
A Low Dose Combination of Withaferin A and Caffeic Acid Phenethyl Ester Possesses Anti-Metastatic Potential In Vitro: Molecular Targets and Mechanisms

 ,
,  , , , , ,
, , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Drug Treatment
2.2. Cell Viability Assay
2.3. Morphological Observations
2.4. In Vitro Scratch/Wound Healing Assay
2.5. In Vitro Cell Invasion Assay
2.6. Tube Formation Assay
2.7. cDNA Array
2.8. Flow Cytometry Analysis
2.9. Western Blot Analysis
2.10. Immunofluorescence
2.11. Immunoprecipitation
2.12. VEGF ELISA
2.13. Apoptosis Assay
2.14. Combination Index (CI) Analysis
2.15. RNA Extraction and Real Time Quantitative Polymerase Chain Reaction (RT-qPCR)
2.16. Statistical Analysis
3. Results
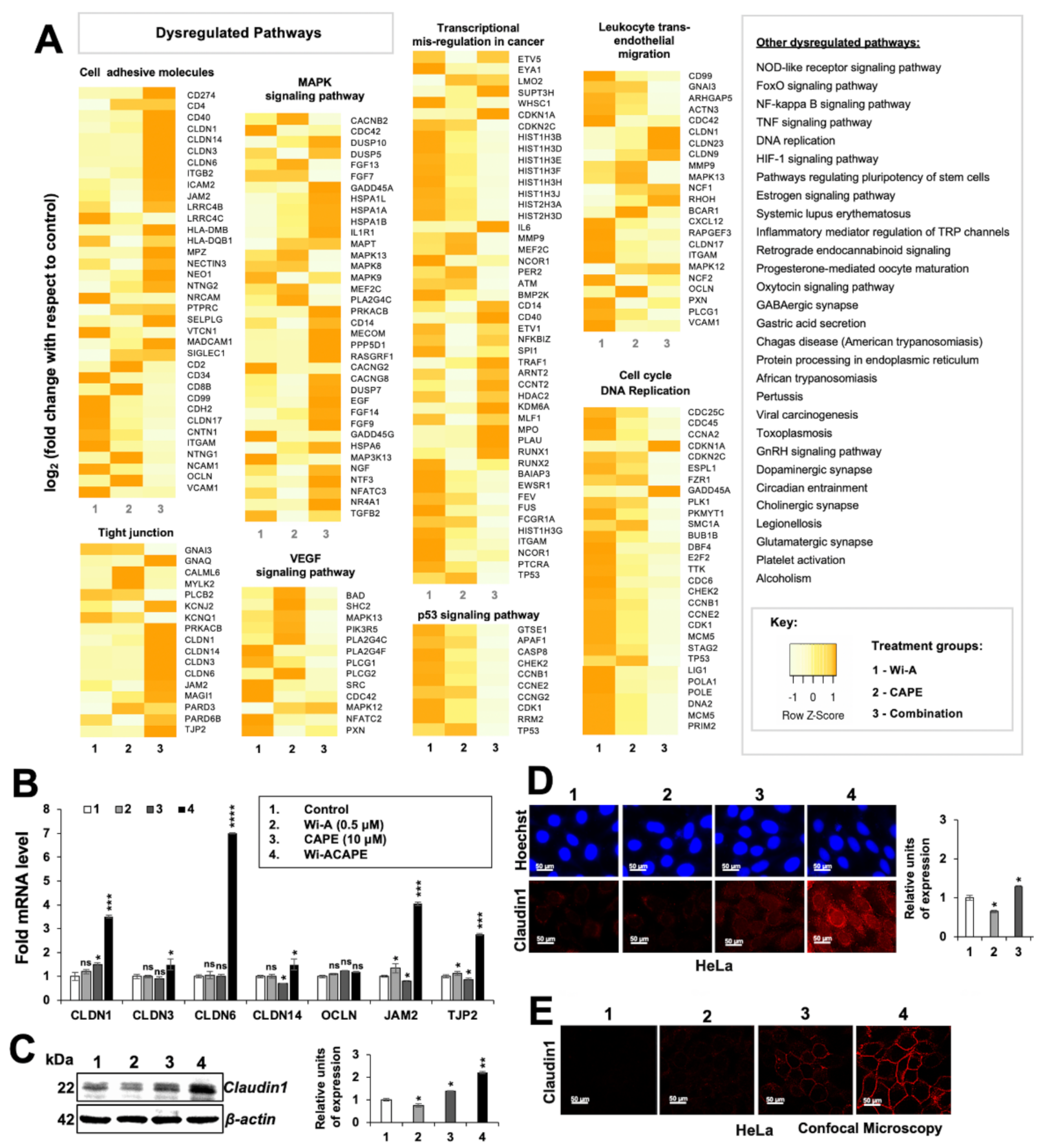
3.1. A Low Dose Combination of Wi-A and CAPE Inhibited Cancer Cell Migration, Invasion and Angiogenesis
3.2. Wi-ACAPE Treated Cells Showed Inactivation of Metastatic Signaling Pathways
3.3. Wi-ACAPE Dysregulated Tight Junction (TJ) Genes
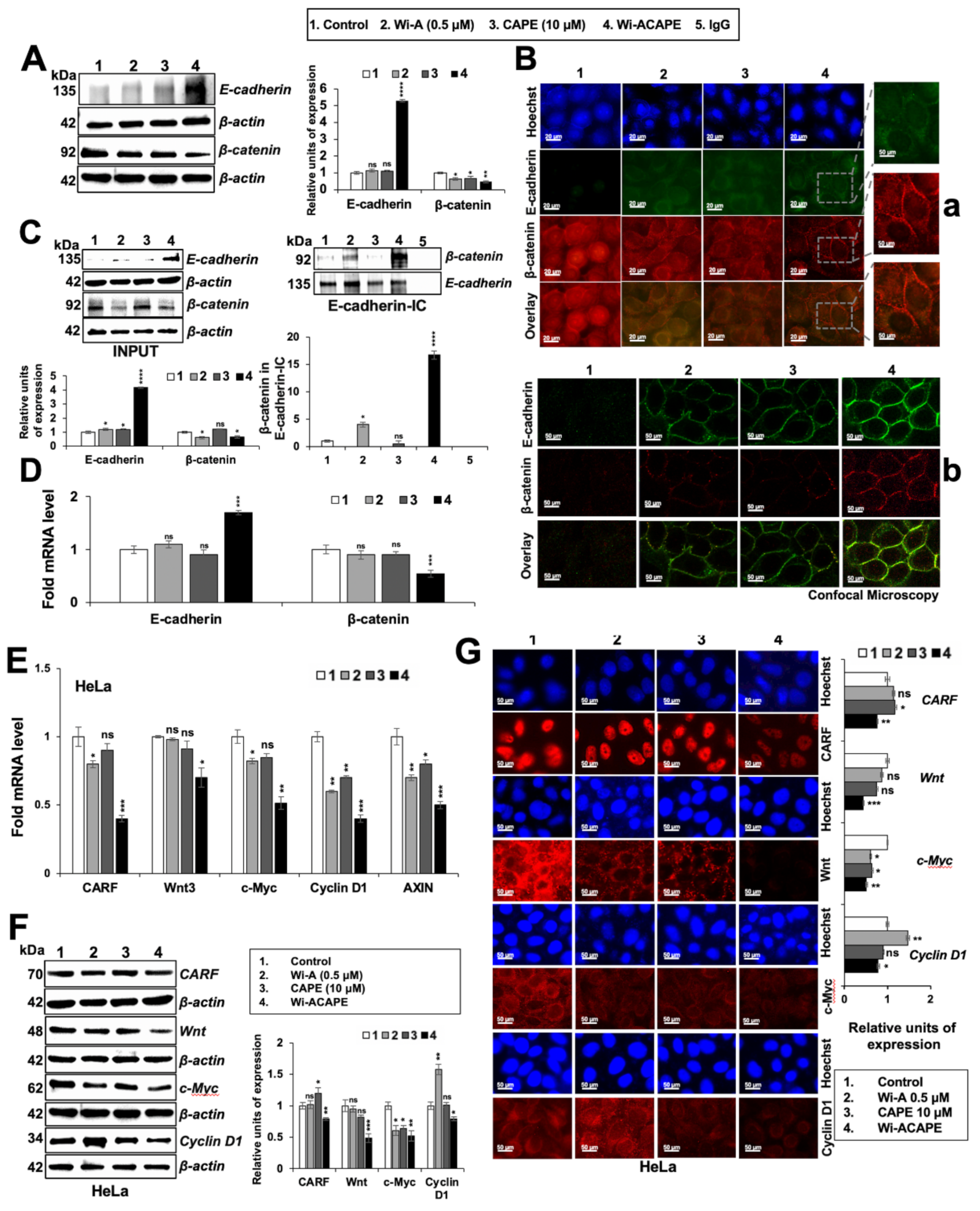
3.4. Wi-ACAPE Treated Cells Showed Upregulation of E-Cadherin and Downregulation of β-Catenin
3.5. Wi-ACAPE Caused Downregulation of Wnt/β-Catenin Mediated EMT Signaling
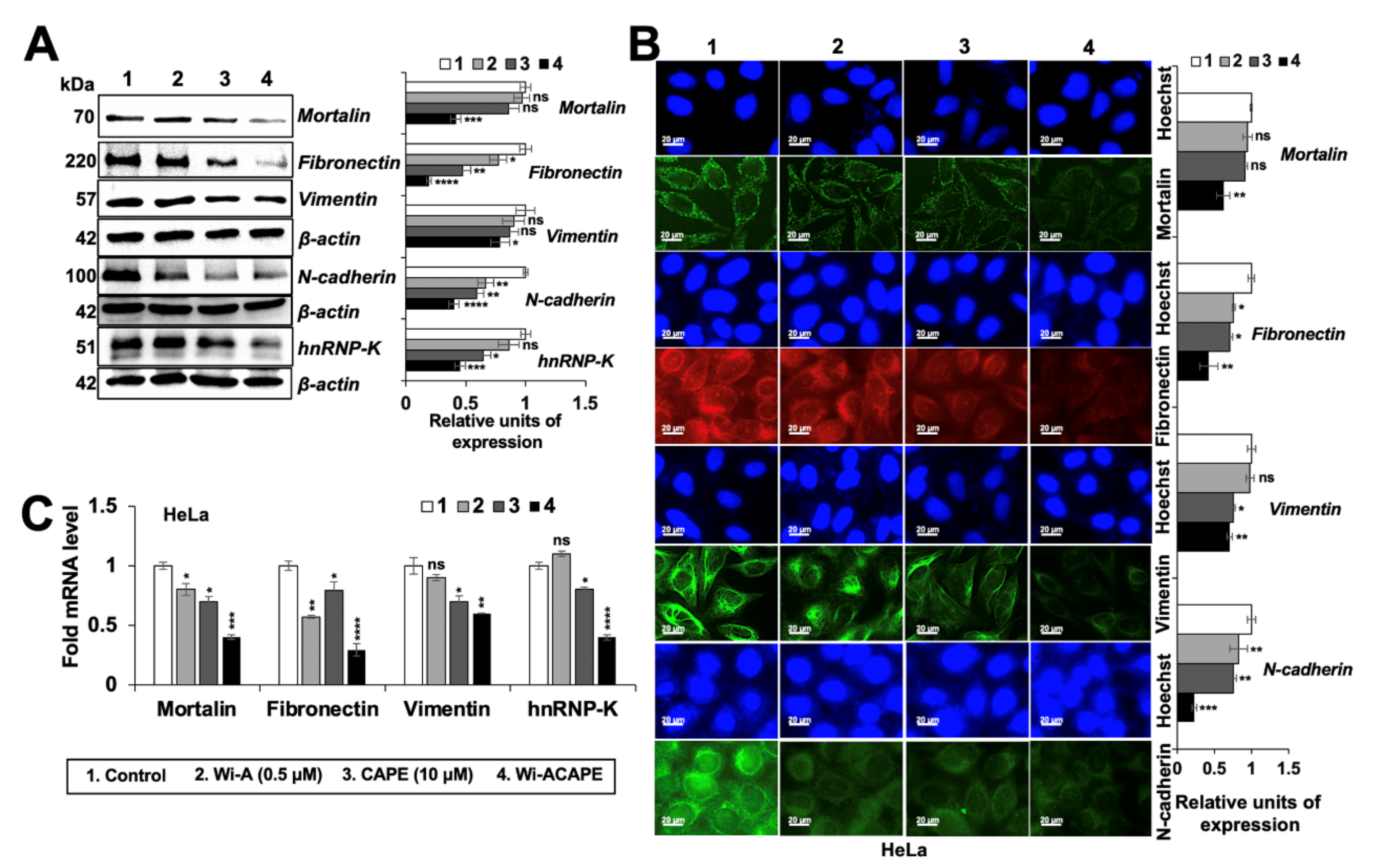
3.6. Wi-ACAPE Treated Cells Showed Reversal of EMT Signaling
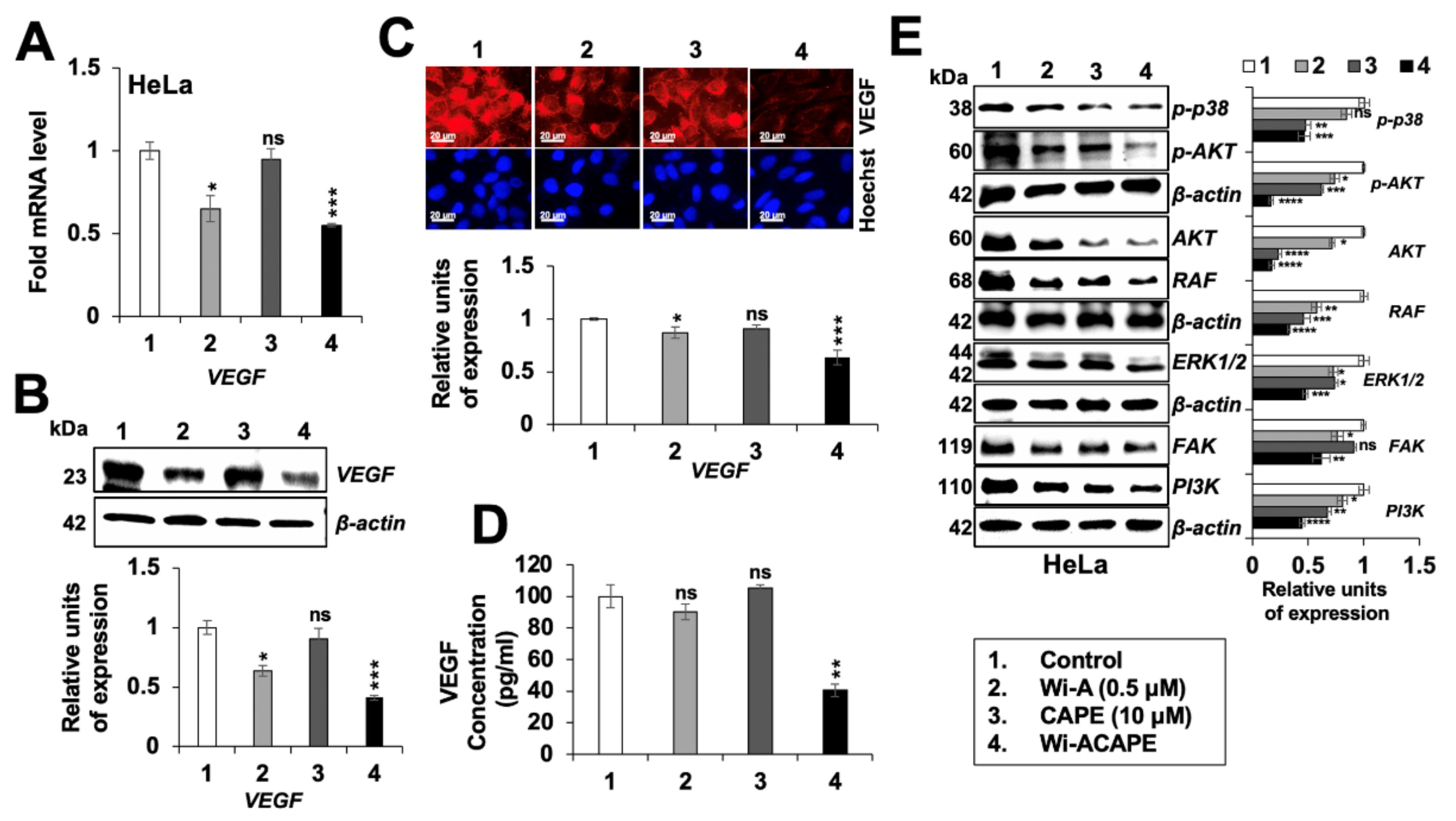
3.7. Wi-ACAPE Treated Cells Showed Inactivation of VEGF Signaling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chang, J.C. Cancer stem cells: Role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine 2016, 95, S20–S25. [Google Scholar] [CrossRef]
- Zirlik, K.; Duyster, J. Anti-angiogenics: Current situation and future perspectives. Oncol. Res. Treat. 2018, 41, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Houthuijzen, J.M.; Daenen, L.G.; Roodhart, J.M.; Voest, E.E. The role of mesenchymal stem cells in anti-cancer drug resistance and tumour progression. Br. J. Cancer 2012, 106, 1901–1906. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.; Piwnica-Worms, D.; Piwnica-Worms, H. Contribution of p53 to metastasis. Cancer Discov. 2014, 4, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Harakandi, C.; Nininahazwe, L.; Xu, H.; Liu, B.; He, C.; Zheng, Y.C.; Zhang, H. Recent advances on the intervention sites targeting USP7-MDM2-p53 in cancer therapy. Bioorg. Chem. 2021, 116, 105273. [Google Scholar] [CrossRef]
- Ciruelos Gil, E.M. Targeting the PI3K/AKT/mTOR pathway in estrogen receptor-positive breast cancer. Cancer Treat. Rev. 2014, 40, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal. Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef]
- Holmes, K.; Roberts, O.L.; Thomas, A.M.; Cross, M.J. Vascular endothelial growth factor receptor-2: Structure, function, intracellular signalling and therapeutic inhibition. Cell. Signal. 2007, 19, 2003–2012. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, T.; Zhang, S.; Wang, J.; Chen, Y.; Zhao, H.; Yang, Y.; Shi, S.; Chen, Q.; Liu, K. The Wnt signaling pathway in tumorigenesis, pharmacological targets, and drug development for cancer therapy. Biomark. Res. 2021, 9, 68. [Google Scholar] [CrossRef]
- Liu, W.; Vivian, C.J.; Brinker, A.E.; Hampton, K.R.; Lianidou, E.; Welch, D.R. Microenvironmental influences on metastasis suppressor expression and function during a metastatic cell’s journey. Cancer Microenviron. 2014, 7, 117–131. [Google Scholar] [CrossRef]
