
Dissecting the Mechanism of Action of Spiperone—A Candidate for Drug Repurposing for Colorectal Cancer

 , , , ,
, , , ,  ,
,  ,
,  ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. siRNA Transfections
2.3. MTT (Thiazolyl Blue Tetrazolium Bromide) Viability Assay and Viability Rescue Experiments
2.4. Extreme Limiting Dilution Assay (ELDA)
2.5. Apoptosis Assay
2.6. Cell Cycle Assay
2.7. Phospholipidosis Assay
2.8. Intracellular Calcium Measurements
2.9. Immunofluorescence Microscopy Analysis
2.10. Western Blotting
2.11. Mitochondrial Membrane Potential Analysis
2.12. Intact Cell Respiration Using High-Resolution Respirometry
2.13. RNA Extraction and Real-Time PCR
2.14. XBP1 Splicing Variant Polymerase Chain Reaction (PCR)
2.15. Transcriptomic Analysis (RNA Extraction, Sequencing, and Bioinformatic Analysis)
2.16. Lipidomic Analysis (Extraction, Analysis, and Processing)
2.17. Depmap Omics Data Analysis
3. Results
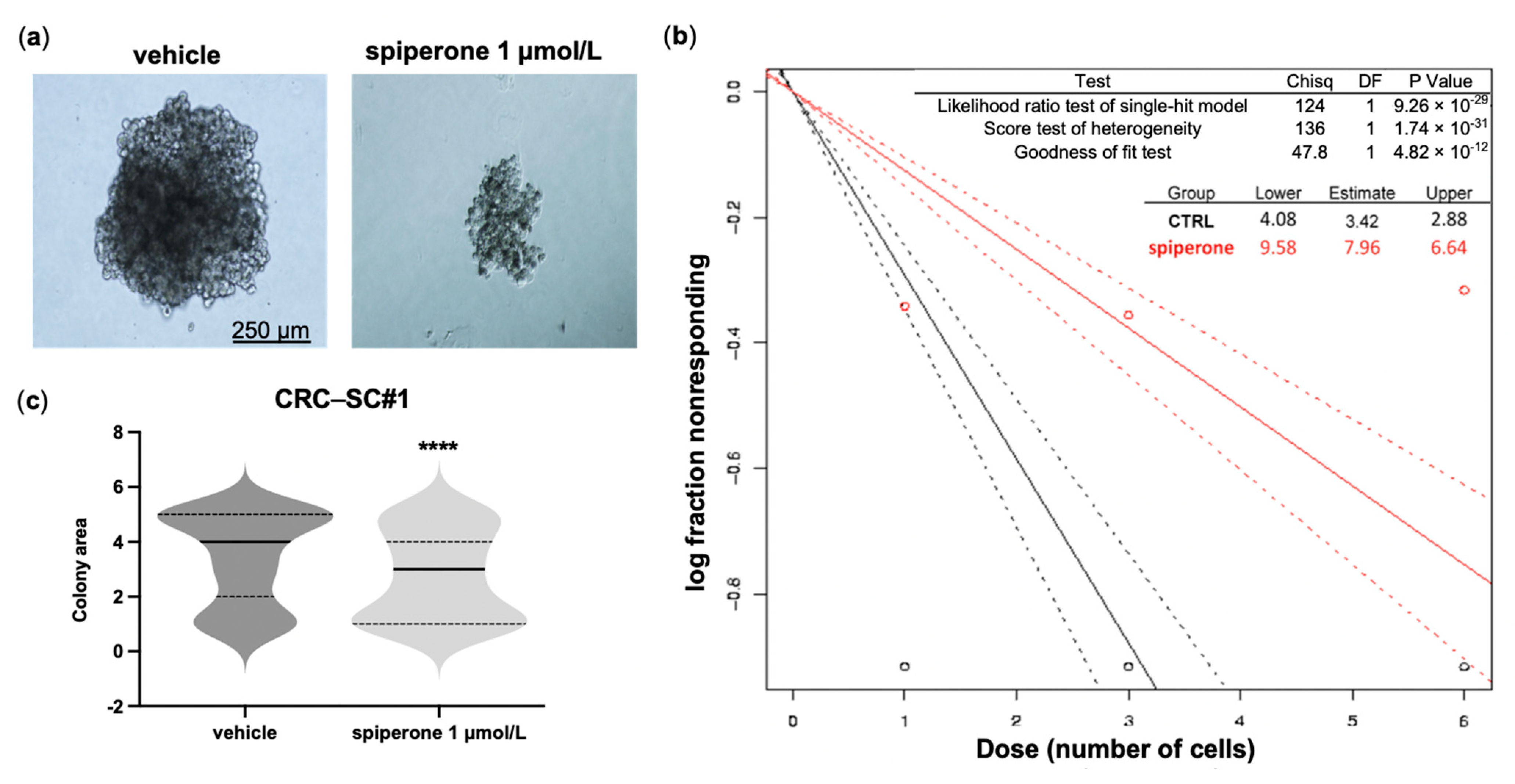
3.1. Spiperone Is Cytotoxic to CRC Cells and Impairs the Clonogenic Potential of CRC-SCs
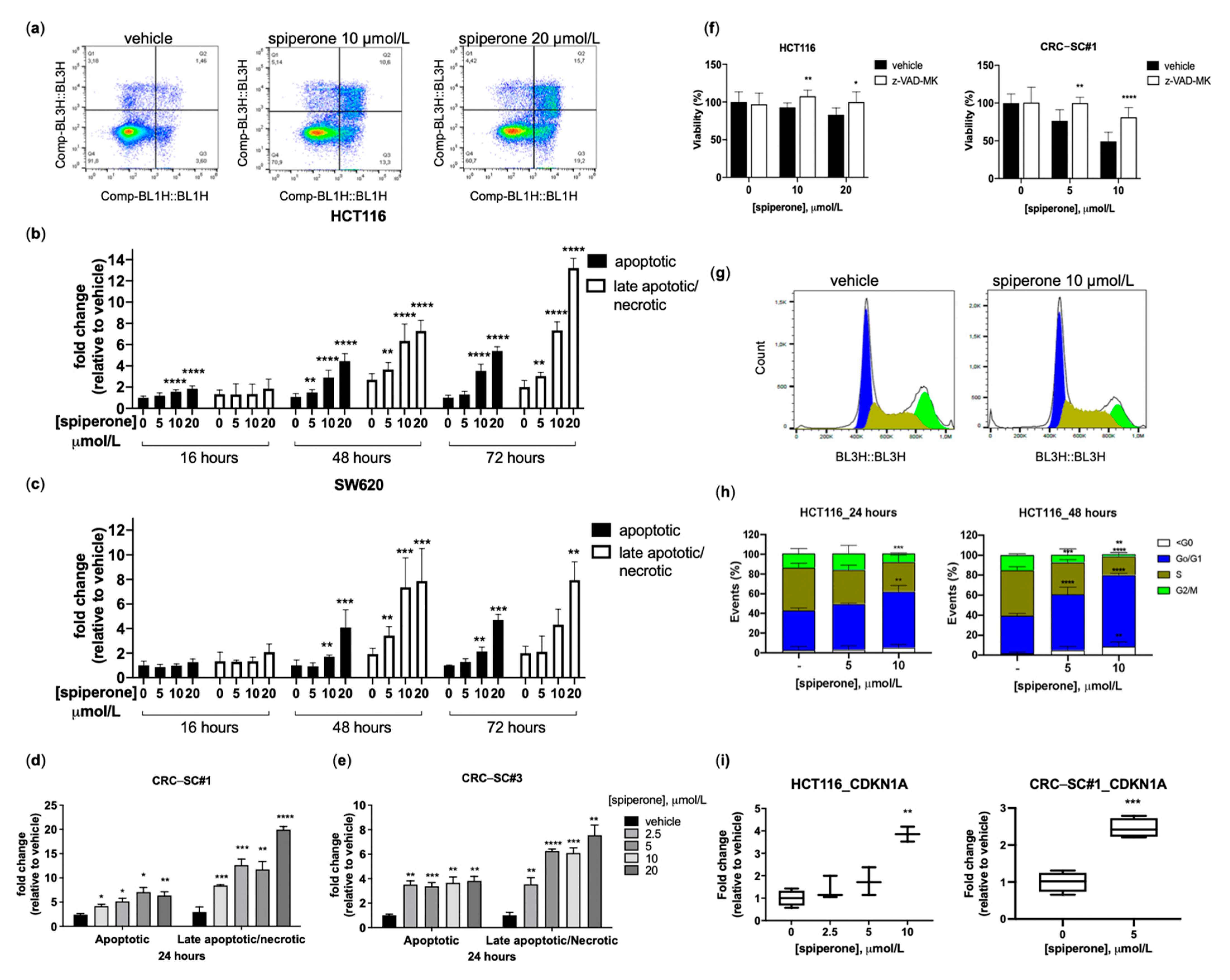
3.2. Spiperone Induces Cell Cycle Arrest and Apoptotic Cell Death
3.3. Spiperone-Treated Cells Maintain Lysosomal Membrane Integrity
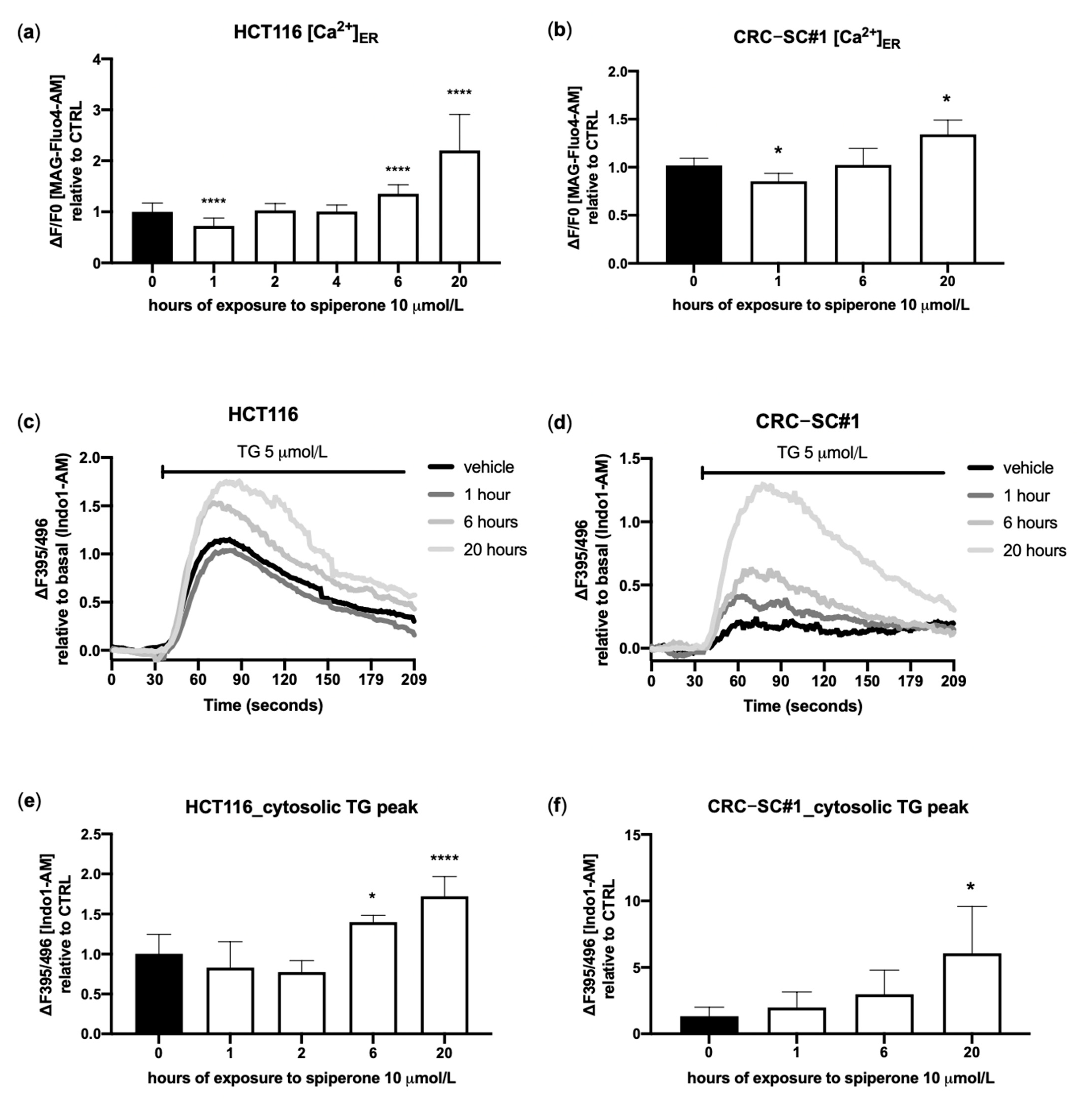
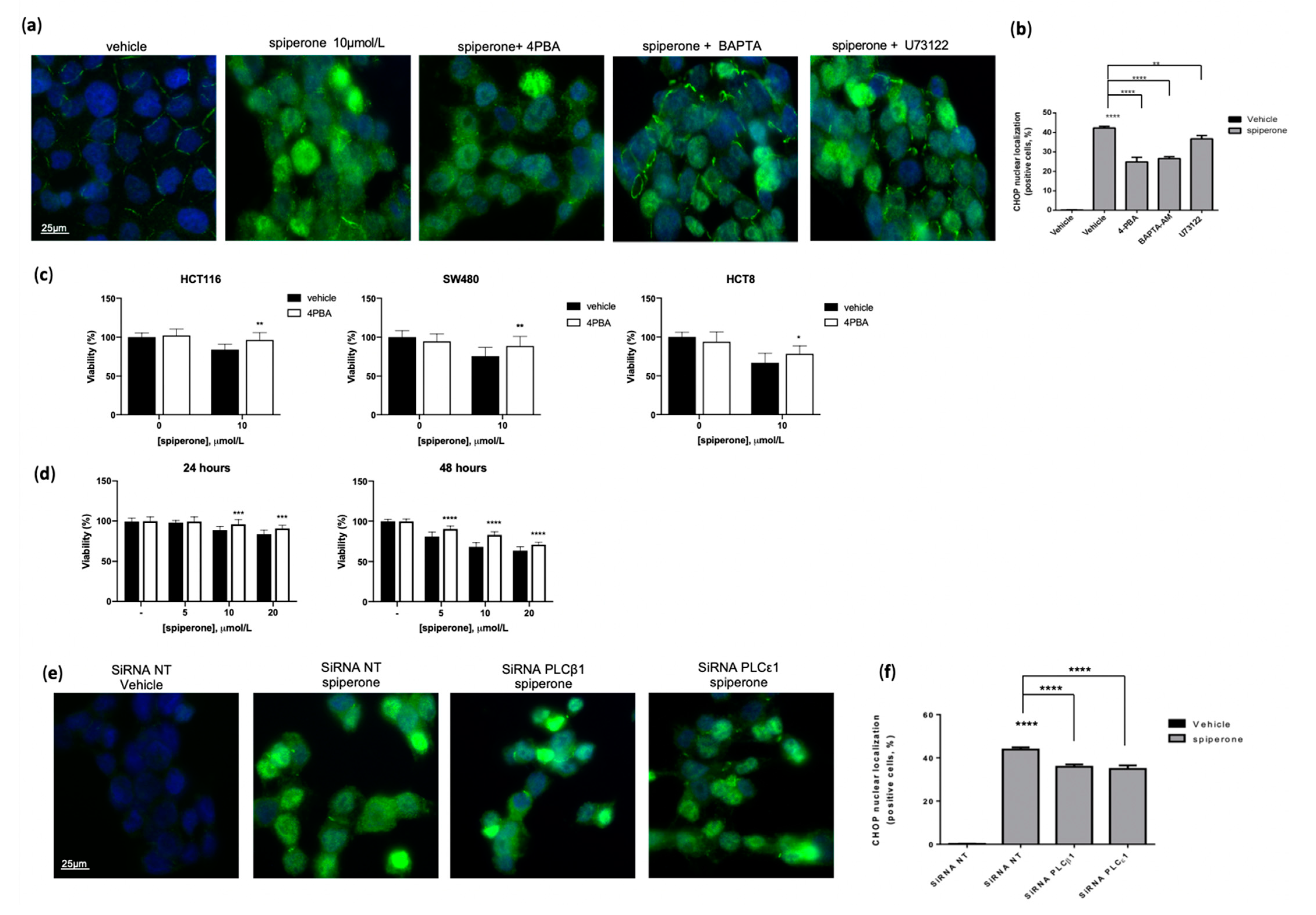
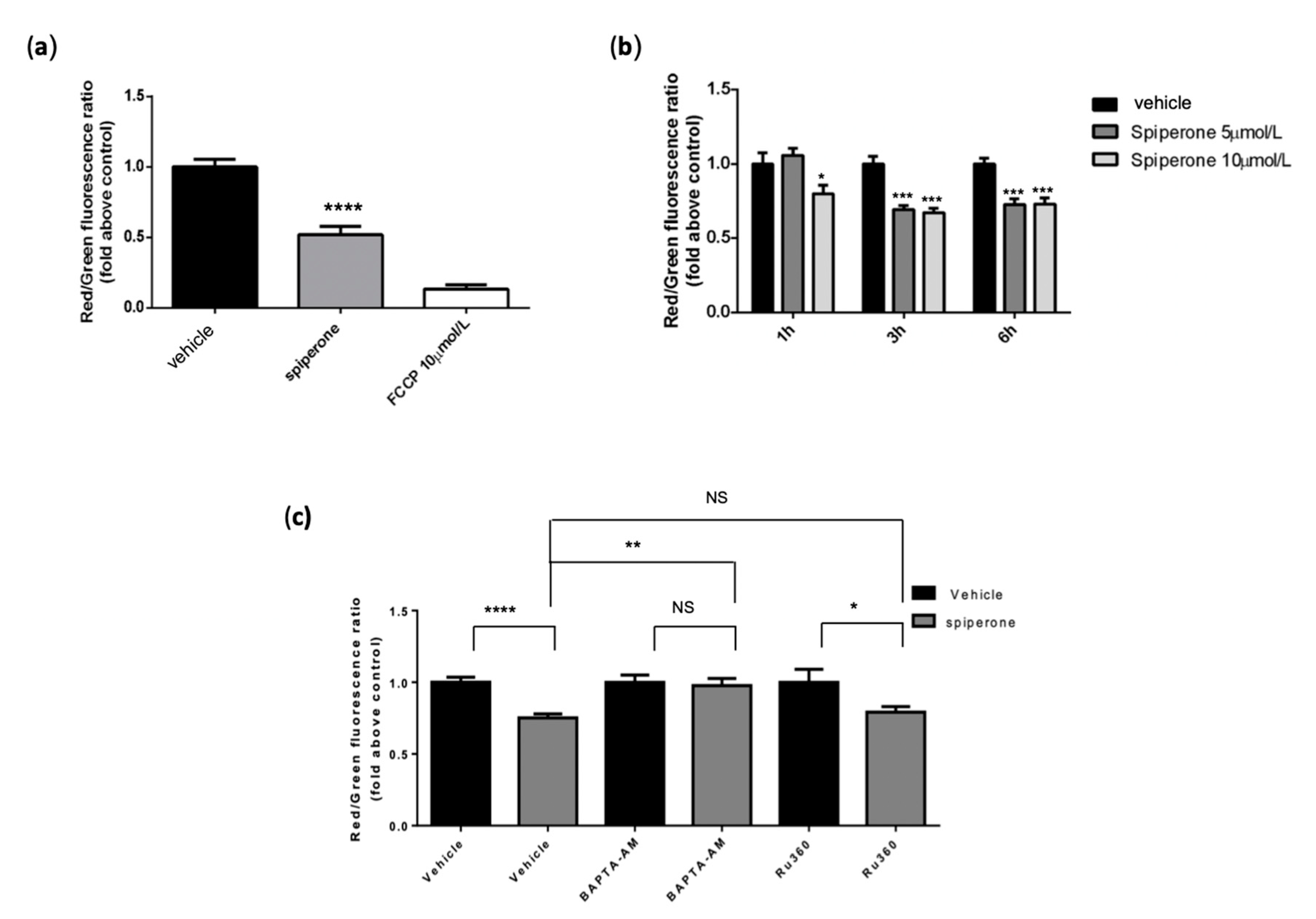
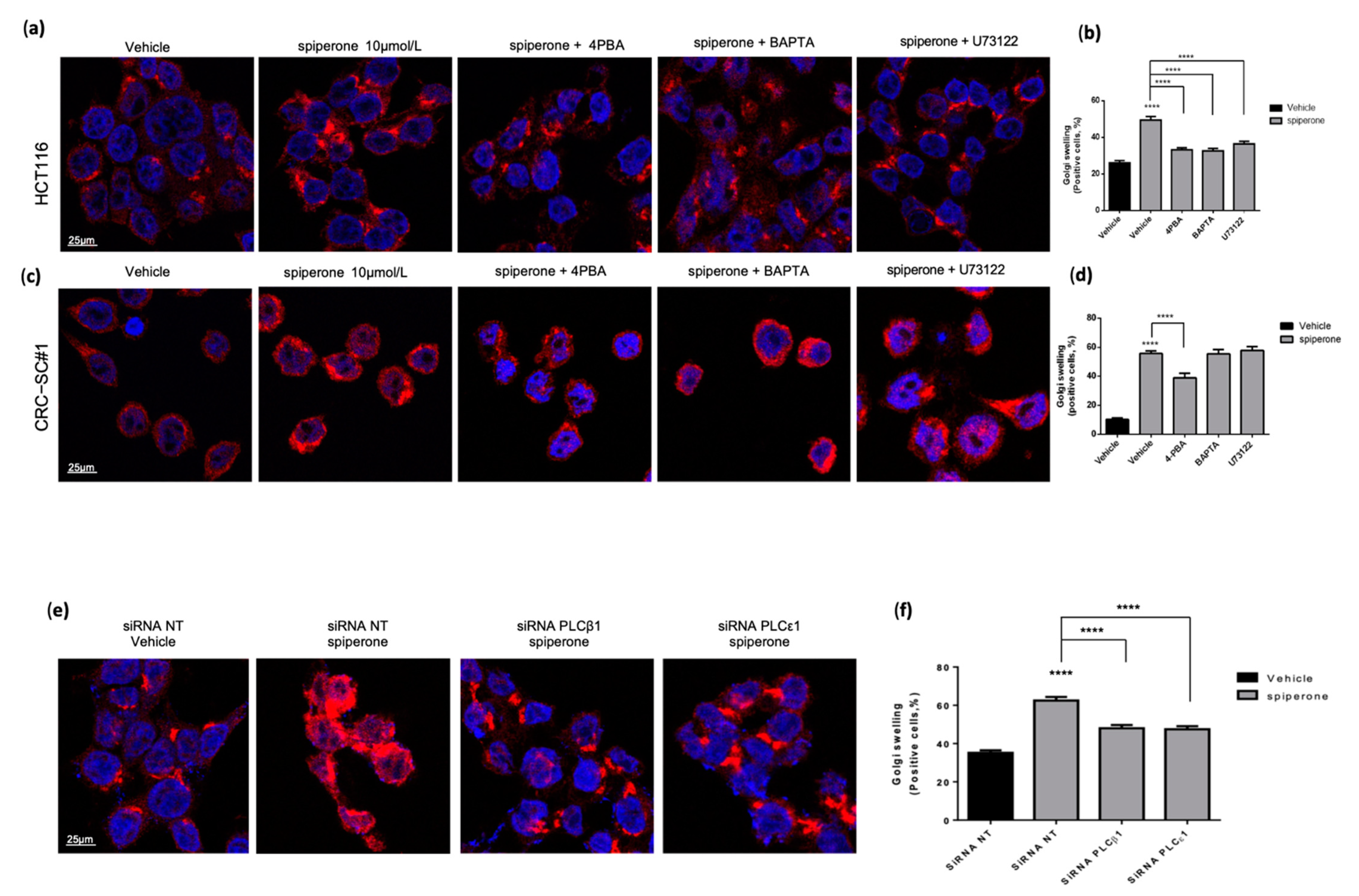
3.4. Spiperone Triggers Acute PLC-Dependent Ca2+ Modulation
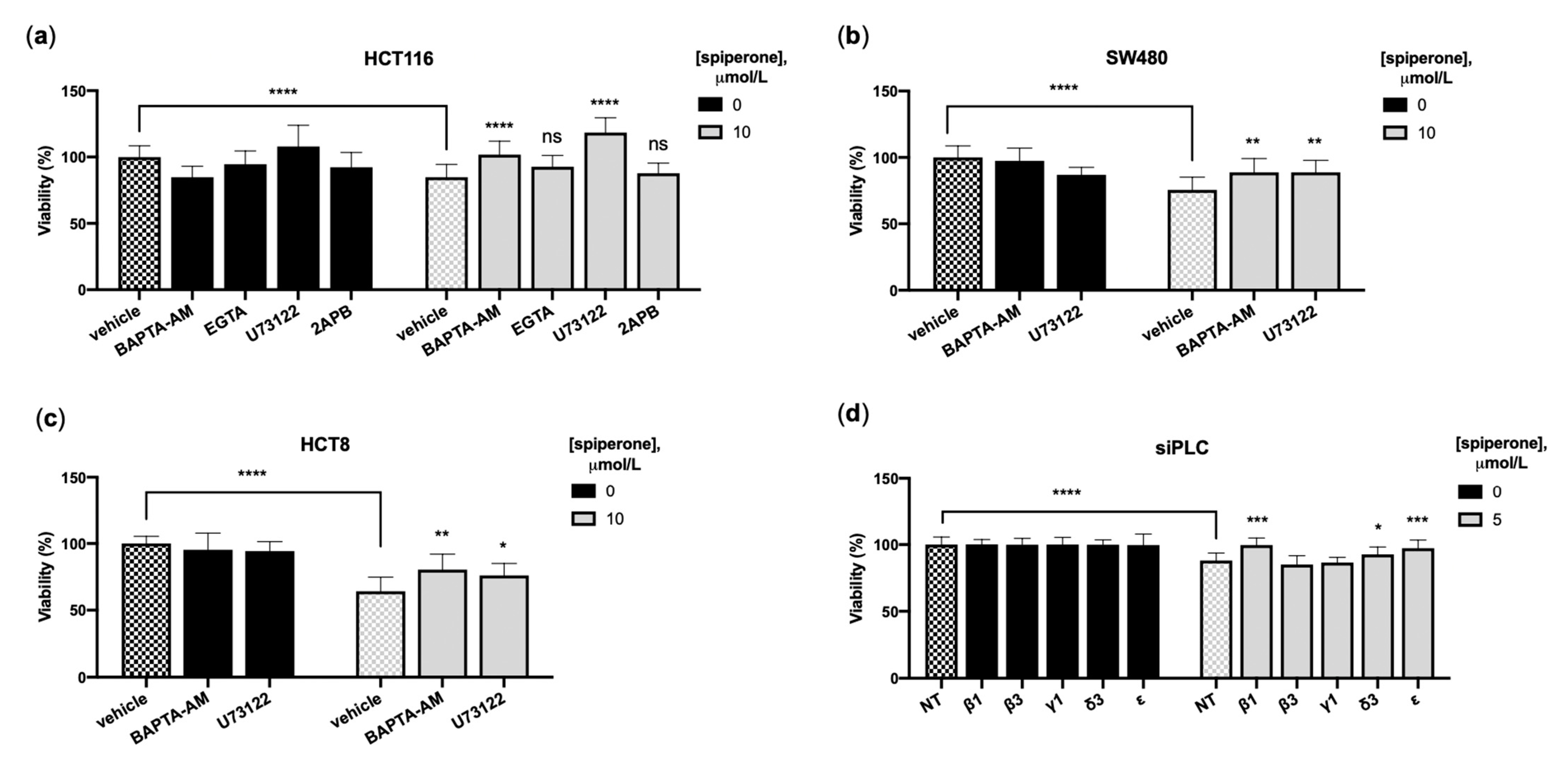
3.5. Spiperone Induces Ca2+/PLC-Dependent Cell Death
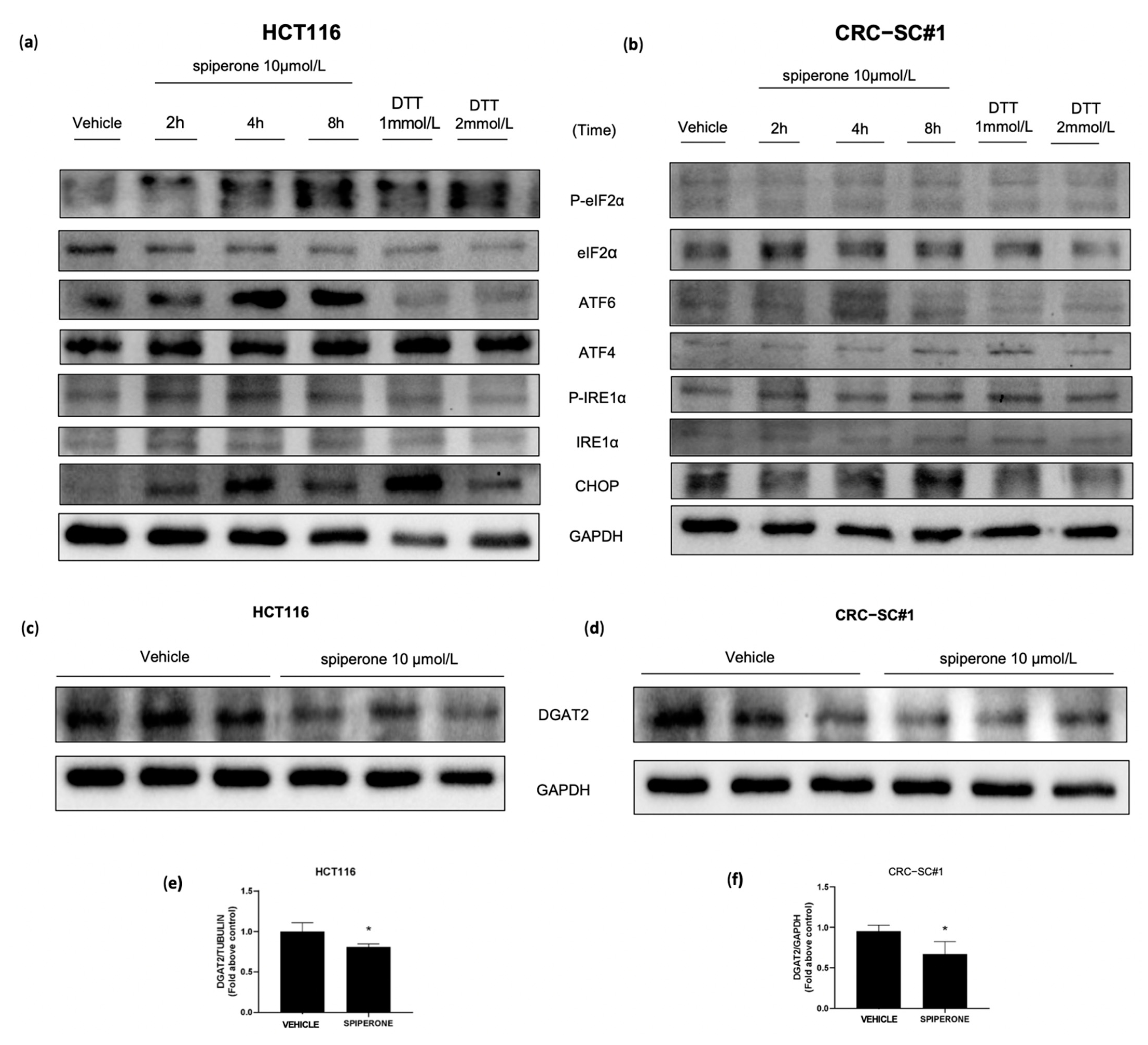
3.6. Spiperone Causes ER-Stress-Induced Cell Death
3.7. Spiperone Induces Mitochondrial Damage
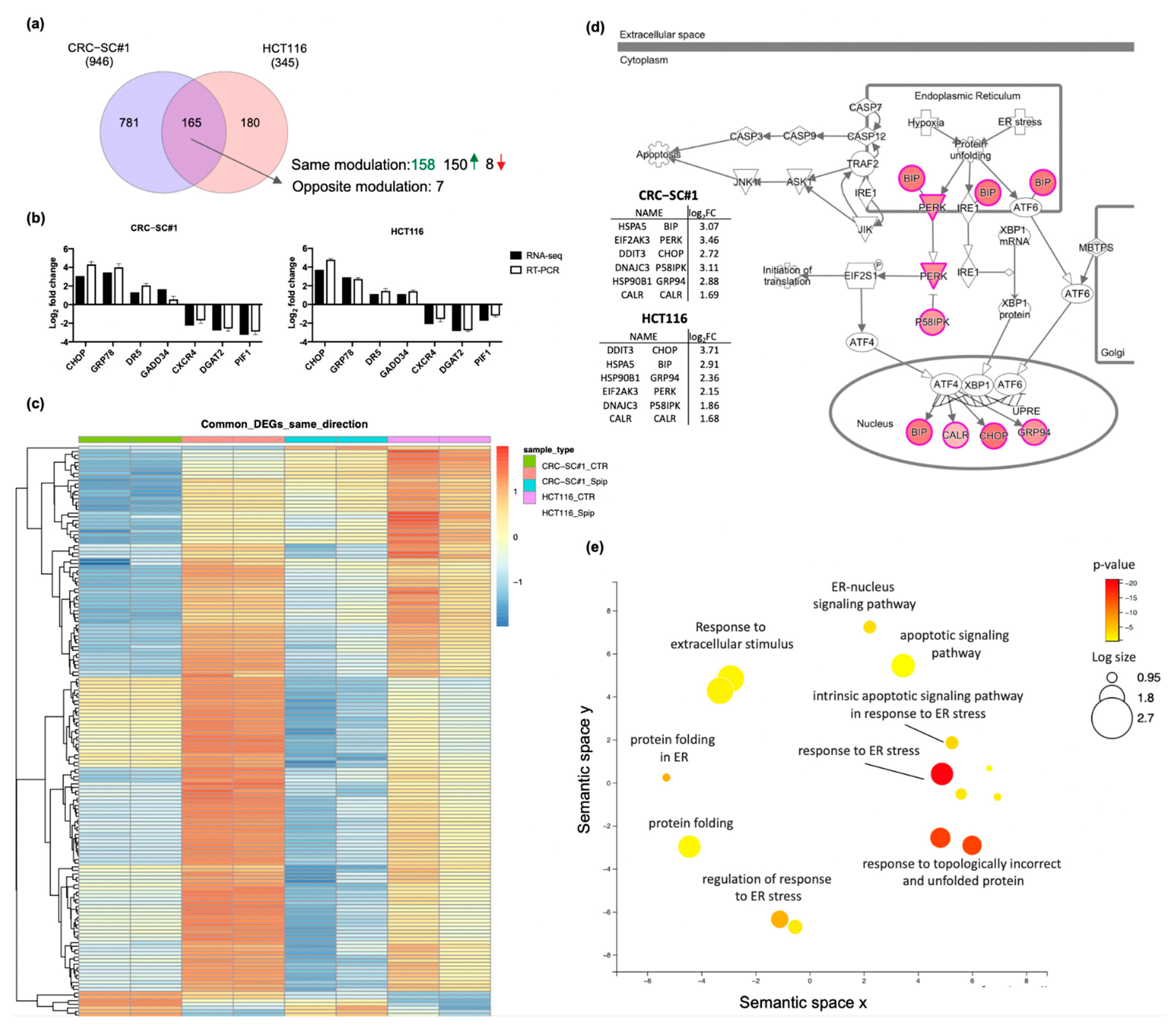
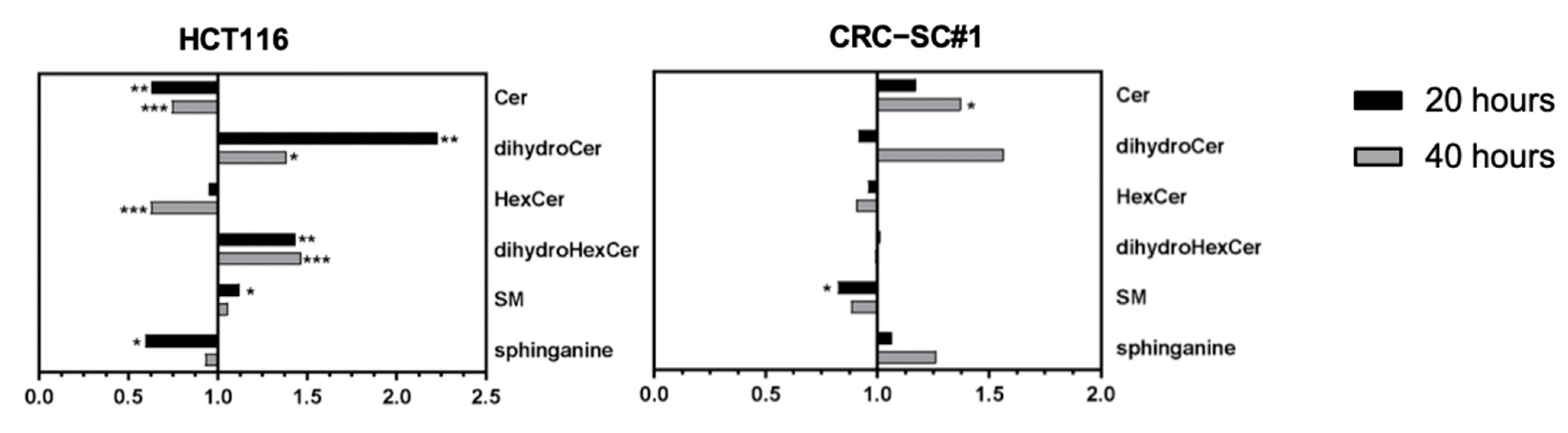
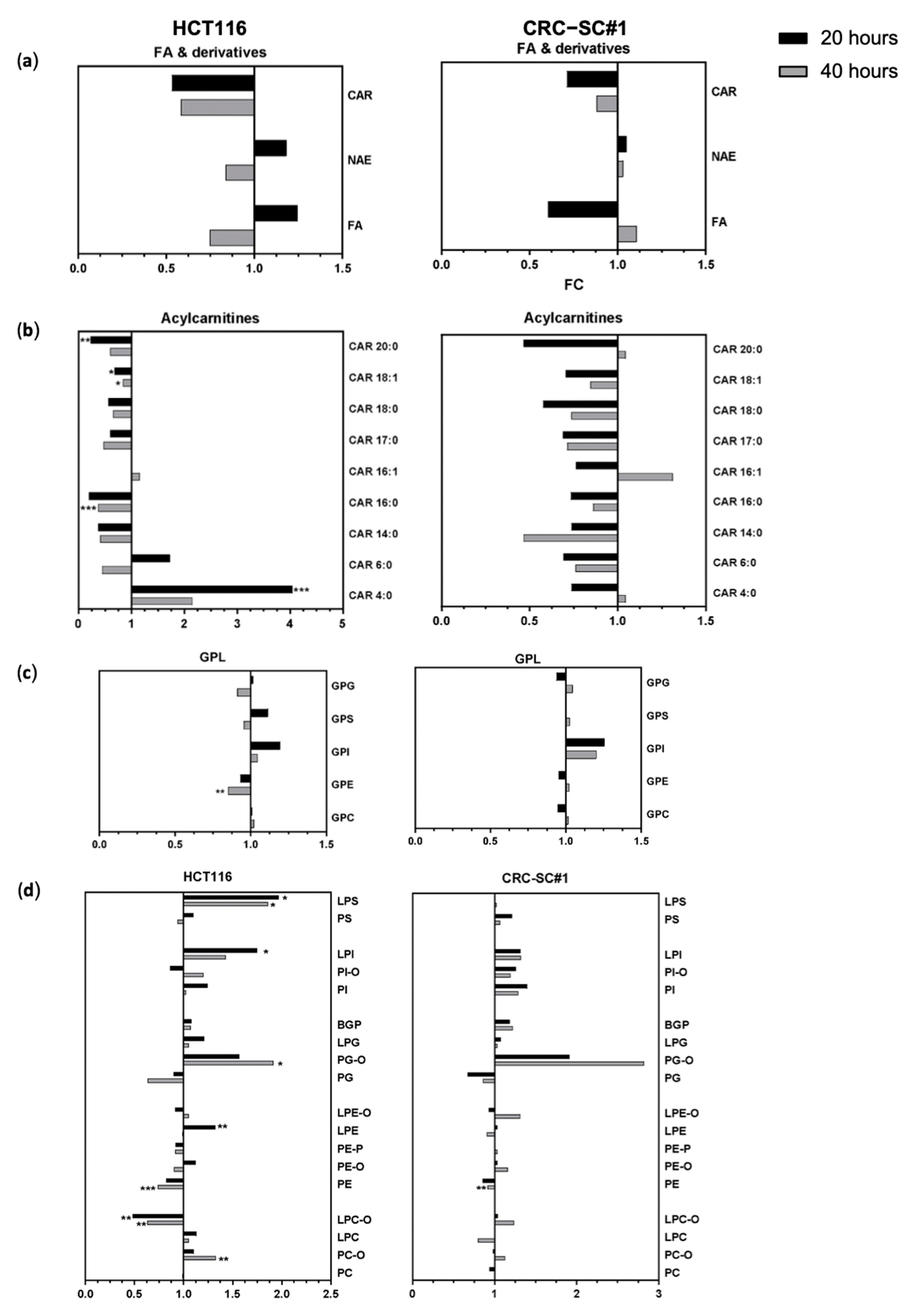
3.8. Spiperone Treatment Disrupts Lipid Metabolism in CRC Cells
3.9. Spiperone Induces Golgi Apparatus Deregulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Van Der Stok, E.P.; Spaander, M.C.W.; Grünhagen, D.J.; Verhoef, C.; Kuipers, E.J. Surveillance after Curative Treatment for Colorectal Cancer. Nat. Rev. Clin. Oncol. 2017, 14, 297–315. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Nogueira, L.; Mariotto, A.B.; Rowland, J.H.; Yabroff, K.R.; Alfano, C.M.; Jemal, A.; Kramer, J.L.; Siegel, R.L. Cancer Treatment and Survivorship Statistics, 2019. CA Cancer J. Clin. 2019, 69, 363–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouakrim, D.A.; Pizot, C.; Boniol, M.; Malvezzi, M.; Boniol, M.; Negri, E.; Bota, M.; Jenkins, M.A.; Bleiberg, H.; Autier, P. Trends in Colorectal Cancer Mortality in Europe: Retrospective Analysis of the WHO Mortality Database. BMJ 2015, 351, h4970. [Google Scholar] [CrossRef] [Green Version]
- Brenner, H.; Bouvier, A.M.; Foschi, R.; Hackl, M.; Larsen, I.K.; Lemmens, V.; Mangone, L.; Francisci, S. Progress in Colorectal Cancer Survival in Europe from the Late 1980s to the Early 21st Century: The EUROCARE Study. Int. J. Cancer 2012, 131, 1649–1658. [Google Scholar] [CrossRef]
- Fakih, M. Metastatic Colorectal Cancer: Current State and Future Directions. J. Clin. Oncol. 2015, 33, 1809–1824. [Google Scholar] [CrossRef]
- Zarour, L.R.; Anand, S.; Billingsley, K.G.; Bisson, W.H.; Cercek, A.; Clarke, M.F.; Coussens, L.M.; Gast, C.E.; Geltzeiler, C.B.; Hansen, L.; et al. Colorectal Cancer Liver Metastasis: Evolving Paradigms and Future Directions. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Walters, T.; Hawkins, N.; Pollock, R.; Colaone, F.; Shergill, S.; Ross, P. Systematic Review and Network Meta-Analyses of Third-Line Treatments for Metastatic Colorectal Cancer. J. Cancer Res. Clin. Oncol. 2020, 146, 2575–2587. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- De Rosa, M.; Pace, U.; Rega, D.; Costabile, V.; Duraturo, F.; Izzo, P.; Delrio, P. Genetics, Diagnosis and Management of Colorectal Cancer (Review). Oncol. Rep. 2015, 34, 1087–1096. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, R.M.; Montagut, C.; Wainberg, Z.A.; Ronga, P.; Audhuy, F.; Taieb, J.; Stintzing, S.; Siena, S.; Santini, D. Optimising the Use of Cetuximab in the Continuum of Care for Patients with Metastatic Colorectal Cancer. ESMO Open 2018, 3, e000353. [Google Scholar] [CrossRef] [Green Version]
- Golshani, G.; Zhang, Y. Advances in Immunotherapy for Colorectal Cancer: A Review. Therap. Adv. Gastroenterol. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.-H.; Chen, Y.-X.; Fang, J.-Y. Comprehensive Review of Targeted Therapy for Colorectal Cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Van Der Jeught, K.; Xu, H.C.; Li, Y.J.; Lu, X.-B.; Ji, G. Drug resistance and new therapies in colorectal cancer. World J. Gastroenterol. 2018, 24, 3834–3848. [Google Scholar] [CrossRef] [PubMed]
- Kuşoğlu, A.; Biray Avcı, Ç. Cancer Stem Cells: A Brief Review of the Current Status. Gene 2019, 681, 80–85. [Google Scholar] [CrossRef]
- Turdo, A.; Veschi, V.; Gaggianesi, M.; Chinnici, A.; Bianca, P.; Todaro, M.; Stassi, G. Meeting the Challenge of Targeting Cancer Stem Cells. Front. Cell Dev. Biol. 2019, 7, 16. [Google Scholar] [CrossRef] [Green Version]
- Hervieu, C.; Christou, N.; Battu, S.; Mathonnet, M. The Role of Cancer Stem Cells in Colorectal Cancer: From the Basics to Novel Clinical Trials. Cancers 2021, 13, 1092. [Google Scholar] [CrossRef]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical Development Success Rates for Investigational Drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Scannell, J.W.; Blanckley, A.; Boldon, H.; Warrington, B. Diagnosing the Decline in Pharmaceutical R&D Efficiency. Nat. Rev. Drug Discov. 2012, 11, 191–200. [Google Scholar]
