Richmond, T.D.; Huang, K.; Barber, D.L. CBL Linker Region and RING Finger Mutations Lead to Enhanced Granulocyte-Macrophage Colony-stimulating Factor (GM-CSF) Signaling via Elevated Levels of JAK2 and LYN. J. Biol. Chem. 2013, 288, 19459–19470. [Google Scholar] [CrossRef]
- Nagao, T.; Oshikawa, G.; Wu, N.; Kurosu, T.; Miura, O. DNA Damage Stress and Inhibition of Jak2-V617F Cause Its Degradation and Synergistically Induce Apoptosis through Activation of GSK3β. PLoS ONE 2011, 6, e27397. [Google Scholar] [CrossRef]
- Bunda, S.; Qin, K.; Kommaraju, K.; Heir, P.; Ohh, M. Juvenile myelomonocytic leukaemia-associated mutation in Cbl promotes resistance to apoptosis via the Lyn-PI3K/AKT pathway. Oncogene 2014, 34, 789–797. [Google Scholar] [CrossRef]
- Ueno, H.; Sasaki, K.; Honda, H.; Nakamoto, T.; Yamagata, T.; Miyagawa, K.; Mitani, K.; Yazaki, Y.; Hirai, H. c-Cbl Is Tyrosine-Phosphorylated by Interleukin-4 and Enhances Mitogenic and Survival Signals of Interleukin-4 Receptor by Linking With the Phosphatidylinositol 3′-Kinase Pathway. Blood 1998, 91, 46–53. [Google Scholar] [CrossRef]
- Belizaire, R.; Koochaki, S.H.J.; Udeshi, N.D.; Vedder, A.; Sun, L.; Svinkina, T.; Hartigan, C.; McConkey, M.; Kovalcik, V.; Bizuayehu, A.; et al. CBL mutations drive PI3K/AKT signaling via increased interaction with LYN and PIK3R1. Blood 2021, 137, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Matsubara, Y. Ras/MAPK syndromes and childhood hemato-oncological diseases. Int. J. Hematol. 2012, 97, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Niemeyer, C.M.; Arico, M.; Basso, G.; Biondi, A.; Rajnoldi, A.C.; Creutzig, U.; Haas, O.; Harbott, J.; Hasle, H.; Kerndrup, G.; et al. Chronic myelomonocytic leukemia in childhood: A retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS). Blood 1997, 89, 3534–3543. [Google Scholar]
- Niemeyer, C.M. JMML genomics and decisions. Hematology 2018, 2018, 307–312. [Google Scholar] [CrossRef]
- Stieglitz, E.; Taylor-Weiner, A.N.; Chang, T.Y.; Gelston, L.C.; Wang, Y.-D.; Mazor, T.; Esquivel, E.; Yu, A.; Seepo, S.; Olsen, S.R.; et al. The genomic landscape of juvenile myelomonocytic leukemia. Nat. Genet. 2015, 47, 1326–1333. [Google Scholar] [CrossRef]
- Loh, M.L.; Sakai, D.S.; Flotho, C.; Kang, M.; Fliegauf, M.; Archambeault, S.; Mullighan, C.G.; Chen, L.; Bergstraesser, E.; Bueso-Ramos, C.E.; et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood 2009, 114, 1859–1863. [Google Scholar] [CrossRef]
- Meynier, S.; Rieux-Laucat, F. After 95 years, it's time to eRASe JMML. Blood Rev. 2020, 43, 100652. [Google Scholar] [CrossRef]
- Niemeyer, C.M.; Flotho, C. Juvenile myelomonocytic leukemia: Who’s the driver at the wheel? Blood 2019, 133, 1060–1070. [Google Scholar] [CrossRef]
- Hecht, A.; Meyer, J.A.; Behnert, A.; Wong, E.; Chehab, F.; Olshen, A.; Hechmer, A.; Aftandilian, C.; Bhat, R.; Choi, S.W.; et al. Molecular and phenotypic diversity of CBL-mutated juvenile myelomonocytic leukemia. Haematologica 2020, 107, 178–186. [Google Scholar] [CrossRef]