- Liu, Q.L.; Zhang, Z.; Wei, X.; Zhou, Z.G. Noncoding RNAs in tumor metastasis: Molecular and clinical perspectives. Cell. Mol. Life Sci. 2021, 78, 6823–6850. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.; Warmoes, M.O.; Shen, X.; Locasale, J.W. Epigenetics and cancer metabolism. Cancer Lett. 2015, 356, 309–314. [Google Scholar] [CrossRef]
- Wadhwa, R.; Takano, S.; Kaur, K.; Deocaris, C.C.; Pereira-Smith, O.M.; Reddel, R.R.; Kaul, S.C. Upregulation of mortalin/mthsp70/Grp75 contributes to human carcinogenesis. Int. J. Cancer 2006, 118, 2973–2980. [Google Scholar] [CrossRef]
- Na, Y.; Kaul, S.C.; Ryu, J.; Lee, J.S.; Ahn, H.M.; Kaul, Z.; Kalra, R.S.; Li, L.; Widodo, N.; Yun, C.O.; et al. Stress chaperone mortalin contributes to epithelial-mesenchymal transition and cancer metastasis. Cancer Res. 2016, 76, 2754–2765. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.O.; Bhargava, P.; Na, Y.; Lee, J.S.; Ryu, J.; Kaul, S.C.; Wadhwa, R. Relevance of mortalin to cancer cell stemness and cancer therapy. Sci. Rep. 2017, 7, 42016. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Ryu, J.; Gao, R.; Yaguchi, T.; Kaul, S.C.; Wadhwa, R.; Yun, C.O. Tumor suppression by apoptotic and anti-angiogenic effects of mortalin-targeting adeno-oncolytic virus. J. Gene Med. 2010, 12, 586–595. [Google Scholar] [CrossRef]
- Tania, M.; Khan, M.A.; Fu, J. Epithelial to mesenchymal transition inducing transcription factors and metastatic cancer. Tumour Biol. 2014, 35, 7335–7342. [Google Scholar] [CrossRef] [PubMed]
- Kalra, R.S.; Chaudhary, A.; Yoon, A.R.; Bhargava, P.; Omar, A.; Garg, S.; Yun, C.O.; Kaul, S.C.; Wadhwa, R. CARF enrichment promotes epithelial-mesenchymal transition via Wnt/beta-catenin signaling: Its clinical relevance and potential as a therapeutic target. Oncogenesis 2018, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.K.; Kwon, Y.; Jang, M.; Park, M.; Kim, J.; Cho, S.; Jang, D.G.; Lee, W.B.; Jung, S.H.; Choi, H.J.; et al. Beta-catenin activation down-regulates cell-cell junction-related genes and induces epithelial-to-mesenchymal transition in colorectal cancers. Sci. Rep. 2019, 9, 18440. [Google Scholar] [CrossRef] [PubMed]
- Kyuno, D.; Takasawa, A.; Kikuchi, S.; Takemasa, I.; Osanai, M.; Kojima, T. Role of tight junctions in the epithelial-to-mesenchymal transition of cancer cells. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183503. [Google Scholar] [CrossRef]
- Yang, N.; Chen, H.; Huang, Y.; Song, X.; Yang, P.; Zhang, S.; Yan, W.; Li, N.; Feng, Z. The role and significance of Wnt5a in regulating epithelial-mesenchymal transition in endometrioid adenocarcinoma. Cancer Manag. Res. 2021, 13, 6527–6535. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Liu, Z.; Niu, B.; Zhang, J.; Tan, T.K.; Lee, S.R.; Zhao, Y.; Harris, D.C.; Zheng, G. E-cadherin/beta-catenin complex and the epithelial barrier. J. Biomed. Biotechnol. 2011, 2011, 567305. [Google Scholar] [CrossRef] [PubMed]
- Mendonsa, A.M.; Na, T.Y.; Gumbiner, B.M. E-cadherin in contact inhibition and cancer. Oncogene 2018, 37, 4769–4780. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.J.; Song, P.P.; Zhou, H.; Shen, X.H.; Wang, J.G.; Ma, X.F.; Gu, Y.J.; Liu, D.D.; Feng, A.N.; Qian, X.Y.; et al. Role of epithelial-mesenchymal transition markers E-cadherin, N-cadherin, beta-catenin and ZEB2 in laryngeal squamous cell carcinoma. Oncol. Lett. 2018, 15, 3472–3481. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.M.; Zhang, H.; Han, X. Role of epithelial to mesenchymal transition proteins in gynecological cancers: Pathological and therapeutic perspectives. Tumor Biol. 2014, 35, 9523–9530. [Google Scholar] [CrossRef]