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandrell, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J.; et al. An Analysis of the Attrition of Drug Candidates from Four Major Pharmaceutical Companies. Nat. Rev. Drug Discov. 2015, 14, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V. Do Cancer Drugs Improve Survival or Quality of Life? BMJ 2017, 359, j4528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilking, N.E.; Jönsson, B.; Lindgren, P.; Wilking, U. Does Access to Cancer Drugs Relate to Survival Benefit? A European Study in Countries with Different Economic Status. J. Clin. Oncol. 2017, 35, 6535. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug Repositioning: Identifying and Developing New Uses for Existing Drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Simsek, M.; Meijer, B.; van Bodegraven, A.A.; de Boer, N.K.H.; Mulder, C.J.J. Finding Hidden Treasures in Old Drugs: The Challenges and Importance of Licensing Generics. Drug Discov. Today 2018, 23, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Novac, N. Challenges and Opportunities of Drug Repositioning. Trends Pharmacol. Sci. 2013, 34, 267–272. [Google Scholar] [CrossRef] [PubMed]
- GNS, H.S.; GR, S.; Murahari, M.; Krishnamurthy, M. An Update on Drug Repurposing: Re-Written Saga of the Drug’s Fate. Biomed. Pharmacother. 2018, 110, 700–716. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug Repurposing: Progress, Challenges and Recommendations. Nat. Rev. Drug Discov. 2018, 18, 41–58. [Google Scholar] [CrossRef]
- Bertolini, F.; Sukhatme, V.P.; Bouche, G. Drug Repurposing in Oncology-Patient and Health Systems Opportunities. Nat. Rev. Clin. Oncol. 2015, 12, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Capistrano, I.R.; De Potter, A.; Vandeborne, L.; Bouche, G. An Open Access Database of Licensed Cancer Drugs. Front. Pharmacol. 2021, 12, 627574. [Google Scholar] [CrossRef]
- Sleire, L.; Førde-Tislevoll, H.E.; Netland, I.A.; Leiss, L.; Skeie, B.S.; Enger, P.Ø. Drug Repurposing in Cancer. Pharmacol. Res. 2017, 124, 74–91. [Google Scholar] [CrossRef]
- Würth, R.; Thellung, S.; Bajetto, A.; Mazzanti, M.; Florio, T.; Barbieri, F. Drug-Repositioning Opportunities for Cancer Therapy: Novel Molecular Targets for Known Compounds. Drug Discov. Today 2016, 21, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming Cancer Therapeutic Bottleneck by Drug Repurposing. Signal Transduct. Target. Ther. 2020, 5, 113. [Google Scholar] [CrossRef]
- Sweetman, S.C. Martindale: The Complete Drug Reference; Pharmaceutical Press: London, UK, 2011; 4142. [Google Scholar]
- Varalda, M.; Antona, A.; Bettio, V.; Roy, K.; Vachamaram, A.; Yellenki, V.; Massarotti, A.; Baldanzi, G.; Capello, D. Psychotropic Drugs Show Anticancer Activity by Disrupting Mitochondrial and Lysosomal Function. Front. Oncol. 2020, 10, 562196. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2-ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A Web-Based Tool for the Analysis of Sets through Venn Diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for Gene List Enrichment Analysis and Candidate Gene Prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [Green Version]
- Ubeda, M.; Wang, X.Z.; Zinszner, H.; Wu, I.; Habener, J.F.; Ron, D. Stress-Induced Binding of the Transcriptional Factor CHOP to a Novel DNA Control Element. Mol. Cell. Biol. 1996, 16, 1479–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Mondragon, J.A.; Riudavets-Puig, R.; Rauluseviciute, I.; Berhanu Lemma, R.; Turchi, L.; Blanc-Mathieu, R.; Lucas, J.; Boddie, P.; Khan, A.; Manosalva Pérez, N.; et al. JASPAR 2022: The 9th Release of the Open-Access Database of Transcription Factor Binding Profiles. Nucleic Acids Res. 2022, 50, D165–D173. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Ohta, E.; Izumi, Y.; Ogiwara, A.; Yukihira, D.; Bamba, T.; Fukusaki, E.; Arita, M. MRM-DIFF: Data Processing Strategy for Differential Analysis in Large Scale MRM-Based Lipidomics Studies. Front. Genet. 2015, 5, 471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laduron, P.M.; Janssen, P.F.M.; Leysen, J.E. Spiperone: A Ligand of Choice for Neuroleptic Receptors: 2. Regional Distribution and in Vivo Displacement of Neuroleptic Drugs. Biochem. Pharmacol. 1978, 27, 317–321. [Google Scholar] [CrossRef]
- Liang, L.; MacDonald, K.; Schwiebert, E.M.; Zeitlin, P.L.; Guggino, W.B. Spiperone, Identified through Compound Screening, Activates Calcium-Dependent Chloride Secretion in the Airway. Am. J. Physiol. Physiol. 2009, 296, C131–C141. [Google Scholar] [CrossRef]
- Taylor, C.W.; Tovey, S.C. IP(3) Receptors: Toward Understanding Their Activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a004010. [Google Scholar] [CrossRef]
- Gresset, A.; Sondek, J.; Harden, T.K. The Phospholipase C Isozymes and Their Regulation. Subcell. Biochem. 2015, 58, 61–94. [Google Scholar] [CrossRef] [Green Version]
- Bill, C.A.; Vines, C.M. Phospholipase C. Adv. Exp. Med. Biol. 2020, 1131, 215–242. [Google Scholar] [CrossRef]
- Maruyama, T.; Kanaji, T.; Nakade, S.; Kanno, T.; Mikoshiba, K. 2APB, 2-Aminoethoxydiphenyl Borate, a Membrane-Penetrable Modulator of Ins(l,4,5)P3-Induced Ca2+ Release. J. Biochem. 1997, 122, 498–505. [Google Scholar] [CrossRef]
- Jin, W.; Lo, T.M.; Loh, H.H.; Thayer, S.A. U73122 Inhibits Phospholipase C-Dependent Calcium Mobilization in Neuronal Cells. Brain Res. 1994, 642, 237–243. [Google Scholar] [CrossRef]