- Muraoka, M.; Okuma, C.; Kanamitsu, K.; Ishida, H.; Kanazawa, Y.; Washio, K.; Seki, M.; Kato, M.; Takita, J.; Sato, Y.; et al. Adults with germline CBL mutation complicated with juvenile myelomonocytic leukemia at infancy. J. Hum. Genet. 2016, 61, 523–526. [Google Scholar] [CrossRef]
- Niemeyer, C.M.; Kang, M.W.; Shin, D.H.; Furlan, I.; Erlacher, M.; Bunin, N.J.; Bunda, S.; Finklestein, J.Z.; Sakamoto, K.M.; Gorr, T.; et al. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat. Genet. 2010, 42, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Pérez, B.; Mechinaud, F.; Galambrun, C.; Ben Romdhane, N.; Isidor, B.; Philip, N.; Derain-Court, J.; Cassinat, B.; Lachenaud, J.; Kaltenbach, S.; et al. Germline mutations of the CBL gene define a new genetic syndrome with predisposition to juvenile myelomonocytic leukaemia. J. Med. Genet. 2010, 47, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Kales, S.C.; Ma, K.; Shoemaker, B.A.; Crespo-Barreto, J.; Cangelosi, A.L.; Lipkowitz, S.; Panchenko, A.R. Balancing Protein Stability and Activity in Cancer: A New Approach for Identifying Driver Mutations Affecting CBL Ubiquitin Ligase Activation. Cancer Res. 2015, 76, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Lipka, D.B.; Witte, T.; Toth, R.; Yang, J.; Wiesenfarth, M.; Nöllke, P.; Fischer, A.; Brocks, D.; Gu, Z.; Park, J.; et al. RAS-pathway mutation patterns define epigenetic subclasses in juvenile myelomonocytic leukemia. Nat. Commun. 2017, 8, 2126. [Google Scholar] [CrossRef]
- Caye, A.; Strullu, M.; Guidez, F.; Cassinat, B.; Gazal, S.; Fenneteau, O.; Lainey, E.; Nouri, K.; Nakhaeirad, S.; Dvorsky, R.; et al. Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat. Genet. 2015, 47, 1334–1340. [Google Scholar] [CrossRef]
- Lasho, T.; Patnaik, M.M. Juvenile myelomonocytic leukemia – A bona fide RASopathy syndrome. Best Pract. Res. Clin. Haematol. 2020, 33, 101171. [Google Scholar] [CrossRef]
- Strullu, M.; Caye, A.; Cassinat, B.; Fenneteau, O.; Touzot, F.; Blauwblomme, T.; Rodriguez, R.; Latour, S.; Petit, A.; Barlogis, V.; et al. In hematopoietic cells with a germline mutation of CBL, loss of heterozygosity is not a signature of juvenile myelo-monocytic leukemia. Leukemia 2013, 27, 2404–2407. [Google Scholar] [CrossRef][Green Version]
- Matsuda, K.; Taira, C.; Sakashita, K.; Saito, S.; Tanaka-Yanagisawa, M.; Yanagisawa, R.; Nakazawa, Y.; Shiohara, M.; Fukushima, K.; Oda, M.; et al. Long-term survival after nonintensive chemotherapy in some juvenile myelomonocytic leukemia patients with CBL mutations, and the possible presence of healthy persons with the mutations. Blood 2010, 115, 5429–5431. [Google Scholar] [CrossRef]
- Locatelli, F.; Niemeyer, C.M. How I treat juvenile myelomonocytic leukemia. Blood 2015, 125, 1083–1090. [Google Scholar] [CrossRef]
- Niemeyer, C.M.; Flotho, C.; Lipka, D.B.; Starý, J.; Rössig, C.; Baruchel, A.; Klingebiel, T.; Micalizzi, C.; Michel, G.; Nysom, K.; et al. Response to upfront azacitidine in juvenile myelomonocytic leukemia in the AZA-JMML-001 trial. Blood Adv. 2021, 5, 2901–2908. [Google Scholar] [CrossRef]
- Inagaki, J.; Fukano, R.; Nishikawa, T.; Nakashima, K.; Sawa, D.; Ito, N.; Okamura, J. Outcomes of immunological interventions for mixed chimerism following allogeneic stem cell transplantation in children with juvenile myelomonocytic leukemia. Pediatr. Blood Cancer 2012, 60, 116–120. [Google Scholar] [CrossRef]