- Zhang, N.; Shao, F.; Jia, W. Upregulation of microfibrillar-associated protein 2 is closely associated with tumor angiogenesis and poor prognosis in hepatocellular carcinoma. Oncol. Lett. 2021, 22, 739. [Google Scholar] [CrossRef]
- Wang, B.; Li, X.; Liu, L.; Wang, M. Beta-catenin: Oncogenic role and therapeutic target in cervical cancer. Biol. Res. 2020, 53, 33. [Google Scholar] [CrossRef]
- Perez-Plasencia, C.; Duenas-Gonzalez, A.; Alatorre-Tavera, B. Second hit in cervical carcinogenesis process: Involvement of wnt/beta catenin pathway. Int. Arch. Med. 2008, 1, 10. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X. Targeting the Wnt/beta-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef]
- Wei, B.; Cao, J.; Tian, J.H.; Yu, C.Y.; Huang, Q.; Yu, J.J.; Ma, R.; Wang, J.; Xu, F.; Wang, L.B. Mortalin maintains breast cancer stem cells stemness via activation of Wnt/GSK3beta/beta-catenin signaling pathway. Am. J. Cancer Res. 2021, 11, 2696–2716. [Google Scholar]
- Chuai, Y.; Rizzuto, I.; Zhang, X.; Li, Y.; Dai, G.; Otter, S.J.; Bharathan, R.; Stewart, A.; Wang, A. Vascular endothelial growth factor (VEGF) targeting therapy for persistent, recurrent, or metastatic cervical cancer. Cochrane Database Syst. Rev. 2021, 3, CD013348. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhao, H.; Liu, Y.; Zuo, Y.; Xu, Q.; Liu, L.; Li, X.; Zhu, H.; Zhang, Y.; Zhang, S.; et al. Chronic stress activates plexinA1/VEGFR2-JAK2-STAT3 in vascular endothelial cells to promote angiogenesis. Front. Oncol. 2021, 11, 709057. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wang, B.; Fu, Z.; Wei, J.; Lu, W. Hypoxic microenvironment induces EMT and upgrades stem-like properties of gastric cancer cells. Technol. Cancer Res. Treat. 2016, 15, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, I.; Thavathiru, E.; Ramalingam, S.; Natarajan, G.; Mills, W.; Benbrook, D.; Zuna, R.; Lightfoot, S.; Reis, A.; Anant, S. Wnt inhibitory factor 1 induces apoptosis and inhibits cervical cancer growth, invasion and angiogenesis in vivo. Oncogene 2012, 31, 2725–2737. [Google Scholar] [CrossRef] [PubMed]
- Widodo, N.; Kaur, K.; Shrestha, B.G.; Takagi, Y.; Ishii, T.; Wadhwa, R.; Kaul, S.C. Selective killing of cancer cells by leaf extract of Ashwagandha: Identification of a tumor-inhibitory factor and the first molecular insights to its effect. Clin. Cancer Res. 2007, 13, 2298–2306. [Google Scholar] [CrossRef]
- Sari, A.N.; Bhargava, P.; Dhanjal, J.K.; Putri, J.F.; Radhakrishnan, N.; Shefrin, S.; Ishida, Y.; Terao, K.; Sundar, D.; Kaul, S.C.; et al. Combination of withaferin-A and CAPE provides superior anticancer potency: Bioinformatics and experimental evidence to their molecular targets and mechanism of action. Cancers 2020, 12, 1160. [Google Scholar] [CrossRef]
- Kyakulaga, A.H.; Aqil, F.; Munagala, R.; Gupta, R.C. Withaferin A inhibits epithelial to mesenchymal transition in non-small cell lung cancer cells. Sci. Rep. 2018, 8, 15737. [Google Scholar] [CrossRef]
- Kakar, S.S.; Parte, S.; Kelsey Carter, I.G.J.; Worth, C.; Rameshwar, P.; Ratajczak, M.Z. Withaferin A (WFA) inhibits tumor growth and metastasis by targeting ovarian cancer stem cells. Oncotarget 2017, 8, 74494–74505. [Google Scholar] [CrossRef]