- Roderick, H.L.; Cook, S.J. Ca2+ Signalling Checkpoints in Cancer: Remodelling Ca2+ for Cancer Cell Proliferation and Survival. Nat. Rev. Cancer 2008, 8, 361–375. [Google Scholar] [CrossRef] [Green Version]
- Marchi, S.; Pinton, P. Alterations of Calcium Homeostasis in Cancer Cells. Curr. Opin. Pharmacol. 2016, 29, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Danese, A.; Bouhamida, E.; Aguiari, G.; Previati, M.; Pinton, P.; Giorgi, C. Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer. Int. J. Mol. Sci. 2020, 21, 8323. [Google Scholar] [CrossRef] [PubMed]
- Ghibelli, L.; Cerella, C.; Diederich, M. The Dual Role of Calcium as Messenger and Stressor in Cell Damage, Death, and Survival. Int. J. Cell Biol. 2010, 2010, 546163. [Google Scholar] [CrossRef] [Green Version]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and Apoptosis: ER-Mitochondria Ca2+ Transfer in the Control of Apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [Green Version]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momeni, H.R. Role of Calpain in Apoptosis. Cell J. 2011, 13, 65–72. [Google Scholar] [PubMed]
- Smith, M.A.; Schnellmann, R.G. Calpains, Mitochondria, and Apoptosis. Cardiovasc. Res. 2012, 96, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Gagou, M.E.; Ganesh, A.; Thompson, R.; Phear, G.; Sanders, C.; Meuth, M. Suppression of Apoptosis by PIF1 Helicase in Human Tumor Cells. Cancer Res. 2011, 71, 4998–5008. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, W.J.; Hsieh, S.C.; Chen, C.C.; Wang, F.F. Human DDA3 Is an Oncoprotein Down-Regulated by P53 and DNA Damage. Biochem. Biophys. Res. Commun. 2008, 369, 567–572. [Google Scholar] [CrossRef]
- Dhawan, P.; Ahmad, R.; Chaturvedi, R.; Smith, J.J.; Midha, R.; Mittal, M.K.; Krishnan, M.; Chen, X.; Eschrich, S.; Yeatman, T.J.; et al. Claudin-2 Expression Increases Tumorigenicity of Colon Cancer Cells: Role of Epidermal Growth Factor Receptor Activation. Oncogene 2011, 30, 3234–3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottaiano, A.; Santorsola, M.; Del Prete, P.; Perri, F.; Scala, S.; Caraglia, M.; Nasti, G. Prognostic Significance of CXCR4 in Colorectal Cancer: An Updated Meta-Analysis and Critical Appraisal. Cancers 2021, 13, 3284. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wang, C.; Hao, M.; Sun, R.; Wang, Y.; Liu, T.; Cong, X.; Liu, Y. CYP24A1 Is a Potential Biomarker for the Progression and Prognosis of Human Colorectal Cancer. Hum. Pathol. 2016, 50, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The Unfolded Protein Response: Controlling Cell Fate Decisions under ER Stress and Beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Schröder, M. Endoplasmic Reticulum Stress Responses. Cell. Mol. Life Sci. 2008, 65, 862–894. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Moncan, M.; Mnich, K.; Blomme, A.; Almanza, A.; Samali, A.; Gorman, A.M. Regulation of Lipid Metabolism by the Unfolded Protein Response. J. Cell. Mol. Med. 2021, 25, 1359–1370. [Google Scholar] [CrossRef]
- Le Thomas, A.; Ferri, E.; Marsters, S.; Harnoss, J.M.; Modrusan, Z.; Li, W.; Rudolph, J.; Wang, W.; Wu, T.D.; Walter, P.; et al. Noncanonical MRNA Decay by the Endoplasmic-Reticulum Stress Sensor IRE1α Promotes Cancer-Cell Survival. bioRxiv 2021, 435520. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER Stress-Induced Cell Death Mechanisms. Biochim. Biophys. Acta-Mol. Cell Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [Green Version]
- Shore, G.C.; Papa, F.R.; Oakes, S.A. Signaling Cell Death from the Endoplasmic Reticulum Stress Response. Curr. Opin. Cell Biol. 2011, 23, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell Death Induced by Endoplasmic Reticulum Stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, T.A.; Chaudhary, A.K.; Kumar, S.; O’Malley, J.; Inigo, J.R.; Kumar, R.; Yadav, N.; Chandra, D. Endoplasmic Reticulum-Mediated Unfolded Protein Response and Mitochondrial Apoptosis in Cancer. Biochim. Biophys. Acta-Rev. Cancer 2017, 1867, 58–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy Is Activated for Cell Survival after Endoplasmic ReticulumStress. Mol. Cell. Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [Green Version]
- Yorimitsu, T.; Nair, U.; Yang, Z.; Klionsky, D.J. Endoplasmic Reticulum Stress Triggers Autophagy. J. Biol. Chem. 2006, 281, 30299–30304. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Suaga, P.; Paillusson, S.; Miller, C.C.J. ER-Mitochondria Signaling Regulates Autophagy. Autophagy 2017, 13, 1250–1251. [Google Scholar] [CrossRef] [Green Version]
- Saikia, R.; Joseph, J. AMPK: A Key Regulator of Energy Stress and Calcium-Induced Autophagy. J. Mol. Med. 2021, 99, 1539–1551. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. ER Stress and Its Functional Link to Mitochondria: Role in Cell Survival and Death. Cold Spring Harb. Perspect. Biol. 2011, 3, a004424. [Google Scholar] [CrossRef]
- Kumar, V.; Maity, S. ER Stress-Sensor Proteins and ER-Mitochondrial Crosstalk—Signaling beyond (ER) Stress Response. Biomolecules 2021, 11, 173. [Google Scholar] [CrossRef]
- Yen, C.-L.E.; Stone, S.J.; Koliwad, S.; Harris, C.; Farese, R.V., Jr. DGAT Enzymes and Triacylglycerol Biosynthesis. J. Lipid Res. 2008, 49, 2283. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Corbacho, M.J.; Obeid, L.M. A Novel Role for DGATs in Cancer. Adv. Biol. Regul. 2019, 72, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, K.; Casiano, C.A.; Hamel, J.C.; Molinaro, C.; Fritzler, M.J.; Chan, E.K. Fragmentation of Golgi Complex and Golgi Autoantigens during Apoptosis and Necrosis. Arthritis Res. 2002, 4, R3. [Google Scholar] [CrossRef] [PubMed]