- Oshrine, B. Primary Graft Failure but Treatment Success: A Case of Reversion to Heterozygosity After Allogeneic Hematopoietic Cell Transplantation with Autologous Hematopoietic Recovery in a Child With CBL-related Juvenile Myelomonocytic Leukemia. J. Pediatr. Hematol. 2020, 43, e426–e428. [Google Scholar] [CrossRef]
- Makishima, H.; Cazzolli, H.; Szpurka, H.; Dunbar, A.; Tiu, R.; Huh, J.; Muramatsu, H.; O'Keefe, C.; Hsi, E.; Paquette, R.L.; et al. Mutations of E3 Ubiquitin Ligase Cbl Family Members Constitute a Novel Common Pathogenic Lesion in Myeloid Malignancies. J. Clin. Oncol. 2009, 27, 6109–6116. [Google Scholar] [CrossRef]
- Grand, F.H.; Hidalgo-Curtis, C.E.; Ernst, T.; Zoi, K.; Zoi, C.; McGuire, C.; Kreil, S.; Jones, A.; Score, J.; Metzgeroth, G.; et al. Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood 2009, 113, 6182–6192. [Google Scholar] [CrossRef]
- Abbas, S.; Rotmans, G.; Löwenberg, B.; Valk, P.J. Exon 8 splice site mutations in the gene encoding the E3-ligase CBL are associated with core binding factor acute myeloid leukemias. Haematologica 2008, 93, 1595–1597. [Google Scholar] [CrossRef]
- Reindl, C.; Quentmeier, H.; Petropoulos, K.; Greif, P.A.; Benthaus, T.; Argiropoulos, B.; Mellert, G.; Vempati, S.; Duyster, J.; Buske, C.; et al. CBL Exon 8/9 Mutants Activate the FLT3 Pathway and Cluster in Core Binding Factor/11q Deletion Acute Myeloid Leukemia/Myelodysplastic Syndrome Subtypes. Clin. Cancer Res. 2009, 15, 2238–2247. [Google Scholar] [CrossRef]
- Park, S.H.; Lee, H.J.; Kim, I.-S.; Kang, J.-E.; Lee, E.Y.; Kim, H.-J.; Kim, Y.-K.; Won, J.-H.; Bang, S.M.; Kim, H.; et al. Incidences and Prognostic Impact ofc-KIT, WT1, CEBPA, and CBLMutations, and Mutations Associated with Epigenetic Modification in Core Binding Factor Acute Myeloid Leukemia: A Multicenter Study in a Korean Population. Ann. Lab. Med. 2015, 35, 288–297. [Google Scholar] [CrossRef]
- Allen, C.; Hills, R.; Lamb, K.; Evans, C.; Tinsley, S.; Sellar, R.; O'Brien, M.; Yin, J.L.; Burnett, A.K.; Linch, D.C.; et al. The importance of relative mutant level for evaluating impact on outcome of KIT, FLT3 and CBL mutations in core-binding factor acute myeloid leukemia. Leukemia 2013, 27, 1891–1901. [Google Scholar] [CrossRef]
- Becker, H.; Yoshida, K.; Blagitko-Dorfs, N.; Claus, R.; Pantic, M.; Abdelkarim, M.; Niemöller, C.; Greil, C.; Hackanson, B.; Shiraishi, Y.; et al. Tracing the development of acute myeloid leukemia in CBL syndrome. Blood 2014, 123, 1883–1886. [Google Scholar] [CrossRef]
- Ali, A.M.; Cooper, J.; Walker, A.; Jones, D.; Saad, A. Adult-onset acute myeloid leukaemia in a patient with germline mutation of CBL. Br. J. Haematol. 2020, 192, 665–667. [Google Scholar] [CrossRef]
- Saito, Y.; Aoki, Y.; Muramatsu, H.; Makishima, H.; Maciejewski, J.P.; Imaizumi, M.; Rikiishi, T.; Sasahara, Y.; Kure, S.; Niihori, T.; et al. Casitas B-cell lymphoma mutation in childhood T-cell acute lymphoblastic leukemia. Leuk. Res. 2012, 36, 1009–1015. [Google Scholar] [CrossRef]