- Nagy, Z.; Cheung, B.B.; Tsang, W.; Tan, O.; Herath, M.; Ciampa, O.C.; Shadma, F.; Carter, D.R.; Marshall, G.M. Withaferin A activates TRIM16 for its anti-cancer activity in melanoma. Sci. Rep. 2020, 10, 19724. [Google Scholar] [CrossRef]
- Ting, L.-L.; Chou, A.S.-B.; Hsieh, C.-H.; Hsiung, S.-C.; Pang, S.-T.; Liao, S.-K. Withaferin A targeting both cancer stem cells and metastatic cancer stem cells in the UP-LN1 carcinoma cell model. J. Cancer Metastasis Treat. 2016, 2, 29–40. [Google Scholar]
- Thaiparambil, J.T.; Bender, L.; Ganesh, T.; Kline, E.; Patel, P.; Liu, Y.; Tighiouart, M.; Vertino, P.M.; Harvey, R.D.; Garcia, A.; et al. Withaferin A inhibits breast cancer invasion and metastasis at sub-cytotoxic doses by inducing vimentin disassembly and serine 56 phosphorylation. Int. J. Cancer 2011, 129, 2744–2755. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.Y.; Yang, H.W.; Chu, Y.H.; Chang, Y.C.; Hsieh, M.J.; Chou, M.Y.; Yeh, K.T.; Lin, Y.M.; Yang, S.F.; Lin, C.W. Caffeic acid phenethyl ester inhibits oral cancer cell metastasis by regulating matrix metalloproteinase-2 and the mitogen-activated protein kinase pathway. Evid. Based Complement. Alternat. Med. 2012, 2012, 732578. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Feng, G.; Wu, L.; Zhong, S.; Gao, X.; Tong, Y.; Cui, W.; Qin, Y.; Xu, W.; Xiao, X.; et al. Caffeic acid phenethyl ester suppressed growth and metastasis of nasopharyngeal carcinoma cells by inactivating the NF-kappaB pathway. Drug Des. Dev. Ther. 2019, 13, 1335–1345. [Google Scholar] [CrossRef] [PubMed]
- Kaul, Z.; Yaguchi, T.; Harada, J.I.; Ikeda, Y.; Hirano, T.; Chiura, H.X.; Kaul, S.C.; Wadhwa, R. An antibody-conjugated internalizing quantum dot suitable for long-term live imaging of cells. Biochem. Cell Biol. 2007, 85, 133–140. [Google Scholar] [CrossRef]
- Wadhwa, R.; Sugihara, T.; Hasan, M.K.; Duncan, E.L.; Taira, K.; Kaul, S.C. A novel putative collaborator of p19ARF. Exp. Gerontol. 2003, 38, 245–252. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Nath, B.; Raza, A.; Sethi, V.; Dalal, A.; Ghosh, S.S.; Biswas, G. Understanding flow dynamics, viability and metastatic potency of cervical cancer (HeLa) cells through constricted microchannel. Sci. Rep. 2018, 8, 17357. [Google Scholar] [CrossRef]
- Li, P.; Mao, Z.; Peng, Z.; Zhou, L.; Chen, Y.; Huang, P.-H.; Truica, C.I.; Drabick, J.J.; El-Deiry, W.S.; Dao, M. Acoustic separation of circulating tumor cells. Proc. Natl. Acad. Sci. USA 2015, 112, 4970–4975. [Google Scholar] [CrossRef]
- Paterlini-Brechot, P.; Benali, N.L. Circulating tumor cells (CTC) detection: Clinical impact and future directions. Cancer Lett. 2007, 253, 180–204. [Google Scholar] [CrossRef]
- Zhao, Y.; Yao, R.; Ouyang, L.; Ding, H.; Zhang, T.; Zhang, K.; Cheng, S.; Sun, W. Three-dimensional printing of Hela cells for cervical tumor model in vitro. Biofabrication 2014, 6, 35001. [Google Scholar] [CrossRef]
- Tu, Y.F.; Kaipparettu, B.A.; Ma, Y.; Wong, L.J. Mitochondria of highly metastatic breast cancer cell line MDA-MB-231 exhibits increased autophagic properties. Biochim. Biophys. Acta 2011, 1807, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chan, T.; Mak, M. Morphodynamic signatures of MDA-MB-231 single cells and cell doublets undergoing invasion in confined microenvironments. Sci. Rep. 2021, 11, 6529. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-L.; Chou, C.-K.; Kim, M.; Vasisht, R.; Kuo, Y.-A.; Ang, P.; Liu, C.; Perillo, E.P.; Chen, Y.-A.; Blocher, K. Assessing metastatic potential of breast cancer cells based on EGFR dynamics. Sci. Rep. 2019, 9, 3395. [Google Scholar]