- Bankaitis, V.A.; Garcia-Mata, R.; Mousley, C.J. Golgi Membrane Dynamics and Lipid Metabolism. Curr. Biol. 2012, 22, R414–R424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testa, U.; Pelosi, E.; Castelli, G. Colorectal Cancer: Genetic Abnormalities, Tumor Progression, Tumor Heterogeneity, Clonal Evolution and Tumor-Initiating Cells. Med. Sci. 2018, 6, 31. [Google Scholar] [CrossRef] [Green Version]
- Wedding, U.; Honecker, F.; Bokemeyer, C.; Pientka, L.; Höffken, K. Tolerance to Chemotherapy in Elderly Patients with Cancer. Cancer Control 2007, 14, 44–56. [Google Scholar] [CrossRef]
- Balducci, L.; Colloca, G.; Cesari, M.; Gambassi, G. Assessment and Treatment of Elderly Patients with Cancer. Surg. Oncol. 2010, 19, 117–123. [Google Scholar] [CrossRef]
- Mortensen, P.B. The Incidence of Cancer in Schizophrenic Patients. J. Epidemiol. Community Health 1989, 43, 43–47. [Google Scholar] [CrossRef]
- Lichtermann, D.; Ekelund, J.; Pukkala, E.; Tanskanen, A.; Lönnqvist, J. Incidence of Cancer Among Persons With Schizophrenia and Their Relatives. Arch. Gen. Psychiatry 2001, 58, 573–578. [Google Scholar] [CrossRef] [Green Version]
- Dalton, S.O.; Mellemkjær, L.; Thomassen, L.; Mortensen, P.B.; Johansen, C. Risk for Cancer in a Cohort of Patients Hospitalized for Schizophrenia in Denmark, 1969–1993. Schizophr. Res. 2005, 75, 315–324. [Google Scholar] [CrossRef]
- Zhuo, C.; Xun, Z.; Hou, W.; Ji, F.; Lin, X.; Tian, H.; Zheng, W.; Chen, M.; Liu, C.; Wang, W.; et al. Surprising Anticancer Activities of Psychiatric Medications: Old Drugs Offer New Hope for Patients with Brain Cancer. Front. Pharmacol. 2019, 10, 1262. [Google Scholar] [CrossRef]
- Dalton, S.O.; Johansen, C.; Poulsen, A.H.; Nørgaard, M.; Sørensen, H.T.; McLaughlin, J.K.; Mortensen, P.B.; Friis, S. Cancer Risk among Users of Neuroleptic Medication: A Population-Based Cohort Study. Br. J. Cancer 2006, 95, 934–939. [Google Scholar] [CrossRef] [Green Version]
- Chubak, J.; Boudreau, D.M.; Rulyak, S.J.; Mandelson, M.T. Colorectal Cancer Risk in Relation to Antidepressant Medication Use. Int. J. Cancer 2011, 128, 227–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correll, C.U. From Receptor Pharmacology to Improved Outcomes: Individualising the Selection, Dosing, and Switching of Antipsychotics. Eur. Psychiatry 2010, 25, S12–S21. [Google Scholar] [CrossRef]
- Cherciu, I.; Bărbălan, A.; Pirici, D.; Mărgăritescu, C.; Săftoiu, A. Stem Cells, Colorectal Cancer and Cancer Stem Cell Markers Correlations. Curr. Health Sci. J. 2014, 40, 153–15361. [Google Scholar] [CrossRef] [PubMed]
- Palve, V.; Liao, Y.; Remsing Rix, L.L.; Rix, U. Turning Liabilities into Opportunities: Off-Target Based Drug Repurposing in Cancer. Semin. Cancer Biol. 2021, 68, 209–229. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Meenakshisundaram, S.; Manickam, M.; Sankaranarayanan, M. A Medicinal Chemistry Perspective of Drug Repositioning: Recent Advances and Challenges in Drug Discovery. Eur. J. Med. Chem. 2020, 195, 112275. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Carson, D.A. Spiperone Enhances Intracellular Calcium Level and Inhibits the Wnt Signaling Pathway. BMC Pharmacol. 2009, 9, 13. [Google Scholar] [CrossRef] [Green Version]
- Marchi, S.; Giorgi, C.; Galluzzi, L.; Pinton, P. Ca2+ Fluxes and Cancer. Mol. Cell 2020, 78, 1055–1069. [Google Scholar] [CrossRef]
- Varghese, E.; Samuel, S.M.; Sadiq, Z.; Kubatka, P.; Liskova, A.; Benacka, J.; Pazinka, P.; Kruzliak, P.; Büsselberg, D. Anti-Cancer Agents in Proliferation and Cell Death: The Calcium Connection. Int. J. Mol. Sci. 2019, 20, 3017. [Google Scholar] [CrossRef] [Green Version]
- Okajima, F. Phosphatidylinositol Turnover and Receptors. In Encyclopedia of Endocrine Diseases; Elsevier: Amsterdam, The Netherlands, 2004; pp. 597–603. [Google Scholar]
- Fiume, R.; Faenza, I.; Sheth, B.; Poli, A.; Vidalle, M.C.; Mazzetti, C.; Abdul, S.H.; Campagnoli, F.; Fabbrini, M.; Kimber, S.T.; et al. Nuclear Phosphoinositides: Their Regulation and Roles in Nuclear Functions. Int. J. Mol. Sci. 2019, 20, 2991. [Google Scholar] [CrossRef] [Green Version]
- Piazzi, M.; Blalock, W.L.; Bavelloni, A.; Faenza, I.; D’Angelo, A.; Maraldi, N.M.; Cocco, L. Phosphoinositide-Specific Phospholipase C β 1b (PI-PLCβ1b) Interactome: Affinity Purification-Mass Spectrometry Analysis of PI-PLCβ1b with Nuclear Protein. Mol. Cell. Proteom. 2013, 12, 2220–2235. [Google Scholar] [CrossRef] [Green Version]
- Aisiku, O.; Dowal, L.; Scarlata, S. Protein Kinase C Phosphorylation of PLCβ1 Regulates Its Cellular Localization. Arch. Biochem. Biophys. 2011, 509, 186–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obeng, E.O.; Rusciano, I.; Marvi, M.V.; Fazio, A.; Ratti, S.; Follo, M.Y.; Xian, J.; Manzoli, L.; Billi, A.M.; Mongiorgi, S.; et al. Phosphoinositide-Dependent Signaling in Cancer: A Focus on Phospholipase C Isozymes. Int. J. Mol. Sci. 2020, 21, 2581. [Google Scholar] [CrossRef] [Green Version]
- Xian, J.; Owusu Obeng, E.; Ratti, S.; Rusciano, I.; Marvi, M.V.; Fazio, A.; De Stefano, A.; Mongiorgi, S.; Cappellini, A.; Ramazzotti, G.; et al. Nuclear Inositides and Inositide-Dependent Signaling Pathways in Myelodysplastic Syndromes. Cells 2020, 9, 697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunney, T.D.; Katan, M. Phospholipase C Epsilon: Linking Second Messengers and Small GTPases. Trends Cell Biol. 2006, 16, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Madukwe, J.C.; Garland-Kuntz, E.E.; Lyon, A.M.; Smrcka, A.V. G Protein Subunits Directly Interact with and Activate Phospholipase C. J. Biol. Chem. 2018, 293, 6387–6397. [Google Scholar] [CrossRef] [Green Version]