- Shiba, N.; Park, M.-J.; Taki, T.; Takita, J.; Hiwatari, M.; Kanazawa, T.; Sotomatsu, M.; Ishii, E.; Arakawa, H.; Ogawa, S.; et al. CBL mutations in infant acute lymphoblastic leukaemia. Br. J. Haematol. 2011, 156, 672–674. [Google Scholar] [CrossRef]
- Nicholson, L.; Knight, T.; Matheson, E.; Minto, L.; Case, M.; Sanichar, M.; Bomken, S.; Vormoor, J.; Hall, A.; Irving, J. Casitas B lymphoma mutations in childhood acute lymphoblastic leukemia. Genes Chromosom. Cancer 2011, 51, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.-H.C.; Krishnaswamy, S.; Nandi, S.; Kanteti, R.; Vora, S.; Onel, K.; Hasina, R.; Lo, F.-Y.; El-Hashani, E.; Cervantes, G.; et al. CBL Is Frequently Altered in Lung Cancers: Its Relationship to Mutations in MET and EGFR Tyrosine Kinases. PLoS ONE 2010, 5, e8972. [Google Scholar] [CrossRef] [PubMed]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the Hallmarks of Lung Adenocarcinoma with Massively Parallel Sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Navid, F.; Hiemenz, M.C.; Kaneko, M.; Zhou, S.; Saitta, S.C.; Biegel, J.A. Embryonal rhabdomyosarcoma in a patient with a germline CBL pathogenic variant. Cancer Genet. 2018, 231–232, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Hanson, H.L.; Wilson, M.J.; Short, J.P.; Chioza, B.; Crosby, A.H.; Nash, R.M.; Marks, K.J.; Mansour, S. GermlineCBLmutation associated with a noonan-like syndrome with primary lymphedema and teratoma associated with acquired uniparental isodisomy of chromosome 11q23. Am. J. Med. Genet. Part A 2014, 164, 1003–1009. [Google Scholar] [CrossRef] [PubMed]
- Daniels, S.R.; Liyasova, M.; Kales, S.; Nau, M.M.; Ryan, P.E.; Green, J.E.; Lipkowitz, S. Loss of function Cbl-c mutations in solid tumors. PLoS ONE 2019, 14, e0219143. [Google Scholar] [CrossRef]
- Lyle, C.L.; Belghasem, M.; Chitalia, V.C. c-Cbl: An Important Regulator and a Target in Angiogenesis and Tumorigenesis. Cells 2019, 8, 498. [Google Scholar] [CrossRef]
- Meyer, R.D.; Husain, D.; Rahimi, N. c-Cbl inhibits angiogenesis and tumor growth by suppressing activation of PLCγ1. Oncogene 2011, 30, 2198–2206. [Google Scholar] [CrossRef]
- Niemeyer, C.M. RAS diseases in children. Haematologica 2014, 99, 1653–1662. [Google Scholar] [CrossRef]
- Martinelli, S.; De Luca, A.; Stellacci, E.; Rossi, C.; Checquolo, S.; Lepri, F.; Caputo, V.; Silvano, M.; Buscherini, F.; Consoli, F.; et al. Heterozygous Germline Mutations in the CBL Tumor-Suppressor Gene Cause a Noonan Syndrome-like Phenotype. Am. J. Hum. Genet. 2010, 87, 250–257. [Google Scholar] [CrossRef]
- Tafazoli, A.; Eshraghi, P.; Pantaleoni, F.; Vakili, R.; Moghaddassian, M.; Ghahraman, M.; Muto, V.; Paolacci, S.; Golyan, F.F.; Abbaszadegan, M.R. Novel mutations and their genotype-phenotype correlations in patients with Noonan syndrome, using next-generation sequencing. Adv. Med. Sci. 2018, 63, 87–93. [Google Scholar] [CrossRef]
- Ngongang, C.; Agenbag, G.; Bope, C.D.; Esterhuizen, A.I.; Wonkam, A. Noonan Syndrome in South Africa: Clinical and Molecular Profiles. Front. Genet. 2019, 10, 333. [Google Scholar] [CrossRef]
- Van Der Burgt, I. Noonan syndrome. Orphanet J. Rare Dis. 2007, 2, 4. [Google Scholar] [CrossRef]