- Zuo, X.-X.; Yang, Y.; Zhang, Y.; Zhang, Z.-G.; Wang, X.-F.; Shi, Y.-G. Platelets promote breast cancer cell MCF-7 metastasis by direct interaction: Surface integrin α2β1-contacting-mediated activation of Wnt-β-catenin pathway. Cell Commun. Signal. 2019, 17, 142. [Google Scholar] [CrossRef] [PubMed]
- Elwakeel, A.; Soudan, H.; Eldoksh, A.; Shalaby, M.; Eldemellawy, M.; Ghareeb, D.; Abouseif, M.; Fayad, A.; Hassan, M.; Saeed, H. Implementation of the Chou-Talalay method for studying the in vitro pharmacodynamic interactions of binary and ternary drug combinations on MDA-MB-231 triple negative breast cancer cells. Synergy 2019, 8, 100047. [Google Scholar] [CrossRef]
- Qadir, A.; Wahid, M.; Asif, M.; Roome, T. Synergistic effect of bevacizumab and celecoxib on angiogenesis in vitro using human umbilical vein endothelial cells. Int. J. Clin. Pharmacol. Ther. 2020, 58, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Yukawa, H.; Suzuki, K.; Aoki, K.; Arimoto, T.; Yasui, T.; Kaji, N.; Ishikawa, T.; Ochiya, T.; Baba, Y. Imaging of angiogenesis of human umbilical vein endothelial cells by uptake of exosomes secreted from hepatocellular carcinoma cells. Sci. Rep. 2018, 8, 6765. [Google Scholar] [CrossRef]
- Huang, B.; Huang, M.; Li, Q. Cancer-associated fibroblasts promote angiogenesis of hepatocellular carcinoma by VEGF-mediated EZH2/VASH1 pathway. Technol. Cancer Res. Treat. 2019, 18, 1533033819879905. [Google Scholar] [CrossRef]
- Chen, Z.; Htay, A.; Dos Santos, W.; Gillies, G.T.; Fillmore, H.L.; Sholley, M.M.; Broaddus, W.C. In vitro angiogenesis by human umbilical vein endothelial cells (HUVEC) induced by three-dimensional co-culture with glioblastoma cells. J. Neurooncol. 2009, 92, 121–128. [Google Scholar] [CrossRef]
- Martin, T.A. The role of tight junctions in cancer metastasis. Semin. Cell Dev. Biol. 2014, 36, 224–231. [Google Scholar] [CrossRef]
- Bhat, A.A.; Uppada, S.; Achkar, I.W.; Hashem, S.; Yadav, S.K.; Shanmugakonar, M.; Al-Naemi, H.A.; Haris, M.; Uddin, S. Tight junction proteins and signaling pathways in cancer and inflammation: A functional crosstalk. Front. Physiol. 2018, 9, 1942. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.C.; Pan, S.H.; Yang, S.C.; Yu, S.L.; Che, T.F.; Lin, C.W.; Tsai, M.S.; Chang, G.C.; Wu, C.H.; Wu, Y.Y.; et al. Claudin-1 is a metastasis suppressor and correlates with clinical outcome in lung adenocarcinoma. Am. J. Respir. Crit. Care Med. 2009, 179, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.A.; Syed, N.; Therachiyil, L.; Nisar, S.; Hashem, S.; Macha, M.A.; Yadav, S.K.; Krishnankutty, R.; Muralitharan, S.; Al-Naemi, H.; et al. Claudin-1, a double-edged sword in cancer. Int. J. Mol. Sci. 2020, 21, 569. [Google Scholar] [CrossRef] [PubMed]
- Heuberger, J.; Birchmeier, W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002915. [Google Scholar] [CrossRef] [PubMed]
- Delmas, A.L.; Riggs, B.M.; Pardo, C.E.; Dyer, L.M.; Darst, R.P.; Izumchenko, E.G.; Monroe, M.; Hakam, A.; Kladde, M.P.; Siegel, E.M. WIF1 is a frequent target for epigenetic silencing in squamous cell carcinoma of the cervix. Carcinogenesis 2011, 32, 1625–1633. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Yu, Y.; Inoue, A.; Widodo, N.; Kaul, S.C.; Wadhwa, R. Heterogeneous nuclear ribonucleoprotein K (hnRNP-K) promotes tumor metastasis by induction of genes involved in extracellular matrix, cell movement, and angiogenesis. J. Biol. Chem. 2013, 288, 15046–15056. [Google Scholar] [CrossRef]