- Kelley, G.G.; Kaproth-Joslin, K.A.; Reks, S.E.; Smrcka, A.V.; Wojcikiewicz, R.J.H. G-Protein-Coupled Receptor Agonists Activate Endogenous Phospholipase Cϵ and Phospholipase Cβ3 in a Temporally Distinct Manner. J. Biol. Chem. 2006, 281, 2639–2648. [Google Scholar] [CrossRef] [Green Version]
- Dusaban, S.S.; Purcell, N.H.; Rockenstein, E.; Masliah, E.; Cho, M.K.; Smrcka, A.V.; Brown, J.H. Phospholipase Cɛ Links G Protein-Coupled Receptor Activation to Inflammatory Astrocytic Responses. Proc. Natl. Acad. Sci. USA 2013, 110, 3609–3614. [Google Scholar] [CrossRef] [Green Version]
- Park, J.B.; Lee, C.S.; Jang, J.H.; Ghim, J.; Kim, Y.J.; You, S.; Hwang, D.; Suh, P.G.; Ryu, S.H. Phospholipase Signalling Networks in Cancer. Nat. Rev. Cancer 2012, 12, 782–792. [Google Scholar] [CrossRef]
- Zhang, R.Y.; Du, W.Q.; Zhang, Y.C.; Zheng, J.N.; Pei, D.S. PLCε Signaling in Cancer. J. Cancer Res. Clin. Oncol. 2016, 142, 715–722. [Google Scholar] [CrossRef]
- Tyutyunnykova, A.; Telegeev, G.; Dubrovska, A. The Controversial Role of Phospholipase C Epsilon (PLCε) in Cancer Development and Progression. J. Cancer 2017, 8, 716–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhou, C.; Qiu, G.; Yang, Y.; Yan, D.; Xing, T.; Fan, J.; Tang, H.; Peng, Z. Phospholipase C Epsilon Plays a Suppressive Role in Incidence of Colorectal Cancer. Med. Oncol. 2012, 29, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.; McCarthy, A.; Baxendale, R.; Guichard, S.; Magno, L.; Kessaris, N.; El-Bahrawy, M.; Yu, P.; Katan, M. Tumor Suppressor Role of Phospholipase Ce in Ras-Triggered Cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 4239–4244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, V.; Swigart, P.; Cheung, R.; Cockcroft, S.; Katan, M. Regulation of Inositol Lipid-Specific Phospholipase Cδ by Changes in Ca2+ Ion Concentrations. Biochem. J. 1997, 327, 545–552. [Google Scholar] [CrossRef]
- Thompson, J.L.; Shuttleworth, T.J. Orai Channel-Dependent Activation of Phospholipase C-δ: A Novel Mechanism for the Effects of Calcium Entry on Calcium Oscillations. J. Physiol. 2011, 589, 5057–5069. [Google Scholar] [CrossRef]
- Kim, Y.H.; Park, T.J.; Lee, Y.H.; Baek, K.J.; Suh, P.G.; Ryu, S.H.; Kim, K.T. Phospholipase C-Δ1 Is Activated by Capacitative Calcium Entry That Follows Phospholipase C-β Activation upon Bradykinin Stimulation. J. Biol. Chem. 1999, 274, 26127–26134. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Golebiewska, U.; D’amico, S.; Scarlata, S. The Small G Protein Rac1 Activates Phospholipase C1 through Phospholipase C2. J. Biol. Chem. 2010, 285, 24999–25008. [Google Scholar] [CrossRef] [Green Version]
- Shao, Q.; Luo, X.; Yang, D.; Wang, C.; Cheng, Q.; Xiang, T.; Ren, G. Phospholipase Cδ1 Suppresses Cell Migration and Invasion of Breast Cancer Cells by Modulating KIF3A-Mediated ERK1/2/β- Catenin/MMP7 Signalling. Oncotarget 2017, 8, 29056–29066. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Qin, Y.R.; Xie, D.; Hu, L.; Kwong, D.L.; Srivastava, G.; Sai, W.T.; Guan, X.Y. Characterization of a Novel Tumor-Suppressor Gene PLCδ1 at 3p22 in Esophageal Squamous Cell Carcinoma. Cancer Res. 2007, 67, 10720–10726. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, K.; Hirata, M.; Yamaguchi, H.; Nakamura, Y.; Fukami, K. Phospholipase Cδ3 Is a Novel Binding Partner of Myosin VI and Functions as Anchoring of Myosin VI on Plasma Membrane. Adv. Enzym. Regul. 2011, 51, 171–181. [Google Scholar] [CrossRef]
- Satow, R.; Hirano, T.; Batori, R.; Nakamura, T.; Murayama, Y.; Fukami, K. Phospholipase Cδ1 Induces E-Cadherin Expression and Suppresses Malignancy in Colorectal Cancer Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 13505–13510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Wen, J.; Lin, B.; Chen, H.; Bhandari, A.; Qi, Y.; Zheng, D.; Wang, O. Phospholipase C Delta 3 Inhibits Apoptosis and Promotes Proliferation, Migration, and Invasion of Thyroid Cancer Cells via Hippo Pathway. Acta Biochim. Biophys. Sin. 2021, 53, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Putney, J.W.; Tomita, T. Phospholipase C Signaling and Calcium Influx. Adv. Biol. Regul. 2012, 52, 152–164. [Google Scholar] [CrossRef] [Green Version]
- Gundlach, A.L.; Largent, B.L.; Snyder, S.H. 125I-Spiperone: A Novel Ligand for D2 Dopamine Receptors. Life Sci. 1984, 35, 1981–1988. [Google Scholar] [CrossRef]
- Im, D.; Inoue, A.; Fujiwara, T.; Nakane, T.; Yamanaka, Y.; Uemura, T.; Mori, C.; Shiimura, Y.; Kimura, K.T.; Asada, H.; et al. Structure of the Dopamine D2 Receptor in Complex with the Antipsychotic Drug Spiperone. Nat. Commun. 2020, 11, 6442. [Google Scholar] [CrossRef] [PubMed]
- Kondej, M.; Stępnicki, P.; Kaczor, A.A. Multi-Target Approach for Drug Discovery against Schizophrenia. Int. J. Mol. Sci. 2018, 19, 3105. [Google Scholar] [CrossRef] [Green Version]
- Cobos, E.; Entrena, J.M.; Nieto, F.R.; Cendán, C.M.; Del Pozo, E. Pharmacology and Therapeutic Potential of Sigma1 Receptor Ligands. Curr. Neuropharmacol. 2008, 6, 344–366. [Google Scholar] [CrossRef]