- Martinelli, S.; Stellacci, E.; Pannone, L.; D'Agostino, D.; Consoli, F.; Lissewski, C.; Silvano, M.; Cencelli, G.; Lepri, F.; Maitz, S.; et al. Molecular Diversity and Associated Phenotypic Spectrum of GermlineCBLMutations. Hum. Mutat. 2015, 36, 787–796. [Google Scholar] [CrossRef]
- Hyakuna, N.; Muramatsu, H.; Higa, T.; Chinen, Y.; Wang, X.; Kojima, S. Germline Mutation ofCBLIs Associated with Moyamoya Disease in a Child with Juvenile Myelomonocytic Leukemia and Noonan Syndrome-Like Disorder. Pediatr. Blood Cancer 2014, 62, 542–544. [Google Scholar] [CrossRef]
- Guey, S.; Grangeon, L.; Brunelle, F.; Bergametti, F.; Amiel, J.; Lyonnet, S.; Delaforge, A.; Arnould, M.; Desnous, B.; Bellesme, C.; et al. De novo mutations in CBL causing early-onset paediatric moyamoya angiopathy. J. Med. Genet. 2017, 54, 550–557. [Google Scholar] [CrossRef]
- Hong, Y.; Keylock, A.; Jensen, B.; Jacques, T.S.; Ogunbiyi, O.; Omoyinmi, E.; Saunders, D.; Mallick, A.A.; Tooley, M.; Newbury-Ecob, R.; et al. Cerebral arteriopathy associated with heterozygous variants in the casitas B-lineage lymphoma gene. Neurol. Genet. 2020, 6, e448. [Google Scholar] [CrossRef]
- Scott, R.M.; Smith, E.R. Moyamoya Disease and Moyamoya Syndrome. N. Engl. J. Med. 2009, 360, 1226–1237. [Google Scholar] [CrossRef]
- Naramura, M.; Jang, I.K.; Kole, H.; Huang, F.; Haines, D.; Gu, H. c-Cbl and Cbl-b regulate T cell responsiveness by promoting ligand-induced TCR down-modulation. Nat. Immunol. 2002, 3, 1192–1199. [Google Scholar] [CrossRef]
- Kitaura, Y.; Jang, I.K.; Wang, Y.; Han, Y.-C.; Inazu, T.; Cadera, E.J.; Schlissel, M.; Hardy, R.R.; Gu, H. Control of the B Cell-Intrinsic Tolerance Programs by Ubiquitin Ligases Cbl and Cbl-b. Immunity 2007, 26, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Coe, R.R.; McKinnon, M.; Tarailo-Graovac, M.; Ross, C.J.; Wasserman, W.; Friedman, J.M.; Rogers, P.C.; van Karnebeek, C.D. A case of splenomegaly in CBL syndrome. Eur. J. Med. Genet. 2017, 60, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Pathak, A.; NCI DCEG Cancer Genomics Research Laboratory; Pemov, A.; McMaster, M.L.; Dewan, R.; Ravichandran, S.; Pak, E.; Dutra, A.; Lee, H.J.; Vogt, A.; et al. Juvenile myelomonocytic leukemia due to a germline CBL Y371C mutation: 35-year follow-up of a large family. Qual. Life Res. 2015, 134, 775–787. [Google Scholar] [CrossRef]
- Wiel, L.C.; Pastore, S.; Taddio, A.; Tommasini, A. A Case of Uveitis in a Patient with Juvenile Myelomonocytic Leukemia Successfully Treated with Adalimumab. J. Pediatr. Hematol. 2019, 42, e373–e376. [Google Scholar] [CrossRef]
- Hadjadj, J.; Aladjidi, N.; Fernandes, H.; Leverger, G.; Magérus-Chatinet, A.; Mazerolles, F.; Stolzenberg, M.-C.; Jacques, S.; Picard, C.; Rosain, J.; et al. Pediatric Evans syndrome is associated with a high frequency of potentially damaging variants in immune genes. Blood 2019, 134, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Tejwani, N.; Tayal, P.; Mehta, A.; Dass, J. Somatic Hemizygous Y371H CBL Mutation with Loss of Heterozygosity Presenting with BENTA Type Lymphoid Proliferation. Indian J. Hematol. Blood Transfus. 2019, 36, 594–596. [Google Scholar] [CrossRef]