- Lohela, M.; Bry, M.; Tammela, T.; Alitalo, K. VEGFs and receptors involved in angiogenesis versus lymphangiogenesis. Curr. Opin. Cell Biol. 2009, 21, 154–165. [Google Scholar] [CrossRef]
- Bielenberg, D.R.; Zetter, B.R. The contribution of angiogenesis to the process of metastasis. Cancer J. 2015, 21, 267–273. [Google Scholar] [CrossRef]
- Gandalovicova, A.; Rosel, D.; Fernandes, M.; Vesely, P.; Heneberg, P.; Cermak, V.; Petruzelka, L.; Kumar, S.; Sanz-Moreno, V.; Brabek, J. Migrastatics-anti-metastatic and anti-invasion drugs: Promises and challenges. Trends Cancer 2017, 3, 391–406. [Google Scholar] [CrossRef]
- Steeg, P.S. Targeting metastasis. Nat. Rev. Cancer 2016, 16, 201–218. [Google Scholar] [CrossRef]
- Howard, S.; Deroo, T.; Fujita, Y.; Itasaki, N. A positive role of cadherin in Wnt/beta-catenin signalling during epithelial-mesenchymal transition. PLoS ONE 2011, 6, e23899. [Google Scholar] [CrossRef] [PubMed]
- Fantozzi, A.; Gruber, D.C.; Pisarsky, L.; Heck, C.; Kunita, A.; Yilmaz, M.; Meyer-Schaller, N.; Cornille, K.; Hopfer, U.; Bentires-Alj, M.; et al. VEGF-mediated angiogenesis links EMT-induced cancer stemness to tumor initiation. Cancer Res. 2014, 74, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, H.; Xia, F.; Zhang, Q.W.; Zhang, Y.Y.; Zhao, Q.; Chao, Z.H.; Jiang, Z.W.; Jiang, C.C. Epithelial-mesenchymal transition is necessary for acquired resistance to cisplatin and increases the metastatic potential of nasopharyngeal carcinoma cells. Int. J. Mol. Med. 2014, 33, 151–159. [Google Scholar] [CrossRef]
- Garg, S.; Huifu, H.; Kumari, A.; Sundar, D.; Kaul, S.C.; Wadhwa, R. Induction of senescence in cancer cells by a novel combination of cucurbitacin B and withanone: Molecular mechanism and therapeutic potential. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.; Garg, S.; Liu, Y.; Afzal, S.; Gao, R.; Yun, C.O.; Kaul, S.C.; Wadhwa, R. Identification and functional characterization of anti-metastasis and anti-angiogenic activities of triethylene glycol derivatives. Front. Oncol. 2018, 8, 552. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, Y.; Cheryan, V.T.; Wu, W.; Cui, C.Q.; Polin, L.A.; Pass, H.I.; Dou, Q.P.; Rishi, A.K.; Wali, A. Withaferin A inhibits the proteasome activity in mesothelioma in vitro and in vivo. PLoS ONE 2012, 7, e41214. [Google Scholar] [CrossRef]
- Lee, J.; Sehrawat, A.; Singh, S.V. Withaferin A causes activation of Notch2 and Notch4 in human breast cancer cells. Breast Cancer Res. Treat. 2012, 136, 45–56. [Google Scholar] [CrossRef]
- Gao, R.; Shah, N.; Lee, J.S.; Katiyar, S.P.; Li, L.; Oh, E.; Sundar, D.; Yun, C.O.; Wadhwa, R.; Kaul, S.C. Withanone-rich combination of Ashwagandha withanolides restricts metastasis and angiogenesis through hnRNP-K. Mol. Cancer Ther. 2014, 13, 2930–2940. [Google Scholar] [CrossRef]
- Chaudhary, A.; Kalra, R.S.; Malik, V.; Katiyar, S.P.; Sundar, D.; Kaul, S.C.; Wadhwa, R. 2, 3-Dihydro-3beta-methoxy withaferin-A lacks anti-metastasis potency: Bioinformatics and experimental evidences. Sci. Rep. 2019, 9, 17344. [Google Scholar] [CrossRef]
- Widodo, N.; Priyandoko, D.; Shah, N.; Wadhwa, R.; Kaul, S.C. Selective killing of cancer cells by Ashwagandha leaf extract and its component withanone involves ROS signaling. PLoS ONE 2010, 5, e13536. [Google Scholar] [CrossRef]