- Aringhieri, S.; Carli, M.; Kolachalam, S.; Verdesca, V.; Cini, E.; Rossi, M.; McCormick, P.J.; Corsini, G.U.; Maggio, R.; Scarselli, M. Molecular Targets of Atypical Antipsychotics: From Mechanism of Action to Clinical Differences. Pharmacol. Ther. 2018, 192, 20–41. [Google Scholar] [CrossRef]
- Hille, B.; Dickson, E.J.; Kruse, M.; Vivas, O.; Suh, B.C. Phosphoinositides Regulate Ion Channels. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2015, 1851, 844–856. [Google Scholar] [CrossRef]
- Telezhkin, V.; Reilly, J.M.; Thomass, A.M.; Tinkers, A.; Brown, D.A. Structural Requirements of Membrane Phospholipids for M-Type Potassium Channel Activation and Binding. J. Biol. Chem. 2012, 287, 10001–10012. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Bauer, C.S.; Zhen, X.-g.; Xie, C.; Yang, J. Dual Regulation of Voltage-Gated Calcium Channels by PtdIns(4,5)P2. Nature 2002, 419, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, H.A.T.; Leblanc, N.; Albert, A.P.; Greenwood, I.A. Inhibitory Role of Phosphatidylinositol 4,5-Bisphosphate on TMEM16A-Encoded Calcium-Activated Chloride Channels in Rat Pulmonary Artery. Br. J. Pharmacol. 2014, 171, 4311–4321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohacs, T. Phosphoinositide Regulation of TRP Channels. Handb. Exp. Pharmacol. 2014, 223, 1143–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Runnels, L.W.; Yue, L.; Clapham, D.E. The TRPM7 Channel Is Inactivated by PIP2 Hydrolysis. Nat. Cell Biol. 2002, 4, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Mahieu, F.; Prenen, J.; Janssens, A.; Owsianik, G.; Vennekens, R.; Voets, T. The Ca2+-Activated Cation Channel TRPM4 Is Regulated by Phosphatidylinositol 4,5-Biphosphate. EMBO J. 2006, 25, 467–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Cha, S.K.; Sun, T.J.; Huang, C.L. PIP2 Activates TRPV5 and Releases Its Inhibition by Intracellular Mg2+. J. Gen. Physiol. 2005, 126, 439–451. [Google Scholar] [CrossRef] [Green Version]
- Haustrate, A.; Prevarskaya, N.; Lehen’kyi, V. Role of the TRPV Channels in the Endoplasmic Reticulum Calcium Homeostasis. Cells 2020, 9, 317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stock, K.; Kumar, J.; Synowitz, M.; Petrosino, S.; Imperatore, R.; Smith, E.S.J.; Wend, P.; Purfürst, B.; Nuber, U.A.; Gurok, U.; et al. Neural Precursor Cells Induce Cell Death of High-Grade Astrocytomas through Stimulation of TRPV1. Nat. Med. 2012, 18, 1232–1238. [Google Scholar] [CrossRef] [Green Version]
- Ip, S.W.; Lan, S.H.; Lu, H.F.; Huang, A.C.; Yang, J.S.; Lin, J.P.; Huang, H.Y.; Lien, J.C.; Ho, C.C.; Chiu, C.F.; et al. Capsaicin Mediates Apoptosis in Human Nasopharyngeal Carcinoma NPC-TW 039 Cells through Mitochondrial Depolarization and Endoplasmic Reticulum Stress. Hum. Exp. Toxicol. 2012, 31, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Wisnoskey, B.J.; Sinkins, W.G.; Schilling, W.P. Activation of Vanilloid Receptor Type I in the Endoplasmic Reticulum Fails to Activate Store-Operated Ca2+ Entry. Biochem. J. 2003, 372, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Albarran, L.; Berna-Erro, A.; Dionisio, N.; Redondo, P.C.; Lopez, E.; Lopez, J.J.; Salido, G.M.; Brull Sabate, J.M.; Rosado, J.A. TRPC6 Participates in the Regulation of Cytosolic Basal Calcium Concentration in Murine Resting Platelets. Biochim. Biophys. Acta-Mol. Cell Res. 2014, 1843, 789–796. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Okawa, H.; Wang, Y.; Liman, E.R. Phosphatidylinositol 4,5-Bisphosphate Rescues TRPM4 Channels from Desensitization. J. Biol. Chem. 2005, 280, 39185–39192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, J.; Sun, B.; Du, J.; Yang, W.; Chen, H.C.; Overton, J.D.; Runnels, L.W.; Yue, L. Phosphatidylinositol 4,5-Bisphosphate (PIP 2) Controls Magnesium Gatekeeper TRPM6 Activity. Sci. Rep. 2011, 1, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bong, A.H.L.; Monteith, G.R. Calcium Signaling and the Therapeutic Targeting of Cancer Cells. Biochim. Biophys. Acta-Mol. Cell Res. 2018, 1865, 1786–1794. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of Cell Death: The Calcium-Apoptosis Link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Romero-Garcia, S.; Prado-Garcia, H. Mitochondrial Calcium: Transport and Modulation of Cellular Processes in Homeostasis and Cancer (Review). Int. J. Oncol. 2019, 54, 1155–1167. [Google Scholar] [CrossRef] [Green Version]
- Bagur, R.; Rgy Hajnó Czky, G. Molecular Cell Review Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [Green Version]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER–Mitochondrial Coupling Promotes Mitochondrial Respiration and Bioenergetics during Early Phases of ER Stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef] [Green Version]
- Hammadi, M.; Oulidi, A.; Gackière, F.; Katsogiannou, M.; Slomianny, C.; Roudbaraki, M.; Dewailly, E.; Delcourt, P.; Lepage, G.; Lotteau, S.; et al. Modulation of ER Stress and Apoptosis by Endoplasmic Reticulum Calcium Leak via Translocon during Unfolded Protein Response: Involvement of GRP78. FASEB J. 2013, 27, 1600–1609. [Google Scholar] [CrossRef]
- Zhai, X.; Sterea, A.M.; El Hiani, Y. Lessons from the Endoplasmic Reticulum Ca2+ Transporters-A Cancer Connection. Cells 2020, 9, 1536. [Google Scholar] [CrossRef]
- Giorgi, C.; Bonora, M.; Sorrentino, G.; Missiroli, S.; Poletti, F.; Suski, J.M.; Ramirez, F.G.; Rizzuto, R.; Di Virgilio, F.; Zito, E.; et al. P53 at the Endoplasmic Reticulum Regulates Apoptosis in a Ca2+-Dependent Manner. Proc. Natl. Acad. Sci. USA 2015, 112, 1779–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca2+ Homeostasis and Endoplasmic Reticulum (ER) Stress: An Integrated View of Calcium Signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Coe, H.; Michalak, M. Calcium Binding Chaperones of the Endoplasmic Reticulum. Gen. Physiol. Biophys. 2009, 28, 96–103. [Google Scholar]