- Seaby, E.G.; Gilbert, R.D.; Andreoletti, G.; Pengelly, R.J.; Mercer, C.; Hunt, D.; Ennis, S. Unexpected Findings in a Child with Atypical Hemolytic Uremic Syndrome: An Example of How Genomics Is Changing the Clinical Diagnostic Paradigm. Front. Pediatr. 2017, 5, 113. [Google Scholar] [CrossRef]
- Quaio, C.R.; Carvalho, J.F.; da Silva, C.A.; Bueno, C.; Brasil, A.S.; Pereira, A.C.; Jorge, A.A.; Malaquias, A.C.; Kim, C.A.; Bertola, D.R. Autoimmune disease and multiple autoantibodies in 42 patients with RASopathies. Am. J. Med. Genet. Part A 2012, 158A, 1077–1082. [Google Scholar] [CrossRef]
- Bülow, L.; Lissewski, C.; Bressel, R.; Rauch, A.; Stark, Z.; Zenker, M.; Bartsch, O. Hydrops, fetal pleural effusions and chylothorax in three patients withCBLmutations. Am. J. Med. Genet. Part A 2014, 167, 394–399. [Google Scholar] [CrossRef]
- Weathington, N.M.; Mallampalli, R.K. Emerging therapies targeting the ubiquitin proteasome system in cancer. J. Clin. Investig. 2014, 124, 6–12. [Google Scholar] [CrossRef]
- Lub, S.; Maes, K.; Menu, E.; De Bruyne, E.; Vanderkerken, K.; Van Valckenborgh, E. Novel strategies to target the ubiquitin proteasome system in multiple myeloma. Oncotarget 2015, 7, 6521–6537. [Google Scholar] [CrossRef]
- Grosicki, S.; Simonova, M.; Spicka, I.; Pour, L.; Kriachok, I.; Gavriatopoulou, M.; Pylypenko, H.; Auner, H.W.; Leleu, X.; Doronin, V.; et al. Once-per-week selinexor, bortezomib, and dexamethasone versus twice-per-week bortezomib and dexamethasone in patients with multiple myeloma (BOSTON): A randomised, open-label, phase 3 trial. Lancet 2020, 396, 1563–1573. [Google Scholar] [CrossRef]
- Bunda, S.; Kang, M.W.; Sybingco, S.S.; Weng, J.; Favre, H.; Shin, D.H.; Irwin, M.S.; Loh, M.L.; Ohh, M. Inhibition of SRC Corrects GM-CSF Hypersensitivity That Underlies Juvenile Myelomonocytic Leukemia. Cancer Res. 2013, 73, 2540–2550. [Google Scholar] [CrossRef]
- Klimowicz, A.C.; Bisson, S.A.; Hans, K.; Long, E.M.; Hansen, H.C.; Robbins, S. The phytochemical piceatannol induces the loss of CBL and CBL-associated proteins. Mol. Cancer Ther. 2009, 8, 602–614. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leardini, D.; Messelodi, D.; Muratore, E.; Baccelli, F.; Bertuccio, S.N.; Anselmi, L.; Pession, A.; Masetti, R. Role of CBL Mutations in Cancer and Non-Malignant Phenotype. Cancers 2022, 14, 839. https://doi.org/10.3390/cancers14030839
Leardini D, Messelodi D, Muratore E, Baccelli F, Bertuccio SN, Anselmi L, Pession A, Masetti R. Role of CBL Mutations in Cancer and Non-Malignant Phenotype. Cancers. 2022; 14(3):839. https://doi.org/10.3390/cancers14030839
Chicago/Turabian StyleLeardini, Davide, Daria Messelodi, Edoardo Muratore, Francesco Baccelli, Salvatore N. Bertuccio, Laura Anselmi, Andrea Pession, and Riccardo Masetti. 2022. "Role of CBL Mutations in Cancer and Non-Malignant Phenotype" Cancers 14, no. 3: 839. https://doi.org/10.3390/cancers14030839
APA StyleLeardini, D., Messelodi, D., Muratore, E., Baccelli, F., Bertuccio, S. N., Anselmi, L., Pession, A., & Masetti, R. (2022). Role of CBL Mutations in Cancer and Non-Malignant Phenotype. Cancers, 14(3), 839. https://doi.org/10.3390/cancers14030839