- Kakar, S.S.; Ratajczak, M.Z.; Powell, K.S.; Moghadamfalahi, M.; Miller, D.M.; Batra, S.K.; Singh, S.K. Withaferin a alone and in combination with cisplatin suppresses growth and metastasis of ovarian cancer by targeting putative cancer stem cells. PloS One 2014, 9, e107596. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, R.; Nigam, N.; Bhargava, P.; Dhanjal, J.K.; Goyal, S.; Grover, A.; Sundar, D.; Ishida, Y.; Terao, K.; Kaul, S.C. Molecular characterization and enhancement of anticancer activity of caffeic acid phenethyl ester by gamma cyclodextrin. J. Cancer 2016, 7, 1755–1771. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Bhargava, P.; Yu, Y.; Sari, A.N.; Zhang, H.; Ishii, N.; Yan, K.; Zhang, Z.; Ishida, Y.; Terao, K.; et al. Novel caffeic acid phenethyl ester-mortalin antibody nanoparticles offer enhanced selective cytotoxicity to cancer cells. Cancers 2020, 12, 2370. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Gao, R.; Shah, N.; Bhargava, P.; Furune, T.; Kaul, S.C.; Terao, K.; Wadhwa, R. Anticancer activity in honeybee propolis: Functional insights to the role of caffeic acid phenethyl ester and its complex with gamma-cyclodextrin. Integr. Cancer Ther. 2018, 17, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Tseng, J.C.; Lin, C.Y.; Su, L.C.; Fu, H.H.; Yang, S.D.; Chuu, C.P. CAPE suppresses migration and invasion of prostate cancer cells via activation of non-canonical Wnt signaling. Oncotarget 2016, 7, 38010–38024. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.Y.; Kao, C.L.; Tsai, P.H.; Tsai, T.H.; Chiou, S.H.; Wu, W.F.; Ku, H.H.; Wong, T.T. Caffeic acid phenethyl ester preferentially enhanced radiosensitizing and increased oxidative stress in medulloblastoma cell line. Childs Nerv. Syst. 2008, 24, 987–994. [Google Scholar] [CrossRef]
- Gheldof, A.; Berx, G. Cadherins and epithelial-to-mesenchymal transition. Prog. Mol. Biol. Transl. Sci. 2013, 116, 317–336. [Google Scholar] [CrossRef]
- Song, Y.; Ye, M.; Zhou, J.; Wang, Z.W.; Zhu, X. Restoring E-cadherin expression by natural compounds for anticancer therapies in genital and urinary cancers. Mol. Ther. Oncolytics 2019, 14, 130–138. [Google Scholar] [CrossRef]
- Chu, S.C.; Yu, C.C.; Hsu, L.S.; Chen, K.S.; Su, M.Y.; Chen, P.N. Berberine reverses epithelial-to-mesenchymal transition and inhibits metastasis and tumor-induced angiogenesis in human cervical cancer cells. Mol. Pharmacol. 2014, 86, 609–623. [Google Scholar] [CrossRef]
- Tyszka-Czochara, M.; Lasota, M.; Majka, M. Caffeic acid and metformin inhibit invasive phenotype induced by TGF-beta1 in C-4I and HTB-35/SiHa human cervical squamous carcinoma cells by acting on different molecular targets. Int. J. Mol. Sci. 2018, 19, 266. [Google Scholar] [CrossRef]
- Han, S.T.; Kim, J.S.; Lee, J.Y.; Kim, M.K.; Yoo, J.S.; Han, B.G.; Choi, S.O.; Yang, J.W. The mechanism of attenuation of epithelial-mesenchymal transition by a phosphodiesterase 5 inhibitor via renal klotho expression. Clin. Exp. Pharmacol. Physiol. 2018, 45, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Pan, S.T.; Qiu, J.X. Effect of plumbagin on epithelial-mesenchymal transition and underlying mechanisms in human tongue squamous cell carcinoma cells. Zhonghua Kou Qiang Yi Xue Za Zhi 2017, 52, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.S.; Shi, M.D.; Chien, C.S.; Shih, Y.W. Pinocembrin suppresses TGF-beta1-induced epithelial-mesenchymal transition and metastasis of human Y-79 retinoblastoma cells through inactivating alphavbeta3 integrin/FAK/p38alpha signaling pathway. Cell Biosci. 2014, 4, 41. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Meng, Z.; Zhang, R.; Wu, X.; Zhang, M.; Zhang, S.; Jin, T. Prognostic value of mortalin correlates with roles in epithelial-mesenchymal transition and angiogenesis in lung adenocarcinoma. Carcinogenesis 2021, bgab081. [Google Scholar] [CrossRef]