- Chan, S.L.; Fu, W.; Zhang, P.; Cheng, A.; Lee, J.; Kokame, K.; Mattson, M.P. Herp Stabilizes Neuronal Ca2+ Homeostasis and Mitochondrial Function during Endoplasmic Reticulum Stress. J. Biol. Chem. 2004, 279, 28733–28743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wang, Q.C.; Sun, Z.; Li, T.; Yang, K.; An, C.; Guo, C.; Tang, T.S. ER Stress Mediated Degradation of Diacylglycerol Acyltransferase Impairs Mitochondrial Functions in TMCO1 Deficient Cells. Biochem. Biophys. Res. Commun. 2019, 512, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Groenendyk, J.; Szabo, E.; Gold, L.I.; Opas, M. Calreticulin, a Multi-Process Calcium-Buffering Chaperone of the Endoplasmic Reticulum. Biochem. J. 2009, 417, 651–666. [Google Scholar] [CrossRef]
- Prell, T.; Lautenschläger, J.; Grosskreutz, J. Calcium-Dependent Protein Folding in Amyotrophic Lateral Sclerosis. Cell Calcium 2013, 54, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Eletto, D.; Dersh, D.; Argon, Y. GRP94 in ER Quality Control and Stress Responses. Semin. Cell Dev. Biol. 2010, 21, 479–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turano, C.; Gaucci, E.; Grillo, C.; Chichiarelli, S. ERp57/GRP58: A Protein with Multiple Functions. Cell. Mol. Biol. Lett. 2011, 16, 539–563. [Google Scholar] [CrossRef]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic Reticulum Stress Signalling—From Basic Mechanisms to Clinical Applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of Endoplasmic Reticulum Stress-Induced Apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2019, 9, 3083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabariès, S.; Annis, M.G.; Lazaris, A.; Petrillo, S.K.; Huxham, J.; Abdellatif, A.; Palmieri, V.; Chabot, J.; Johnson, R.M.; Van Laere, S.; et al. Claudin-2 Promotes Colorectal Cancer Liver Metastasis and Is a Biomarker of the Replacement Type Growth Pattern. Commun. Biol. 2021, 4, 657. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Pan, X.; Hu, X.; Zhang, Y.-H.; Wang, S.; Huang, T.; Cai, Y.-D. Gene Expression Differences among Different MSI Statuses in Colorectal Cancer. Int. J. Cancer 2018, 143, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.B.; Dias, S.M.G.; Dang, C.V.; et al. Targeting Mitochondrial Glutaminase Activity Inhibits Oncogenic Transformation. Cancer Cell 2010, 18, 207–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- So, J.S.; Hur, K.Y.; Tarrio, M.; Ruda, V.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Lichtman, A.H.; Iwawaki, T.; Glimcher, L.H.; et al. Silencing of Lipid Metabolism Genes through Ire1α-Mediated Mrna Decay Lowers Plasma Lipids in Mice. Cell Metab. 2012, 16, 487–499. [Google Scholar] [CrossRef] [Green Version]
- Giussani, P.; Tringali, C.; Riboni, L.; Viani, P.; Venerando, B. Sphingolipids: Key Regulators of Apoptosis and Pivotal Players in Cancer Drug Resistance. Int. J. Mol. Sci. 2014, 15, 4356–4392. [Google Scholar] [CrossRef] [Green Version]
- del Solar, V.; Lizardo, D.Y.; Li, N.; Hurst, J.J.; Brais, C.J.; Ekin, G.; Correspondence, A.-G.; Del Solar, V.; Ekin Atilla-Gokcumen, G. Differential Regulation of Specific Sphingolipids in Colon Cancer Cells during Staurosporine-Induced Apoptosis. Chem. Biol. 2015, 22, 1662–1670. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of Bioactive Lipid Signalling: Lessons from Sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Young, M.M.; Kester, M.; Wang, H.-G. Sphingolipids: Regulators of Crosstalk between Apoptosis and Autophagy. J. Lipid Res. 2013, 54, 5–19. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Guardiola, P.; Casas, J.; Megías-Roda, E.; Solé, S.; Perez-Montoyo, H.; Yeste-Velasco, M.; Erazo, T.; Diéguez-Martínez, N.; Espinosa-Gil, S.; Muñoz-Pinedo, C.; et al. The Anti-Cancer Drug ABTL0812 Induces ER Stress-Mediated Cytotoxic Autophagy by Increasing Dihydroceramide Levels in Cancer Cells. Autophagy 2021, 17, 1349–1366. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Maurer, B.J.; Liu, Y.Y.; Wang, E.; Allegood, J.C.; Kelly, S.; Symolon, H.; Liu, Y.; Merrill, A.H.; Gouazé-Andersson, V.; et al. N-(4-Hydroxyphenyl)Retinamide Increases Dihydroceramide and Synergizes with Dimethylsphingosine to Enhance Cancer Cell Killing. Mol. Cancer Ther. 2008, 7, 2967–2976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.; Rao, X.; Kim, C.Y.; Freiser, H.; Zhang, Q.; Jiang, Z.; Li, G. Gamma-Tocotrienol Induces Apoptosis and Autophagy in Prostate Cancer Cells by Increasing Intracellular Dihydrosphingosine and Dihydroceramide. Int. J. Cancer 2012, 130, 685–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morad, S.A.F.; Cabot, M.C. Ceramide-Orchestrated Signalling in Cancer Cells. Nat. Rev. Cancer 2013, 13, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, M.; Ogretmen, B. The Role of Ceramide Metabolism and Signaling in the Regulation of Mitophagy and Cancer Therapy. Cancers 2021, 13, 2475. [Google Scholar] [CrossRef]
- Stiban, J.; Caputo, L.; Colombini, M. Ceramide Synthesis in the Endoplasmic Reticulum Can Permeabilize Mitochondria to Proapoptotic Proteins. J. Lipid Res. 2008, 49, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Hendrich, A.; Michalak, K. Lipids as a Target for Drugs Modulating Multidrug Resistance of Cancer Cells. Curr. Drug Targets 2005, 4, 23–30. [Google Scholar] [CrossRef]
- Barceló-Coblijn, G.; Martin, M.L.; De Almeida, R.F.M.; Antònia Noguera-Salvà, M.; Marcilla-Etxenike, A.; Guardiola-Serrano, F.; Lüth, A.; Kleuser, B.; Halver, J.E.; Escribá, P.V. Sphingomyelin and Sphingomyelin Synthase (SMS) in the Malignant Transformation of Glioma Cells and in 2-Hydroxyoleic Acid Therapy. Proc. Natl. Acad. Sci. USA 2011, 108, 19569–19574. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Park, D.W.; Stigler, W.S.; Creighton, J.; Ravi, S.; Darley-Usmar, V.; Zmijewski, J.W. Mitochondria and AMP-Activated Protein Kinase-Dependent Mechanism of Efferocytosis. J. Biol. Chem. 2013, 288, 26013–26026. [Google Scholar] [CrossRef] [Green Version]
- Lemasters, J.J. Variants of Mitochondrial Autophagy: Types 1 and 2 Mitophagy and Micromitophagy (Type 3). Redox Biol. 2014, 2, 749–754. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zhang, J.; Liu, C.; Wang, Q.; Yan, J.; Hui, L.; Jia, Q.; Shan, H.; Tao, L.; Zhang, M. The Role of Mitophagy in Regulating Cell Death. Oxid. Med. Cell. Longev. 2021, 2021, 6617256. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, M.; Cocco, T.; Lorusso, M. Ceramide Interaction with the Respiratory Chain of Heart Mitochondria. Biochemistry 2000, 39, 6660–6668. [Google Scholar] [CrossRef] [PubMed]
- Delierneux, C.; Kouba, S.; Shanmughapriya, S.; Potier-Cartereau, M.; Trebak, M.; Hempel, N. Mitochondrial Calcium Regulation of Redox Signaling in Cancer. Cells 2020, 9, 432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassi, S.; Chiricozzi, E.; Mauri, L.; Sonnino, S.; Prinetti, A. Sphingolipids and Neuronal Degeneration in Lysosomal Storage Disorders. J. Neurochem. 2019, 148, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Fugio, L.B.; Coeli-Lacchini, F.B.; Leopoldino, A.M. Sphingolipids and Mitochondrial Dynamic. Cells 2020, 9, 581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and Their Metabolism in Physiology and Disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Van Meer, G.; Lisman, Q. Sphingolipid Transport: Rafts and Translocators. J. Biol. Chem. 2002, 277, 25855–25858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridevi, P.; Alexander, H.; Laviad, E.L.; Min, J.; Mesika, A.; Hannink, M.; Futerman, A.H.; Alexander, S. Stress-Induced ER to Golgi Translocation of Ceramide Synthase 1 Is Dependent on Proteasomal Processing. Exp. Cell Res. 2010, 316, 78–91. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene Symbol | Sense | Antisense |
|---|---|---|
| PLCB1 | GGACUUACGUGGAAGUAGAtt | UCUACUUCCACGUAAGUCCca |
| PLCB3 | CGUCCUUUGUGGAGACCAAtt | UUGGUCUCCACAAAGGACGac |
| PLCG1 | GGGUGAAAAAGAUCCGUGAtt | UCACGGAUCUUUUUCACCCag |
| PLCD3 | GGUUUGUGGUGGAAGAUUAtt | UAAUCUUCCACCACAAACCgg |
| PLCE1 | GCAGGAAAAUUCAUCCUUAtt | UAAGGAUGAAUUUUCCUGCac |
| DDIT3 | GUAGUGAAUUGAUCUAGAUtt | AUCUAGAUCAAUUCACUACca |
| Target Gene | Forward | Reverse |
|---|---|---|
| CDKN1A | CCTCATCCCGTGTTCTCCTTT | GTACCACCCAGCGGACAAGT |
| CXCR4 | CAGCAGGTAGCAAAGTGACG | ATAGTCCCCTGAGCCCATTT |
| CHOP | CATCACCACACCTGAAAGCA | TCAGCTGCCATCTCTGCAG |
| DGAT2 | AAAGAATGGGAGTGGCAATG | TCCTCGAAGATCACCTGCTT |
| DR5 | GAGCTAAGTCCCTGCACCAC | AATCACCGACCTTGACCATC |
| PIF2 | CCCTGGATTGTGTGGAGATT | ACTCCAGACTGAGGCTCCTG |
| GADD34 | CTCAAGCGCCCAGAAACC | CTCCTGGGCCTGGGTGAT |
| GRP78 | GTTCTTGCCGTTCAAGGTGG | TGGTACAGTAACAACTGCATG |
| GUSB | ATCGCCATCAACAACAC | CTTGGGATACTTGGAGGTG |
| HPRT | AAGGACCCCACGAAGTGTTG | GGCTTTGTATTTTGCTTTTCC |
| PLCB1 | GAGGCTAGAAGAAGCGCAAA | ATTGCTGTCTTCACTGATCTTTCCT |
| PLCB3 | GCCTCAGAAGTCTCTGGGTG | GGACATCTCCTCAGTGGCAT |
| PLCG1 | TGTCCCACAGACCAACGC | ATTCCGCTTCCGCACCAG |
| PLCE1 | ACGTCTGTCAGAAGCCCAGT | GCTTTAAGCATGGACCAACG |
| PLCD3 | CCAGAACCACTCTCAGCATCCA | TTGAAGCCATTGTTGAGCAC |
| Cell Line | IC50 72 h (95% CI) (μmol/L) |
|---|---|
| HCT116 | 7.10 (6.61–7.64) |
| HCT8 | 5.26 (4.83–5.72) |
| SW620 | 9.23 (8.55–9.97) |
| SW480 | 9.87 (8.97–10.9) |
| CRC-SC#1 | 3.76 (3.38–4.18) |
| CRC-SC#2 | 8.14 (7.38–8.98) |
| CRC-SC#3 | 4.13 (3.72–4.58) |
| CRC-SC#4 | 3.51 (3.16–3.92) |
| dd-HCT116 1 | 5.62 (5.17–6.12) |
| PBMC | 31.6 (30.3–32.9) |
| vAT-MSC | 61.7 (53.3–71.4) |
| hDF1 | 98.0 (85.2–113) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antona, A.; Varalda, M.; Roy, K.; Favero, F.; Mazzucco, E.; Zuccalà, M.; Leo, G.; Soggia, G.; Bettio, V.; Tosi, M.; et al. Dissecting the Mechanism of Action of Spiperone—A Candidate for Drug Repurposing for Colorectal Cancer. Cancers 2022, 14, 776. https://doi.org/10.3390/cancers14030776
Antona A, Varalda M, Roy K, Favero F, Mazzucco E, Zuccalà M, Leo G, Soggia G, Bettio V, Tosi M, et al. Dissecting the Mechanism of Action of Spiperone—A Candidate for Drug Repurposing for Colorectal Cancer. Cancers. 2022; 14(3):776. https://doi.org/10.3390/cancers14030776
Chicago/Turabian StyleAntona, Annamaria, Marco Varalda, Konkonika Roy, Francesco Favero, Eleonora Mazzucco, Miriam Zuccalà, Giovanni Leo, Giulia Soggia, Valentina Bettio, Martina Tosi, and et al. 2022. "Dissecting the Mechanism of Action of Spiperone—A Candidate for Drug Repurposing for Colorectal Cancer" Cancers 14, no. 3: 776. https://doi.org/10.3390/cancers14030776
APA StyleAntona, A., Varalda, M., Roy, K., Favero, F., Mazzucco, E., Zuccalà, M., Leo, G., Soggia, G., Bettio, V., Tosi, M., Gaggianesi, M., Riva, B., Reano, S., Genazzani, A., Manfredi, M., Stassi, G., Corà, D., D’Alfonso, S., & Capello, D. (2022). Dissecting the Mechanism of Action of Spiperone—A Candidate for Drug Repurposing for Colorectal Cancer. Cancers, 14(3), 776. https://doi.org/10.3390/cancers14030776