- Hasan, M.K.; Yaguchi, T.; Sugihara, T.; Kumar, P.K.; Taira, K.; Reddel, R.R.; Kaul, S.C.; Wadhwa, R. CARF is a novel protein that cooperates with mouse p19ARF (human p14ARF) in activating p53. J. Biol. Chem. 2002, 277, 37765–37770. [Google Scholar] [CrossRef]
- Hasan, M.K.; Yaguchi, T.; Minoda, Y.; Hirano, T.; Taira, K.; Wadhwa, R.; Kaul, S.C. Alternative reading frame protein (ARF)-independent function of CARF (collaborator of ARF) involves its interactions with p53: Evidence for a novel p53-activation pathway and its negative feedback control. Biochem. J. 2004, 380, 605–610. [Google Scholar] [CrossRef]
- Hasan, M.K.; Yaguchi, T.; Harada, J.I.; Hirano, T.; Wadhwa, R.; Kaul, S.C. CARF (collaborator of ARF) interacts with HDM2: Evidence for a novel regulatory feedback regulation of CARF-p53-HDM2-p21WAF1 pathway. Int. J. Oncol. 2008, 32, 663–671. [Google Scholar] [CrossRef]
- Cheung, C.T.; Singh, R.; Yoon, A.R.; Hasan, M.K.; Yaguchi, T.; Kaul, S.C.; Yun, C.O.; Wadhwa, R. Molecular characterization of apoptosis induced by CARF silencing in human cancer cells. Cell Death Differ. 2011, 18, 589–601. [Google Scholar] [CrossRef]
- Eskander, R.N.; Tewari, K.S. Targeting angiogenesis in advanced cervical cancer. Ther. Adv. Med. Oncol. 2014, 6, 280–292. [Google Scholar] [CrossRef]
- Lee, N.; Kim, S.I.; Lee, M.; Kim, H.S.; Kim, J.W.; Park, N.H.; Song, Y.S. Bevacizumab efficacy and recurrence pattern of persistent and metastatic cervical cancer. In Vivo 2019, 33, 863–868. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sari, A.N.; Dhanjal, J.K.; Elwakeel, A.; Kumar, V.; Meidinna, H.N.; Zhang, H.; Ishida, Y.; Terao, K.; Sundar, D.; Kaul, S.C.; et al. A Low Dose Combination of Withaferin A and Caffeic Acid Phenethyl Ester Possesses Anti-Metastatic Potential In Vitro: Molecular Targets and Mechanisms. Cancers 2022, 14, 787. https://doi.org/10.3390/cancers14030787
Sari AN, Dhanjal JK, Elwakeel A, Kumar V, Meidinna HN, Zhang H, Ishida Y, Terao K, Sundar D, Kaul SC, et al. A Low Dose Combination of Withaferin A and Caffeic Acid Phenethyl Ester Possesses Anti-Metastatic Potential In Vitro: Molecular Targets and Mechanisms. Cancers. 2022; 14(3):787. https://doi.org/10.3390/cancers14030787
Chicago/Turabian StyleSari, Anissa Nofita, Jaspreet Kaur Dhanjal, Ahmed Elwakeel, Vipul Kumar, Hazna Noor Meidinna, Huayue Zhang, Yoshiyuki Ishida, Keiji Terao, Durai Sundar, Sunil C. Kaul, and et al. 2022. "A Low Dose Combination of Withaferin A and Caffeic Acid Phenethyl Ester Possesses Anti-Metastatic Potential In Vitro: Molecular Targets and Mechanisms" Cancers 14, no. 3: 787. https://doi.org/10.3390/cancers14030787
APA StyleSari, A. N., Dhanjal, J. K., Elwakeel, A., Kumar, V., Meidinna, H. N., Zhang, H., Ishida, Y., Terao, K., Sundar, D., Kaul, S. C., & Wadhwa, R. (2022). A Low Dose Combination of Withaferin A and Caffeic Acid Phenethyl Ester Possesses Anti-Metastatic Potential In Vitro: Molecular Targets and Mechanisms. Cancers, 14(3), 787. https://doi.org/10.3390/cancers14030787